
Abstract
Our initial understanding of immune-regulatory cells was based on the discovery of suppressor cells that assure peripheral T-cell tolerance and promote immune homeostasis. Research has particularly focused on the importance of regulatory T cells (Tregs) for immune modulation, e.g. directing host responses to tumours or inhibiting autoimmunity development. However, recent studies report the discovery of self-reactive pro-inflammatory T cells—termed anti-regulatory T cells (anti-Tregs)—that target immune-suppressive cells. Thus, regulatory cells can now be defined as both cells that suppress immune reactions as well as effector cells that counteract the effects of suppressor cells and support immune reactions. Self-reactive anti-Tregs have been described that specifically recognize human leukocyte antigen-restricted epitopes derived from proteins that are normally expressed by regulatory immune cells, including indoleamine 2,3-dioxygenase (IDO), tryptophan 2,6-dioxygenase (TDO), programmed death-ligand 1 (PD-L1), and forkhead box P3 (Foxp3). These proteins are highly expressed in professional antigen-presenting cells under various physiological conditions, such as inflammation and stress. Therefore, self-reactive T cells that recognize such targets may be activated due to the strong activation signal given by their cognate targets. The current review describes the existing knowledge regarding these self-reactive anti-Tregs, providing examples of antigen-specific anti-Tregs and discussing their possible roles in immune homeostasis and their potential future clinical applications.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In 1974, the Danish immunologist Niels Jerne proposed that the immune system functions as a network regulated via interactions between the variable parts of lymphocytes and their secreted molecules—a theory that earned him the Nobel prize in 1984. Jerne believed that the variable region (the idiotype) of the B-cell or T-cell receptor was recognizable by other lymphocytes (anti-idiotypes), which were in turn recognizable by other lymphocytes and so on. Although many parts of his model were incorrect or oversimplified, he was the first to determine that the immune system relies on a tightly regulated set of cells and molecules that recognize each other even in the absence of foreign antigens. It is now clear that the immune system comprises a complex network of cells that react towards each other to preserve the organism’s integrity while eliminating all elements that are deemed dangerous. Various regulatory mechanisms function to terminate immune responses to antigens, to return the immune system to a basal state after clearing an antigen, and to maintain unresponsiveness (e.g. tolerance) to self-antigens.
The realization that T cells could assist in antibody responses led to the obverse finding that T cells could also inhibit these responses. Recognition of suppressor T cells, now called regulatory T cells (Tregs), highlighted the importance of their functions in maintaining immunological self-tolerance and immune homeostasis. Tregs assure peripheral T-cell tolerance and downregulate immune responses under various inflammatory circumstances [1]. Accordingly, the Treg deficiency and/or altered function can lead to autoimmunity [2]. Tregs suppress effector T cell activity in many different ways [3], including through cell surface expression of inhibitory molecules (e.g. CTLA-4), as by the production of immunosuppressive cytokines (e.g. IL-10 and TGF-β) [4–6]. Importantly, Tregs are not the only regulatory cells responsible for immune suppression. Several cell types participate in the elaborate network of central and peripheral tolerance mechanisms under physiological conditions, including regulatory B cells, dendritic cell subtypes, and myeloid-derived suppressor cells (MDSCs).
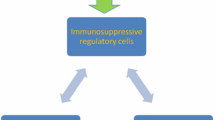
We recently reported that self-reactive pro-inflammatory T cells can specifically target immune-suppressive cells in the periphery, suggesting that the immune system includes mechanisms to counteract the immune-suppressive feedback signals mediated by regulatory cells. We have identified self-reactive T cells that recognized human leukocyte antigen (HLA)-restricted epitopes derived from proteins normally expressed by regulatory immune cells—including indoleamine 2,3-dioxygenase (IDO), tryptophan 2,6-dioxygenase (TDO), programmed death-ligand 1 (PD-L1), and forkhead box P3 (Foxp3). Since these T cells can react against regulatory immune cells, I have proposed that these cells should be termed anti-regulatory T cells (anti-Tregs) [7]. Such anti-Tregs may serve to directly suppress the function of regulatory immune cells within immune-regulatory networks, potentially “supporting” effector T cells by directly eliminating regulatory cells. Anti-Tregs may further assist T cells by secreting pro-inflammatory cytokines at inflammation sites (Fig. 1).

Anti-Tregs target cancer cells as well as immune-regulatory cells. Effector T cells are able to recognize HLA-restricted epitopes (red) derived from mutated or overexpressed proteins in tumour cells. Regulatory immune cells (red), e.g. regulatory T cells, different dendritic cell subtypes, myeloid derived suppressor cells, and M2 macrophages in contrast to control or terminate immune responses. The regulatory arm secures the unresponsiveness or tolerance to self-antigens. Regulatory immune cells suppress immunity through a number of different cellular and extracellular factors (red arrows), including the stimulation of inhibitory T-cell pathways (e.g., PD-1 and CTLA-4); the release of immune-suppressive cytokines, like TGF-β and IL-10; and the expression metabolic enzymes, like IDO and arginase. These immune-tolerance mechanisms may also be exploited by cancer cells to achieve immune escape, which becomes more pronounced with disease progression. Hence, many of the mechanisms considered helpful in autoimmune settings are used by tumours to suppress immune responses towards malignant cells in cancerous settings. Thus, cancer cells (a) as well as other regulatory immune cells (b) (e.g., tumour-associated dendritic cells and MDSCs) express checkpoint inhibitors (e.g. PD-L1), inhibitory cytokines, and metabolic enzymes (e.g. IDO) that restrain the anti-tumor activity of anti-tumour specific T cells in the tumour microenvironment. Specific anti-Tregs recognizing HLA-restricted derived epitopes (yellow), which are generated from intracellular degraded antigens, are able to eliminate (black arrows) regulatory immune cells as well as cancer cells. Anti-Tregs can promote local immune suppression by the secretion of effector cytokines (blue arrows) or by directly eliminating regulatory immune cells (black arrows). It must be assumed that anti-Tregs themselves are hampered by the suppressive effects of their targets (red arrows). Hence, under normal physiological conditions, equilibrium between immune activation and suppression may indeed be necessary to maintain immune homeostasis. The role of self-reactive effector and suppressor cells in immune-regulatory networks may thus be miscellaneous. The activation of anti-Tregs by vaccination may directly target immune inhibitory pathways in the tumour microenvironment, modulate immune regulation, and potentially alter tolerance to tumour antigens. Because immune-suppressive cells might antagonize the desired effects of therapeutic cancer vaccines, the addition of anti-Treg antigens would consequently be easily implementable and highly synergistic
Self-reactive T cells that can recognize disease-causing T cells could be a mechanism of limiting autoimmune reactions and may be involved in recovery from antigen-induced autoimmune disease [8]. New findings support the view of regulatory T cells as having both suppressor and effector capabilities. Presumably, self-reactive effector T cells (i.e. anti-Tregs) may contribute to immune homeostasis, representing yet another cellular subtype that participates in the network that supports immune system function without creating pathogenic autoimmune conditions. The present review will describe how anti-Treg activation can strongly influence immune reactions through both direct and indirect mechanisms.
Antigen-specific anti-Tregs
Metabolic enzymes
Altered tumour metabolism results in essential nutrient depletion and may lead to accumulation of immune-suppressive metabolites. l-tryptophan is an essential amino acid required for protein synthesis, and tryptophan metabolism clearly influences immune responses. T cells sense low tryptophan levels via the serine/threonine-protein kinase GCN2, triggering proliferative arrest [9]. Such enzymatic immunosuppressive effects are mediated through local tryptophan depletion, as well as by direct immune-suppressive tryptophan metabolites [10].
The heme dioxygenases IDO, IDO2, and TDO catalyse the degradation of l- and d-tryptophan to N-formylkynurenine. IDO is upregulated by inflammatory cytokines, such as type I and II interferons, and is thus considered an important counter-regulatory enzyme in controlling disproportionate immune responses [11]. By distinct mechanisms, TDO and IDO each catalyse the first and rate-limiting step of tryptophan oxidation, yielding kynurenine [12]. IDO expression has been repeatedly described in cancer [11, 13], and IDO-transfected tumour cells become resistant to immune eradication [14], making IDO the focus of much research [15]. IDO also exerts effects mediated through enhancement of local Treg-mediated immune suppression. Constitutive IDO expression in DCs attracts T cells with regulatory properties [11]. We recently described spontaneous T cell-mediated immune reactivity against IDO, IDO2, and TDO in cancer patients [16–21]. Moreover, direct ex vivo assays revealed spontaneous T-cell reactivity towards IDO in patients suffering from unrelated tumour types, i.e. renal cell carcinoma, melanoma, and breast cancer. Such IDO-reactive T cells effectively lysed IDO+ cancer cell lines of different origin, including directly ex vivo enriched leukaemia cells. IDO2- and TDO-specific T cells also recognize malignant cells of different origin. IDO-driven immune suppression is a common mechanism that has been described in a diversity of human cancers, and anti-IDO immune responses also seem relevant in cancers of unrelated origin. We further found that IDO-specific cytotoxic T lymphocytes (CTLs) recognized and killed IDO+ dendritic cells, demonstrating their ability to react to non-malignant immune cells.
Interestingly, we have also detected circulating IDO-specific T cells in healthy donors, although not as frequently as in patients with cancer [17, 19, 21]. Moreover, we have identified specific reactivity towards IDO2 and TDO, which are also involved in tryptophan catabolism in healthy individuals. Unlike IDO-specific immunity, TDO-/IDO2-specific immunity is as frequent in healthy donors as in patients with malignant disease [17, 19]. Additionally, the functional phenotype of TDO-specific T-cell responses differs depending on the condition of the host. In healthy subjects, these cells predominately comprise Th1 cells that produce interferon (IFN)-γ and tumour necrosis factor (TNF)-α. On the other hand, cancer patients show greater prevalence of TDO-reactive CD4+ T-cells that release not only IFNγ and TNFα but also interleukin (IL)-17 and IL-10 in response to TDO-derived class II HLA-restricted peptides. Hence, healthy donors show a predominant Th1 helper response, while CD4+ T-cell responses in cancer patients are skewed towards a Treg response.
Notably, IDO was recently reported to be a crucial factor in the MDSC-mediated suppression of anti-tumour immune responses [22, 23], suggesting that therapeutic immunological targeting of IDO could also suppress the effects of MDSCs. MDSCs inhibit the activation, proliferation, and cytotoxicity of effector T cells and natural killer cells, as well as induce Treg differentiation and expansion. Both cancer cells and MDSCs can suppress T cells by manipulating l-arginine metabolism via the enzymes nitric oxide synthase (NOS) and arginase. Many tumours exhibit increased expressions of arginase and inducible NOS (iNOS), leading to arginine depletion from the tumour microenvironment [24]. Several studies emphasize the importance of this altered tumour arginine metabolism in the suppression of tumour-specific T-cell responses, and it was recently demonstrated that AML blasts show an arginase-dependent ability to inhibit T-cell proliferation and haematopoietic stem cells. Furthermore, arginase and iNOS inhibitors reduce the suppressive activity of AML [25].
Checkpoint inhibitors
T-cell receptor co-stimulatory pathways regulate T-cell activation, playing a central role in maintaining immune system homeostasis. Members of the CD28 family of receptors—including CD28, CTLA-4, ICOS, and programmed death 1 (PD-1)—are key elements of the immunological synapse. Upon interaction with their corresponding ligands, these receptors can generate potent co-stimulatory or inhibitory signals in T cells.
PD-1 is a vital regulatory molecule that delivers inhibitory signals to T cells, making them functionally silent against their antigens. The PD-1 ligands PD-L1 (B7-H1) and PD-L2 (B7-H2) are expressed on antigen-presenting cells (APCs), placental cells, and non-haematopoietic cells found in inflammatory microenvironments, and expressions of these ligands can be induced by interferons. PD-1 and its ligands play central roles in creating the immune inhibitory tumour microenvironment that protects cancer cells from immune-cell-mediated death. Notably, PD-L1 helps protect malignant cells from immune destruction and is expressed by cancer cells in many different malignancies [26–34]. Blockade of either PD-1 or PD-L1 using monoclonal antibodies has produced outstanding clinical responses [35, 36], and the FDA approved the anti-PD-1 antibodies pembrolizumab and nivolumab in September and December of 2014, respectively.
Our group was the first to describe spontaneous CD8+ and CD4+ T-cell reactivity against PD-L1 in peripheral blood from both patients with various cancers and healthy donors [37, 38]. PD-L1-specific anti-Tregs can recognize PD-L1-expressing non-malignant cells in a PD-L1-dependent manner, representing yet another example of the immune system’s ability to directly react to the immune-suppressive mechanisms of cancerous cells. We additionally found that PD-L1-specific anti-Tregs can kill PD-L1-expressing melanoma cells and cutaneous T-cell lymphoma cells [8–12], and Minami et al. described the lysis of PD-L1+ HLA-A24+ renal carcinoma cells by HLA-A24-restricted PD-L1-specific T cells [39]. Notably, humeral recognition of PD-L1 has also been described in rheumatoid arthritis [40].
Forkhead box P3
Foxp3 expression is strongly associated with Tregs [1]. Gilboa and colleagues demonstrated that mice vaccinated against FoxP3 showed a FoxP3-specific T-cell response that eliminated FoxP3+ Tregs and enhanced antitumor immunity [41]. Similarly, a FoxP3-specific T-cell response in an atherosclerosis model led to a substantial decrease in the number of FoxP3+ Tregs, with correspondingly increased atherosclerotic lesion formation [42]. We recently reported that humans also show natural CD8 reactivity towards FoxP3 [43]. FoxP3-specific anti-Tregs recognized Tregs and killed malignant T cells expressing high FoxP3 levels, suggesting that vaccination against FoxP3 could be useful in lymphoma patients with FoxP3+ malignant T cells.
Chemokine CCL22
CCL22 secretion by tumour cells and tumour-associated macrophages attracts and recruits Tregs to the tumour microenvironment, leading to suppression of anti-cancer immunity [44, 45]. Solid tumour CCL22 production reportedly causes Treg accumulation in many cancers, including ovarian, prostate, oesophageal, gastric, and breast carcinomas and glioblastomas [46, 47]. On the other hand, tumours lacking CCL22 expression are not infiltrated by Tregs, even when producing other CCR4-binding chemokines (e.g. CCL17)—suggesting that Tregs are recruited to the tumour environment via the CCL22/CCR4 axis [46]. We recently demonstrated that specific T cells can target CCL22-expressing cells [48]. Analysis of the CCL22 protein signal sequence revealed an HLA-A2-restricted peptide epitope, which we used for in vitro stimulation of peripheral blood mononuclear cells (PBMCs) to expand CCL22-specific T-cell populations. Even though CCL22 is secreted out of the cell, CCL22-expressing cells can be identified by T cells that recognize an epitope derived from the CCL22 signal peptide. CCL22-specific T cells recognized and killed CCL22-expressing solid cancer cells, including breast and colon cancer cells, and lysed acute monocytic leukaemia cells in a manner dependent on CCL22 expression. We identified spontaneous T-cell responses against the CCL22-derived epitope in both cancer patients and healthy donors. In cultures, addition or activation of CCL22-specific T cells decreases the CCL22 level in the microenvironment. Hence, activating CCL22-specific T cells (e.g. by vaccination) could directly target cancer cells and tumour-associated macrophages, thereby modulating Treg recruitment into the tumour environment and augmenting anti-cancer immunity.
NF-kappa-B inhibitor alpha
Nuclear factor-κB (NF-κB) is constitutively active in most cancers and controls multiple cellular processes, including proliferation, invasion, and drug resistance. NF-κB is primarily regulated through associations with inhibitory proteins termed inhibitors of NF-κB (IκBs). Increased NF-κB activity in tumour cells is correlated with decreased stability of IκB proteins, particularly IκB-α. Following activation by a large number of inducers, IB proteins are degraded by the proteasome. In target cells, proteasomal degradation generates HLA-restricted antigenic peptides that are recognized by cytotoxic T cells.
We have detected naturally occurring IκB-α-specific T cells in peripheral blood samples from patients with unrelated tumour types [49], suggesting that the increased IκB-α proteasomal degradation in cancer induces IκB-α-specific cytotoxic T cells. NF-κB plays a major role in coordinating the expressions of a wide variety of genes that control immune responses, and influences various cells involved in both adaptive and innate immune responses. Accordingly, IκB-α-specific cytotoxic T cells may act as regulatory cells and play a more general role in inflammation processes and in immune system regulation. Constitutive NF-κB activation is additionally associated with multiple pathogenic settings other than cancer, including inflammatory diseases such as rheumatoid arthritis, inflammatory bowel disease, multiple sclerosis, and asthma. However, any potential role of IκB-α-reactive T cells in these settings has yet to be explored.
Anti-Tregs expand at sites of inflammation
Immunologists generally believe that the host immune system avoids self-reactive T cells via thymic selection, which prevents autoimmune reactions. Long-standing hypotheses have proposed that self-tolerance is maintained through clonal deletion of self-reactive T cells harbouring T-cell receptors with high affinity to a target/HLA complex [50]. However, recent evidence suggests that thymic selection only prunes self-reactive T-cell clones rather than eliminating them. Upon activation in the periphery, self-reactive T cells show a lower activation profile compared to foreign-reactive T cells; however, blood samples from healthy humans contain self-peptide-specific CD8 T cells at frequencies similar to those specific for non-self-antigens.
Professional APCs highly express proteins such as PD-L1 and IDO, which are induced by interferons expressed at inflammation sites. To verify that interferons expand populations of PD-L1-specific anti-Tregs, we performed two subcutaneous IFNg injections in C57 mice. The mice were sacrificed 1 week later, and the spleens of the IFNg-treated mice showed a strong PD-L1-specific T-cell response. Similarly, 3 days of treating C57 mice with the allergen DNFB led to an influx (or expansion) of PD-L1-specific T cells at the inflammation site (unpublished data). We have also demonstrated that addition of known IDO inducers (e.g. IFN-γ and CpG oligodeoxynucleotides) leads to expansion of IDO-specific anti-Tregs among human PBMCs without additional stimulation [17]. This evidence confirms that anti-Tregs are activated and proliferate due to a strong activation signal from their cognate targets (i.e. professional APCs) at inflammation sites, which may explain the substantial reactivity observed in both cancer patients and healthy individuals.
Cytomegalovirus (CMV) is likely one of the most immune-dominant antigens encountered by the human immune system [51]. CMV infection induces IDO expression in monocytes, which purportedly confers an advantage to CMV-infected monocytes with regards to escaping T-cell responses [52]. Notably, the presence of IDO-specific anti-Tregs correlates to the presence of CMV responses [19], suggesting that IDO-specific anti-Treg cell responses may develop as support in cases of constitutive anti-CMV T-cell responses.
It was recently suggested that immune surveillance may involve T cells specific for the self-protein OCT4, which were found to be reduced in patients with germ-cell cancers compared to healthy individuals. Interestingly, patients with immunodeficiencies show increased risk of developing these cancers [53]. Furthermore, chemotherapy reportedly leads to induction of anti-OCT4 immunity in patients with cancer. However, compared to healthy individuals, we found that patients with cancer showed increased levels of anti-Tregs that specifically recognized self-antigens expressed in immune cells. Specifically, IDO-, PD-L1-, and Foxp3-specific anti-Tregs were detected more frequently in patients with cancer than in healthy individuals. The frequency of TDO-specific T-cell reactivity was similar between healthy donors and patients with cancer. However, we have observed that a trend towards improved overall survival among patients with cancer who exhibited a TDO-specific IL-17 response and impaired survival in patients with IL-10-producing TDO-reactive CD4+ T cells [54].
Anti-Tregs impact immunity
To characterize the importance of anti-Tregs in immune reactions, we examined their effects on other adaptive immune cells. We demonstrated that IDO-specific CD8+ T cells can enhance other T-cell responses through their direct and indirect reactions to IDO+ cells. Tryptophan levels are elevated in the presence of IDO-specific T cells, directly corresponding to decreased IDO activity. Furthermore, IDO-specific T cells increase the overall production of pro-inflammatory cytokines (e.g. TNF-α and IL-6) while decreasing IL-10 production. Tryptophan metabolites are directly toxic to CD8+ T cells and CD4+ Th1 cells [55], but not to Th2 cells. Hence, increased IDO activity seems to incrementally direct helper T-cell polarization towards a Th2 phenotype [56]. In contrast, activation of IDO-specific cytotoxic T cells may shift the Th response in a Th1 direction. Moreover, kynurenine may effectively hamper immune responses via binding of the aryl hydrocarbon receptor, which favours local Treg formation. Overall, targeting of IDO-positive cells should decrease the number of Tregs. Indeed, the activation of IDO-specific T cells leads to a decreased frequency of Tregs. Notably, in a phase I clinical trial of IDO vaccination in patients with NCSLC, all treated patients showed a significantly reduced Treg population after the sixth vaccination [57]. Taken together, the data clearly suggest that activation of IDO-specific T cells influences adaptive immune responses by suppressing the effects of IDO activity [17].
To further characterize the effects of anti-Tregs on adaptive immune responses, we stimulated PBMCs in vitro with viral epitopes and then added PD-L1-specific anti-Tregs 1 week later. This resulted in an immense increase in the number of virus-specific CD8+ T cells [58]. Confirming these findings, we observed significantly increased numbers of virus-specific T cells in cultures that were co-stimulated with a known HLA-restricted PD-L1 peptide epitope compared to cultures co-stimulated with an irrelevant HIV epitope [59]. Hence, PD-L1-specific T cells may support the effector phase of an immune response by removing PD-L1-expressing regulatory immune cells that inhibit PD-1+ effector T cells. Supporting this possibility, we observed significantly increased T-cell reactivity towards a DC-based cancer vaccine following co-stimulation with long-peptide epitopes derived from PD-L1 [60]. While CD4+ T-cell reactivity increased the most, CD8+ T cell reactivity was also significantly boosted by co-stimulation with the PD-L1 epitope.
Tregs and anti-Tregs recognize similar targets
As discussed above, peripheral blood samples from both cancer patients and healthy donors contain CD4+ anti-Treg cells that release IFNγ, TNFα, and IL-17. Furthermore, the CD4+ anti-Treg cells from some donors directly suppressed IL-10 production in cultured PBMCs upon stimulation with their cognate antigens [19, 61]. Thus, IDO- and PD-L1-specific CD4+ anti-Tregs likely participate in immune-regulatory networks by acting as specific helper T cells to sustain immune response at sites of inflammation. However, the presently available data do not exclude the possibility that some CD4+ T-cells recognizing similar targets may act as immune-suppressive Tregs. Indeed, following stimulation with both PD-L1 and IDO peptides, we have detected IL-10, which is mainly released by Tregs. Therefore, the induction of specific CD4+ T cells may not always be beneficial in cases of cancer. Moreover, the presence of PD-L1 autoantibodies was first described in patients with rheumatoid arthritis [40]. These issues should obviously be considered when attempting to target PD-L1 in the clinical setting.
In the clearest example of the ability of Tregs to promote immunological tolerance, ablation of natural regulatory T cells (nTregs) leads to massive lymphoproliferation and myeloproliferation. It is assumed that nTregs are more responsive to self-antigens compared to naïve T cells. Although no self-peptide ligands for naturally occurring nTregs have yet been described [62], we have observed a very strong immune-suppressive effect exerted by naturally occurring HLA-A2-restricted CD8+ T cells specific for the anti-inflammatory molecule heme oxygenase-1 (HO-1) [63]. Since HO-1 is expressed in the late phase of inflammatory reactions, such cells likely contribute to the contraction phase of immune responses. It remains debatable whether T-cell receptor-mediated signals are relevant to Treg function. However, it is believed that Treg generation and maintenance requires the presence of specific target antigens [64]. Although it is established that at least some naturally occurring Tregs develop in the thymus [1], the signals within the thymus that confer lineage specificity during T-cell differentiation have not yet been fully determined.
Clinical applications
To evaluate the efficiency and safety of IDO-based vaccinations, we conducted a phase I first-in-man clinical vaccination trial. Fifteen patients with advanced non-small cell lung cancer (NSCLC) were vaccinated with an IDO-derived peptide in Montanide adjuvant (www.clinicaltrials.gov; NCT01219348) [57]. The median overall survival (OS) was >2 years, which was higher than expected for this patient group. Moreover, of the seven patients with stable disease (SD), six are still alive. One patient was classified as exhibiting an objective response (PR) after showing continuous tumour regression with 1 year of vaccine treatment. He and one additional patient are continuing to receive the vaccine every second month at 4 years after inclusion.
The tested vaccine comprised an HLA-A2-restricted epitope of IDO. In a subsequent study, we compared the vaccinated HLA-A2+ patients with the HLA-A2− patients from the intent-to-treat population who were excluded due to HLA type. Median OS was 25.9 months (778 days) among the vaccine-treated HLA-A2+ patients, and 7.7 months (237 days) in the HLA-A2− group of patients who did not receive the vaccine (P = 0.03). Importantly, a large study recently demonstrated that HLA-A2 was an unfavourable prognostic factor among stage I NSCLC patients [65], which is supported by data showing higher in situ T-cell infiltration among non-vaccinated patients compared to vaccinated patients prior to inclusion (Gerome Galon, personal communication). This highlights the potential importance of the significantly longer OS observed in vaccinated HLA-A2+ NSCLC patients compared to unvaccinated HLA-A2− NSCLC patients, although these data remain to be confirmed in larger clinical trials. At our centre at Herlev, we are currently initiating several clinical trials to test vaccines based on IDO and/or PD-L1 for several indications.
Cancer vaccines represent a promising means of eliminating minimal residual disease without inducing significant toxicity or secondary malignancies. However, to date, they have largely failed to significantly improve patient outcomes. This likely reflects malignant cells’ ability to suppress the functions of the induced immune cells. Activating anti-Tregs—for example, by IDO or PD-L1 vaccination—may work synergistically with other immunotherapy. The addition of epitopes like PDL1 and IDO to current cancer vaccine strategies would be easy to implement and is likely to be highly beneficial. Such antigens could serve as a widely applicable target for immunotherapeutic strategies, showing a completely different function and expression pattern compared to previously described antigens.
Several different therapeutic strategies are utilized to target immunosuppression in cancer, including blocking inhibitory pathways, such as the PD-1/PD-L1 pathway. Antibodies that target inhibitory checkpoints can reportedly elicit impressive, dynamic, and durable tumour regression. A major difference between targeting immune checkpoints with monoclonal antibodies versus utilizing anti-Tregs is that anti-Tregs can both decrease the direct immunoregulatory effects of their targets and inhibit other routes of immune suppression that are mediated by their cognate target cells. Accordingly, a vaccine targeting anti-Tregs should attract specific pro-inflammatory T cells to the tumour microenvironment. Anti-Tregs can directly support anti-cancer immunity by killing target cells and indirectly boost anti-cancer immunity by releasing pro-inflammatory cytokines into the microenvironment. Thus, an anti-Treg vaccine should be viewed as complementing rather than competing with other forms of immunotherapy. For example, vaccine-activated PD-L1-specific T cells could be further boosted by PD-L1 blockade since PD-L1 mAbs target the same cells as vaccine-induced T cells. Therefore, this therapeutic strategy may make cells more vulnerable targets. It must also be considered that anti-Tregs may influence immune-regulatory pathways other than those directly mediated by their targets. Immune-suppressive cells simultaneously utilize a number of different immune-suppressive mechanisms to inhibit immune responses, including arginase, IDO, PD-L1, and immune-regulatory cytokines (e.g. IL-10 and TGF-β). For example, PD-L1-specific anti-Tregs not only suppress the immune-regulatory effects of PD-L1 but also target other routes of immune suppression mediated by PD-L1+ target cells.
In summary, the use of anti-Tregs for cancer vaccination represents a completely novel immuno-oncological therapeutic approach. By definition, almost any successful immune therapy strategy aims to induce immunological activation and inflammation. Since immune-suppressive cells can antagonize the desired effects of immunotherapeutic approaches, it may be an effective, easily implementable, and highly synergistic approach to additionally target such cells, e.g. by vaccination. Boosting specific T cells that recognize immune-regulatory proteins like IDO or PD-L1 could directly modulate immune regulation and potentially alter tolerance to tumour antigens. Targeting antigens with immune-regulatory functions represents a novel and specific concept in immunotherapy, in contrast to the specific depletion of cells, which is not limited to targeting products expressed on the cell surface.
Conclusions
We have characterized self-reactive T cells, termed anti-Tregs, that specifically recognize proteins expressed in regulatory immune cells, including IDO, PD-L1, and Foxp3 [58, 66]. PD-L1-specific T cells are a particularly interesting example of the immune system’s ability to influence adaptive immune responses by directly reacting against the immune-suppressive mechanisms employed by cancerous cells. Self-reactive T cells may escape thymic selection to directly participate in the fight against pathogens and to provide the immune system with an additional layer of immune regulation to support immune homeostasis. Boosting the anti-Tregs that recognize regulatory immune cells may directly modulate immune regulation, potentially altering tolerance to tumour antigens. However, it remains unknown how and when these self-reactive T cells are induced or become activated during autoimmune conditions and to what extent they might impact autoimmune disease pathogenesis and development.
Niels Jerne was more right than he knew when he somewhat 40 years ago proposed that the immune system functions as a network regulated via interactions between lymphocytes even in the absence of foregin antigens.
References
Sakaguchi S (2006) Regulatory T cells. Springer Semin Immunopathol 28:1–2
Zhou X, Jeker LT, Fife BT, Zhu S, Anderson MS, McManus MT et al (2008) Selective miRNA disruption in T reg cells leads to uncontrolled autoimmunity. J Exp Med 205:1983–1991
Brusko TM, Putnam AL, Bluestone JA (2008) Human regulatory T cells: role in autoimmune disease and therapeutic opportunities. Immunol Rev 223:371–390
Roncarolo MG, Gregori S, Battaglia M, Bacchetta R, Fleischhauer K, Levings MK (2006) Interleukin-10-secreting type 1 regulatory T cells in rodents and humans. Immunol Rev 212:28–50
Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N et al (2003) Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med 198:1875–1886
Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM et al (2007) The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature 450:566–569
Andersen MH (2015) Immune regulation by self-recognition: novel possibilities for anticancer immunotherapy. J Natl Cancer Inst 107:154
Kumar V, Sercarz EE (1993) The involvement of T cell receptor peptide-specific regulatory CD4+ T cells in recovery from antigen-induced autoimmune disease. J Exp Med 178:909–916
Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D et al (2005) GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity 22:633–642
Platten M, Ho PP, Youssef S, Fontoura P, Garren H, Hur EM et al (2005) Treatment of autoimmune neuroinflammation with a synthetic tryptophan metabolite. Science 310:850–855
Munn DH, Mellor AL (2007) Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest 117:1147–1154
Batabyal D, Yeh SR (2007) Human tryptophan dioxygenase: a comparison to indoleamine 2,3-dioxygenase. J Am Chem Soc 19:15690–15701
Prendergast GC (2008) Immune escape as a fundamental trait of cancer: focus on IDO. Oncogene 27:3889–3900
Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N et al (2003) Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med 9:1269–1274
Romani L, Bistoni F, Perruccio K, Montagnoli C, Gaziano R, Bozza S et al (2006) Thymosin alpha1 activates dendritic cell tryptophan catabolism and establishes a regulatory environment for balance of inflammation and tolerance. Blood 108:2265–2274
Sorensen RB, Berge-Hansen L, Junker N, Hansen CA, Hadrup SR, Schumacher TN et al (2009) The immune system strikes back: cellular immune responses against indoleamine 2,3-dioxygenase. PLoS One 4:e6910
Sorensen RB, Hadrup SR, Svane IM, Hjortso MC, thor Straten P, MH A (2011) Indoleamine 2,3-dioxygenase specific, cytotoxic T cells as immune regulators. Blood 117:2200–2210
Sorensen RB, Kollgaard T, Andersen RS, van den Berg JH, Svane IM, thor Straten P et al (2011) Spontaneous cytotoxic T-cell reactivity against indoleamine 2,3-dioxygenase-2. Cancer Res 71:2038–2044
Munir S, Larsen SK, Iversen TZ, Donia M, Klausen TW, Svane IM et al (2012) Natural CD4(+) T-cell responses against indoleamine 2,3-dioxygenase. PLoS One 7:e34568
Andersen MH (2012) The specific targeting of immune regulation: T-cell responses against indoleamine 2,3-dioxygenase. Cancer Immunol Immunother 61:1289–1297
Andersen MH (2012) CD4 responses against IDO. Oncoimmunology 1:1211–1212
Smith C, Chang MY, Parker KH, Beury DW, DuHadaway JB, Flick HE et al (2012) IDO is a nodal pathogenic driver of lung cancer and metastasis development. Cancer Discov 2:722–735
Yu J, Du W, Yan F, Wang Y, Li H, Cao S et al (2013) Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J Immunol 190:3783–3797
Bronte V, Zanovello P (2005) Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol 5:641–654
Mussai F, De SC, Abu-Dayyeh I, Booth S, Quek L, McEwen-Smith RM et al (2013) Acute myeloid leukemia creates an arginase-dependent immunosuppressive microenvironment. Blood 122:749–758
Kozako T, Yoshimitsu M, Fujiwara H, Masamoto I, Horai S, White Y et al (2009) PD-1/PD-L1 expression in human T-cell leukemia virus type 1 carriers and adult T-cell leukemia/lymphoma patients. Leukemia 23:375–382
Atanackovic D, Luetkens T, Kroger N (2013) Coinhibitory molecule PD-1 as a potential target for the immunotherapy of multiple myeloma. Leukemia. doi:10.1038/leu.2013.310
Yang H, Bueso-Ramos C, Dinardo C, Estecio MR, Davanlou M, Geng QR et al (2014) Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia 28:1280–1288
Krejsgaard T, Odum N, Geisler C, Wasik MA, Woetmann A (2012) Regulatory T cells and immunodeficiency in mycosis fungoides and Sezary syndrome. Leukemia 26:424–432
Kollgaard T, Petersen SL, Hadrup SR, Masmas TN, Seremet T, Andersen MH et al (2005) Evidence for involvement of clonally expanded CD8+ T cells in anticancer immune responses in CLL patients following nonmyeloablative conditioning and hematopoietic cell transplantation. Leukemia 19:2273–2280
Ame-Thomas P, Le PJ, Yssel H, Caron G, Pangault C, Jean R et al (2012) Characterization of intratumoral follicular helper T cells in follicular lymphoma: role in the survival of malignant B cells. Leukemia 26:1053–1063
van de Donk NW, Kamps S, Mutis T, Lokhorst HM (2012) Monoclonal antibody-based therapy as a new treatment strategy in multiple myeloma. Leukemia 26:199–213
Tamura H, Ishibashi M, Yamashita T, Tanosaki S, Okuyama N, Kondo A et al (2013) Marrow stromal cells induce B7-H1 expression on myeloma cells, generating aggressive characteristics in multiple myeloma. Leukemia 27:464–472
Greaves P, Gribben JG (2013) The role of B7 family molecules in hematologic malignancy. Blood 121:734–744
Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P et al (2012) Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 366:2455–2465
Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF et al (2012) Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 366:2443–2453
Munir S, Andersen GH, Met O, Donia M, Frosig TM, Larsen SK et al (2013) HLA-restricted cytotoxic T cells that are specific for the immune checkpoint ligand PD-L1 occur with high frequency in cancer patients. Cancer Res 73:1674–1776
Munir S, Andersen GH, Woetmann A, Odum N, Becker JC, Andersen MH (2013) Cutaneous T cell lymphoma cells are targets for immune checkpoint ligand PD-L1-specific, cytotoxic T cells. Leukemia 27:2251–2253
Minami T, Minami T, Shimizu N, Yamamoto Y, De VM, Nozawa M et al (2015) Identification of programmed death ligand 1-derived peptides capable of inducing cancer-reactive cytotoxic T lymphocytes from HLA-A24+ patients with renal cell carcinoma. J Immunother 38:285–291
Dong H, Strome SE, Matteson EL, Moder KG, Flies DB, Zhu G et al (2003) Costimulating aberrant T cell responses by B7-H1 autoantibodies in rheumatoid arthritis. J Clin Invest 111:363–370
Nair S, Boczkowski D, Fassnacht M, Pisetsky D, Gilboa E (2007) Vaccination against the forkhead family transcription factor Foxp3 enhances tumor immunity. Cancer Res 67:371–380
van Es T, van Puijvelde GH, Foks AC, Habets KL, Bot I, Gilboa E et al (2010) Vaccination against Foxp3(+) regulatory T cells aggravates atherosclerosis. Atherosclerosis 209:74–80
Larsen SK, Munir S, Woetmann A, Froesig TM, Odum N, Svane IM et al (2013) Functional characterization of Foxp3-specific spontaneous immune responses. Leukemia 27:2332–2340
Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P et al (2004) Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med 10:942–949
Ishida T, Ishii T, Inagaki A, Yano H, Komatsu H, Iida S et al (2006) Specific recruitment of CC chemokine receptor 4-positive regulatory T cells in Hodgkin lymphoma fosters immune privilege. Cancer Res 66:5716–5722
Gobert M, Treilleux I, Bendriss-Vermare N, Bachelot T, Goddard-Leon S, Arfi V et al (2009) Regulatory T cells recruited through CCL22/CCR4 are selectively activated in lymphoid infiltrates surrounding primary breast tumors and lead to an adverse clinical outcome. Cancer Res 69:2000–2009
Cao L, Hu X, Zhang J, Huang G, Zhang Y (2014) The role of the CCL22-CCR4 axis in the metastasis of gastric cancer cells into omental milky spots. J Transl Med 12:267. doi:10.1186/s12967-014-0267-1.:267-0267
Martinenaite E, Ahmad SM, Hansen M, Met O, Westergaard MW, Larsen SK, et al. (2016) CCL22-specific T cells: modulating the immunosuppressive tumor microenvironment. In Press ed.
Munir S, Frosig TM, Hansen M, Svane IM, Andersen MH (2012) Characterization of T-cell responses against IkappaBalpha in cancer patients. Oncoimmunology 1:1290–1296
Yu W, Jiang N, Ebert PJ, Kidd BA, Muller S, Lund PJ et al (2015) Clonal deletion prunes but does not eliminate self-specific alphabeta CD8(+) T lymphocytes. Immunity 42:929–941
Sylwester AW, Mitchell BL, Edgar JB, Taormina C, Pelte C, Ruchti F et al (2005) Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J Exp Med 202:673–685
Furset G, Floisand Y, Sioud M (2008) Impaired expression of indoleamine 2, 3-dioxygenase in monocyte-derived dendritic cells in response to Toll-like receptor-7/8 ligands. Immunology 123:263–271
Lim ST, Levine AM (2005) Non-AIDS-defining cancers and HIV infection. Curr Infect Dis Rep 7:227–234
Hjortso MC, Larsen SK, Kongsted P, Met O, Frosig TM, Andersen GH et al (2015) Tryptophan 2,3-dioxygenase (TDO)-reactive T cells differ in their functional characteristics in health and cancer. Oncoimmunology 4:e968480
Frumento G, Rotondo R, Tonetti M, Damonte G, Benatti U, Ferrara GB (2002) Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J Exp Med 196:459–468
Xu H, Oriss TB, Fei M, Henry AC, Melgert BN, Chen L et al (2008) Indoleamine 2,3-dioxygenase in lung dendritic cells promotes Th2 responses and allergic inflammation. Proc Natl Acad Sci U S A 105:6690–6695
Iversen TZ, Engell-Noerregaard L, Ellebaek E, Andersen R, Larsen SK, Bjoern J et al (2014) Long-lasting disease stabilization in the absence of toxicity in metastatic lung cancer patients vaccinated with an epitope derived from indoleamine 2,3 dioxygenase. Clin Cancer Res 20:221–232
Ahmad SM, Larsen SK, Svane IM, Andersen MH (2014) Harnessing PD-L1-specific cytotoxic T cells for anti-leukemia immunotherapy to defeat mechanisms of immune escape mediated by the PD-1 pathway. Leukemia 28:236–238
Ahmad SM, Svane IM, Andersen MH (2014) The stimulation of PD-L1-specific cytotoxic T lymphocytes can both directly and indirectly enhance antileukemic immunity. Blood Cancer J 4:230–233
Ahmad SM, Martinenaite E, Hansen M, Junker N, Borch TH, Met O, et al. (2016) PD-L1 peptide co-stimulation increases immunogenicity of a dendritic cell-based cancer vaccine. In press ed.
Munir S, Andersen GH, Svane IM, Andersen MH (2013) The immune checkpoint regulator PD-L1 is a specific target for naturally occurring CD4+ T cells. Oncoimmunology 2:e23991
Liston A, Nutsch KM, Farr AG, Lund JM, Rasmussen JP, Koni PA et al (2008) Differentiation of regulatory Foxp3+ T cells in the thymic cortex. Proc Natl Acad Sci U S A 105:11903–11908
Andersen MH, Sorensen RB, Brimnes MK, Svane IM, Becker JC, thor Straten P (2009) Identification of heme oxygenase-1-specific regulatory CD8+ T cells in cancer patients. J Clin Invest 119:2245–2256
Samy ET, Parker LA, Sharp CP, Tung KS (2005) Continuous control of autoimmune disease by antigen-dependent polyclonal CD4+CD25+ regulatory T cells in the regional lymph node. J Exp Med 202:771–781
Nagata Y, Hanagiri T, Mizukami M, Kuroda K, Shigematsu Y, Baba T et al (2009) Clinical significance of HLA class I alleles on postoperative prognosis of lung cancer patients in Japan. Lung Cancer 65:91–97
Andersen MH (2013) FOXP3-specific immunity. Oncoimmunology 2:e26247
Acknowledgments
This study was supported by the Danish Cancer Society, the Danish Council for Independent Research, Toyota Foundation, and Herlev Hospital. The funders did not have a role in the writing of the article or the decision to submit the article for publication.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
MHA is an author of different patent applications based on the use of CCL22, PD-L1, TDO, or IDO for vaccination. The rights of the patent applications have been transferred to Copenhagen University Hospital, Herlev, according to the Danish Law of Public Inventions at Public Research Institutions. MHA is a shareholder and board member of the company IO Biotech ApS that has the purpose of developing commercial IDO and PD-L1 vaccines for cancer treatment.
Additional information
This article is a contribution to the special issue on Cancer and Autoimmunity - Guest Editor: Mads Hald Andersen
Rights and permissions
About this article

Cite this article
Andersen, M.H. Anti-regulatory T cells. Semin Immunopathol 39, 317–326 (2017). https://doi.org/10.1007/s00281-016-0593-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-016-0593-x