
Abstract
During the last decade, the treatment for many cancer indications has evolved due to intensive clinical research into anti-tumor agents’ combination. In most instances, new combination treatments consist of an add-on to the standard of care (SOC), which then demonstrate a substantial gain in efficacy and no detrimental effect in tolerability. In the era of targeted therapies, for which maximum tolerated dose (MTD)-based dosing strategies are no longer applicable, early stage studies exploring new combinations are often conducted in the population of interest, expediting the collection of preliminary safety data, to be promptly expanded to collect preliminary efficacy data. Nevertheless, rule-based dose-finding studies are still a prevailing approach for early stage cancer, especially for chemotherapy (CT)-containing combinations. Pharmacokinetic (PK) assessments play a key role throughout the clinical development of drug combinations, informing potential PK interactions. But most importantly, they allow the development of innovative exposure–response (E–R) models aimed at exploring the contribution of each agent to the overall effect of the combination therapy. This review identifies 81 new drug combinations approved by the United States Food and Drug Administration (FDA) for hemato-oncology during the 2011–2021 period and summarizes the main design features of clinical trials and the role of PK assessments.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Drug combinations are important foundations for the pharmacotherapy for cancer. As a result of evolving mutations and altered molecular pathways in tumor cells resulting in multiple mechanisms of drug resistance, drug combinations are necessary to provide long-term tumor control for most patients.
The first successful drug combinations appeared in the early 1960s, when the quadruple combination programs VAMP (vincristine, amethopterin, 6-mercaptopurine, and prednisone) and MOMP (nitrogen mustard, vincristine, methotrexate, and prednisone) demonstrated efficacy in acute childhood leukemia and advanced Hodgkin’s disease [1, 2]. Since then, drug combinations have been used to treat many types of solid and hematologic tumors; some were empirically implemented, and are being used since then, while others entered in clinical practice and gained United States (US) Food and Drug Administration (FDA) approval through pivotal clinical trials. Today, over 350 drug combinations are used to treat solid tumors and hematologic malignancies [2], and the number of new FDA-approved combinations significantly increases every year.
Despite evidence of the improved benefit when anticancer agents are combined, the majority are still developed as monotherapy, which may underlie the substantial failure rates observed as they are unlikely to demonstrate significant benefit over standard of care (SOC) at late phases of the disease. In this sense, the FDA recently issued a guidance document on regulatory policies specific to combination regimes composed of experimental drugs [3].
The design of clinical trials of new combination therapies carries many practical challenges. One of these is the inclusion and extent of pharmacokinetic (PK) assessments, since patient’s venipuncture, sample logistics, and bioanalysis add complexity and expense to clinical studies, while their short- and long-term utility may not always become apparent. Nevertheless, PK data become even more relevant when drugs are part of a combination treatment.
Therefore, our objectives were to evaluate first the extent of recently FDA-approved hemato-oncology drug combinations, and second their main features of clinical development, with a focus on PK assessments. This study may help drug developers in general, and clinical pharmacologists, when deciding on the design of future combination studies in oncology, and the usage of PK assessments to enhance clinical development.
Methods
Search strategy
A comprehensive list of FDA approvals related to new combination therapies for hemato-oncology granted between 2011 and 2021 was assembled from several sources. The FDA’s Hematology/Oncology (Cancer) Approvals & Safety Notifications website was cross-checked against CenterWatch, a database that allows searching of FDA-approved drugs by therapeutic area (here, oncology) [4, 5]. Additionally, US labels of innovative drugs (i.e., drugs owned by the primary commercial sponsor of the pivotal study) from each combination were downloaded from Drugs@FDA to verify approval status [6].
Information about the clinical development of selected combinations was extracted from the corresponding Clinical Pharmacology and Biopharmaceutics (CP&B) Review published on the FDA’s Approved Drug Products website [7]. However, these reviews are made publicly available only when the innovative drug is a new molecular entity (NME)- or a new biological entity (NBE)-containing combination. If FDA reviews were not available, information was extracted from published articles corresponding to clinical studies retrieved from PubMed (filtering by article type = Clinical Trial) or from Clinicaltrials.gov. Generic names of combined agents and/or NCT study code, as shown in the FDA’s Approvals Notifications website, were used as search terms. For pharmacometric studies, “population pharmacokinetic” and “exposure–response” (E–R) were used as search terms, without language, date, or publication status restrictions. Data not found by any of these sources is referred to throughout the ‘Results’ section as ‘unknown’.
Classification of clinical trials and agents
Since clinical trials could be amended throughout their duration, the clinical trial phase (e.g., I, Ib, Ib/II) at Clinicaltrials.gov did not always match that of the corresponding publication. However, this review classified clinical studies according to their final role in regulatory approval, as dose-finding (evaluation of optimal dose and appropriate schedule), proof of concept (PoC) (initial test of a therapy of interest in a defined population or indication [8]), or pivotal (adequate and well-controlled investigation, which is the basis for reaching a conclusion that there is substantial evidence of effectiveness to decide whether to approve a drug [9]).
The system of classifying agents into therapeutic classes was taken from Lu et al. [10]: small molecules (SM) were classified into chemotherapy (CT) agents, kinase inhibitors (KIs), and non-KI small molecule-targeted agents (non-KI targeted agents), while large molecules (LM) were sub-classified into monoclonal antibodies (mAbs), immunotherapy (IO) agents, antibody drug conjugates (ADCs), and fusion proteins.
Inclusion and exclusion criteria
The selection was focused on hemato-oncology approved combination therapies in which both drugs are active independently, but the combination is expected to be more effective than either agent alone. The resulting list was triaged to remove combinations based on the following exclusion criteria: a drug combined with radiotherapy, and a drug combined with another drug that enhances its activity or safety profile but without intrinsic antineoplastic activity (e.g., abiraterone is combined with prednisone based on its positive effect of reducing hypotension; Inqovi® is a fixed-dose combination that includes cedazuridine, which acts as a pharmacoenhancer for decitabine). Even though ADCs are composed of two conjugated antineoplastic drugs, they were selected only if combined with an additional drug. Finally, FDA approvals consisting of label extensions of already approved combinations were excluded from the analyses, since they did not entail a full development.
Search features
The following characteristics from each combination were used in the search: targeted tumor type, therapeutic classes combined, design of main clinical studies (i.e., dose-finding, PoC and pivotal) and dosage used when compared to monotherapy, type of PK assessments, PK evaluations of potential drug–drug interactions between combined agents, and pharmacometric analyses [i.e., population PK (PopPK) and E–R analyses].
Results
Description of the sample
From January 2011 to December 2021, 403 notifications were uploaded to the FDA’s Hematology/Oncology website [11] (Fig. 1). Of these, 7 corresponded to approvals of diagnostics, and 11 to notifications not involving new approvals (e.g., updates of prescribing information, granting regular approval after an accelerated approval, etc.). The remaining 385 were cancer therapy approvals, from which 237 were single-agent therapies, 26 were supportive therapies, and 122 were combination therapies. Among the latter, 10 had at least one agent with no anti-tumor activity (e.g., pharmacoenhancers), and 14 were ADCs. Additionally, 5 approved combinations were detected while reviewing the label of innovative agents from screened combinations. Finally, 22 were label extensions of already approved combinations.

Flowchart of search strategy. Graphics software used: Microsoft Visio
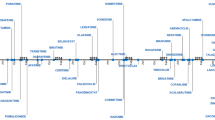
Therefore, 81 applications related to hemato-oncology combination therapies that gained FDA approval for the first time from 2011 to 2021 were identified according to the selection criteria described in the ‘Methods’ section. These are listed in Table 1 and Fig. 2 by year of approval and tumor type.

FDA-approved combinations from 2011 to 2021, organized by chronological order and tumor type. Color of boxes denote the therapeutic class of main agent (in bold), as shown in bottom legend. Chemotherapeutic agents are in white letters. Blue curved lines link label extensions or new indications of approved combinations. 5FU 5-fluorouracil, CTX Cyclophosphamide, FDA Food and Drug Administration, FOLFIRI FOLinic acid-Fluorouracil-IRInotecan, HNSCC head and neck squamous cell cancer, STS soft-tissue sarcoma. *Including immunomodulators, proteasome inhibitors, and hormonal therapy. Graphics software used: Microsoft Visio
Main features of combinations
Of the approved combinations, 50 out of 81 (62%) were indicated for solid tumors (Table 2). The remaining 31 (38%) were indicated for hematologic malignancies, mainly (16 out of 31) for multiple myeloma (MM). The majority (54 out of 81; 67%) of combinations contained drugs previously approved as single agent, while for the remaining 27 out of 81 (33%), the main agent was an NME/NBE. Of note, one of these combinations contained two NMEs: encorafenib and binimetinib, co-developed to treat unresectable or metastatic melanoma (mM).
These combinations involved 83 different agents: 58 were SM and 25 were LM (Table S1). SM comprised 21 CT agents, 20 KIs, and 17 non-KI targeted agents. LM comprised 16 mAbs, 6 IOs, two ADCs and one fusion protein.
Combinations of therapeutic classes
Table 3 summarizes the most frequent combinations of therapeutic classes, regardless of the number of agents in each combination. According to tumor class (solid vs. hematologic), combinations approved for solid tumors (50 out of 81; 62%) consisted in most instances of one or more mAbs plus one or more CT agent (14 out of 50; 28%), followed by an IO plus a CT agent (8 out of 50; 16%). For hematologic malignancies (31 out of 81; 38%), the most common combination types were one mAb plus one or more non-KI targeted agents (10 out of 31; 32%), followed by two or more non-KI targeted agents (7 out of 31; 23%).
The majority of approved combinations were doublets (56%), although an important number of combinations consisted of three (triplets) (37%) or 4 (tetraplets) (7%) agents (Table 3). Doublets, triplets, and tetraplets contains, in turn, one, three, and six pairs of agents combined, respectively. Therefore, each doublet, triplet, and tetraplet results in one, three, and six pairs of agents, respectively. Accordingly, for solid tumors, most frequent pairs of therapeutic classes consisted of a CT agent plus a mAb (32%), two CT agents (21%), and a CT agent plus an IO (19%) (Table 3). For hematologic malignancies, most frequent pairs consisted of two non-KI targeted agents (34%), a non-KI targeted agent plus a mAb (30%), and two CT agents (11%). These frequencies are represented in Fig. 3 by the width of bands linking sectors of therapeutic classes.

Circos plots of frequencies of pairs of therapeutic classes combined, by type of malignancy. Bands linking therapeutic class are duplicated, although with different color depending on class. Width of bands are indicative of the frequencies of pairs of therapeutic classes combined. One doublet implies one pair of therapeutic classes, one triplet three pairs, and one tetraplet six pairs. Graphics software used: R Studio
Half of the combinations (41 out 81; 51%) contained at least one CT, more frequently for solid (29 out of 50; 58%) than for hematologic malignancies (12 out of 31; 39%). For these, most combinations (21 out of 31; 68%) included non-KI targeted agents.
Dosage of combined agents as compared with monotherapy
In most combinations (75 out of 81; 93%), at least one agent was approved as monotherapy for the same tumor type. In 60 of these 75 combinations (80%), dose/s were the same for monotherapy, while in the remaining 15 out of 75 (20%) combinations, either the innovative agent (one out of 15) or the secondary agent (14 out of 15) was given at a lower dose. These were mainly CT agents (8 out of 14), but also two KIs, a non-KI targeted agent, an IO, and a mAb (Table S2).
Main clinical development strategy features
Studies were designed according to the classical paradigm (phase I, II, and III) to most recent streamlined approaches, such as multiple extension phases, seamless studies, etc. In fact, PoC studies were mostly conducted as phase I–II studies or as expansion phases of an amended phase I study. In such cases, they were split based on their role for approval (i.e., dose-finding, PoC, and pivotal studies), as shown below and in Fig. 4. Nevertheless, Supplementary spreadsheet lists them individually.

Summary of main design features of clinical trials. *One study included additional agent in the control arm (A + B vs. A + B + C); ** One study also included one arm with one of the agents as monotherapy. A: innovative agent; B: secondary agent; C: Other secondary agent; SOC: standard of care. Graphics software used: Microsoft Visio
Dose definition studies
Among the 81 combination approvals, 21 (26%) did not include a dose-finding study. Of note, the innovative agent was an LM in 16 of these 21 (76%) combinations, either already approved (11 out of 16; 69%) or an NBE (5 out of 16; 31%). The remaining 5 out of 21 (24%) combinations with no dose-finding study corresponded to: (i) olaparib plus bevacizumab for advanced ovarian cancer; a previous phase II study in relapsed platinum-sensitive ovarian cancer had already explored the addition of an antiangiogenic agent to a poly-ADP-ribose polymerase (PARP) inhibitor (niraparib) [11], (ii) selinexor plus dexamethasone for fifth line MM; the single arm pivotal study in penta-refractory MM included an initial part, in which the dose regimen of dexamethasone was optimized [12], (iii) lenalidomide plus rituximab for follicular lymphoma; lenalidomide is approved at 25 mg day 1–21 every 4 weeks (q4wk) for other hematologic tumors; however, the phase II study ALLIANCE (NCT00238238) of the lenalidomide/rituximab combination included an intrapatient dose escalation of lenalidomide from 15 mg on cycle one, 20 mg on cycle two and 25 mg on cycle three and beyond, depending on tolerability [13]; finally, the dose selected for the pivotal study was 20 mg, (iv) everolimus–exemestane, and (v) palbociclib-fulvestrant; the dose of everolimus and palbociclib in combination with another estrogen antagonist (letrozole) had been already defined in early phase studies [14, 15].
For the 60 out of 81 (74%) combinations with a dose-finding study, 48 out of 60 (80%) were phase Ib (in the intended tumor type). Twenty-one out of 60 (35%) did not include an escalation but just used the dose levels of the agents as monotherapy; in most of these (16 out of 21; 76%), the innovative agent was an LM. In the 39 out of 60 65%) studies with escalation, this was mostly sequential (34 out of 39; 87%), either traditional (e.g., 3 + 3) in 28 studies, or with an adaptive (e.g., Bayesian) design in 6 studies. One study (nivolumab/cabozantinib combination (NCT02496208) used a rolling 6 escalation design [16]. Finally, in 5 out of 39 (13%) studies with escalation, a parallel design was used.
Proof of concept studies
One-third (28) of the approved combinations included a PoC study, either as part of a phase I–II, 12 out of 28 (43%) or as a stand-alone, 16 out of 28 (57%) (Fig. 4). Thirteen out of 28 (46%) were single arm studies, while the remaining 15 out of 28 (54%) were randomized studies, with one of the combined agents as monotherapy, 10 out of 15 (67%), or with two or more dose levels, 5 out of 15 (33%). One PoC study included three arms: two dose levels of the combination and one with one agent in monotherapy (phase II part of study COMBI-d (NCT01072175) with dabrafenib plus trametinib for melanoma) [17]. Finally, one PoC study (phase II part of study NCT01719380 [18]) in colorectal cancer compared the triplet encorafenib plus cetuximab plus alpelisib defined in the phase I study with the doublet encorafenib plus cetuximab, that was finally approved.
Pivotal studies
The study design is about 81 pivotal studies. Among them, 77 (95%) were randomized controlled studies. Most combinations consisted of an add-on therapy to the SOC. As a result, 61 out of the 77 (79%) randomized pivotal studies compared the innovative agent (A) of the combination when added to a secondary agent/s (B) vs. the secondary agent/s (A + B vs. B) (Fig. 4). In 9 out of the 77 (12%) randomized studies, the innovative agent (A) was replacing another agent (C) of an already approved combination (A + B vs. C + B). In 5 out of 77 (7%) studies, none of the combined agents were part of the SOC (A + B vs. C). The remaining two out of 77 (3%) randomized pivotal trials corresponded to packed-formulation combinations; study NCT01696084 CLTR0310-301 compared CPX-351 (cytarabine and daunorubicin) liposome vs. cytarabine plus daunorubicin (A + B vs. A + B) [19], and study ASCERTAIN (NCT03306264) compared ASTX727 tablet on cycle one and decitabine on cycle two vs. the reverse sequence (A + B → A vs. A → A + B) [20].
When an agent was present at both arms of a pivotal study (i.e., A + B vs. B or A + B vs. C + B), it was given at the same dose in most instances, 64 out of 71 (90%). This provided unarguably evidence that the superior efficacy observed in the experimental arm was driven by the innovative agent. However, there were 7 out of 71 (10%) studies in which the dose of one agent was different, thus compromising the assessment of the contribution of each agent to the overall anti-tumor effect (Table S3). Interestingly, study COLUMBUS (NCT01909453) in melanoma initially compared encorafenib 450 mg od plus binimetinib 45 mg vs. encorafenib 300 mg, since encorafenib in combination resulted in a higher maximum tolerated dose than in monotherapy [21]. Consequently, the FDA requested to amend the protocol and use the same dose of encorafenib (300 mg) in both arms, so that the contribution of binimetinib to the overall effect of the combination could be isolated.
Finally, there were 4 out of 81 (5%) combinations approved based on single arm pivotal studies: STORM (NCT02336815) with selinexor plus dexamethasone for penta-refractory MM [22], L-MIND (NCT02399085) with tafasitamab-cxix plus lenalidomide for large B-cell lymphoma [23], EQUULEUS (NCT01998971) with daratumumab plus pomalidomide plus dexamethasone for MM [24], and KEYNOTE-146 (NCT02501096) with pembrolizumab plus lenvatinib for endometrial carcinoma [25].
Main features of pharmacokinetics and pharmacometrics analyses
Blood sampling for PK assessment was performed in 44 out of 60 (73%), 14 out of 28 (50%), and 55 out of 81 (68%) of dose-finding, PoC, and pivotal studies, respectively. Type of sampling (intensive vs. sparse) and agents (innovative and/or secondary) involved in PK assessment Evaluations of the effect of the innovative agent over are described in Table 4.
A PopPK model of the innovative agent with PK data when given in combination was found for 51 of the 81 (63%) approved combinations; in the remaining 30 combinations, a PopPK model was either not performed (26%) or unknown (11%) (Table 5). Most PopPK models (26 out of 51; 51%) pooled PK data from studies in which the innovative agent was given as single agent and in combination, while 18 PopPK models pooled combination studies, and 7 were limited to the pivotal study.
E–R analyses for efficacy and/or safety of the innovative agent in combination were available in half (48%) of the approved combinations, being more prevalent in NME/NBE-containing combinations (20), than in combinations of already approved agents (19). In the other half (42 out 81; 52%), E–R analyses were either not done (35%) or not found (17%). This is the case of panobinostat, bortezomib, and dexamethasone combination as third line for MM, in which PK exposure data was available for the escalation study only. Due to the lack of dose–response (D–R)/E–R data for efficacy from the pivotal study, and the increased rate of serious adverse events and deaths in the experimental arm, it was not possible to determine if a lower dose of panobinostat would provide a better benefit–risk profile [26]. The FDA finally recommended the Sponsor to conduct a dose-finding trial as a post-marketing requirement to adequately characterize the D–R relationship of panobinostat.
Pharmacokinetic evaluation of interactions between combined agents
Evidence regarding the assessment of potential PK interactions between the innovative (A) and secondary agent/s (B) could be retrieved for 46 out of 81 (57%) combinations only, since they were rarely available before the FDA CP&B Review was released.
Evaluation of PK interactions varied largely according to the specificities of each combination. Within-study evaluations of the impact of the secondary agent/s on the PK of the innovative agent (B → A) were performed in 19 out of 46 (41%) combinations; 8 of them in crossover dose-finding studies and 11 in parallel (dose-finding, PoC or pivotal) studies (Fig. 5). Historically controlled evaluations were identified for 27 out of 46 (59%) combinations; in most instances involving pooled PopPK analyses.

Summary of PK evaluations of interactions between combined agents; number of A → B (innovative agent over secondary agent) and B → A (secondary agent/s over innovative agent) evaluations, either as intra-study (crossover or parallel) or as inter-study (historical and/or population PK) Graphics software used: R Studio
Evaluation of the effect of the innovative agent over the PK of secondary agent/s (A → B) was found for 26 combinations. Of these, 21 were within-study evaluations (14 in parallel and 7 in crossover studies), and 5 were historically controlled evaluations.
Nevertheless, evaluation of PK interactions were not necessarily restricted to one approach (within-study or historical). A noteworthy example is bevacizumab plus atezolizumab plus CT (carboplatin and paclitaxel); the phase study IMpower150 included three parallel arms and intensive PK sampling for all agents, allowing the PK effects to be characterized among each of the agents of the tetraplet. Additionally, a PopPK model of atezolizumab explored the potential impact of CT with or without bevacizumab coadministration on the PK of atezolizumab [27].
B → A interactions were more often evaluated/reported than A → B interactions (57% vs. 33%), with the exception of parallel evaluations conducted in phase III studies; since most of these studies had a parallel A + B vs. B design, they could easily explore A → B interactions. Additionally, PK interactions were more frequently explored when agents were either two SM or two LM (63%), than when one was an SM, and the other was an LM (37%).
Discussion
The past decade has witnessed prolific research into clinical hemato-oncology. Among other achievements, a significant number of new drug combinations were demonstrated to have substantial benefit over the SOC, and thus were granted US FDA approval. The search for common patterns or even trends among these combinations is challenging, based on the heterogeneity of the therapeutic classes combined and the tumor types at several line settings. Added to this are the evolving strategies of clinical research, which result in many different clinical trial designs, and PK and pharmacometric evaluations. Nevertheless, this review identified some of these patterns.
First, CT-containing combinations were the most prevalent approvals. CT remains the backbone of treatment to achieve durable responses in many tumor types at early and late stages, despite the low rate of FDA approvals of new agents in this therapeutic class during the same period of time. There were 4 first-time approved agents: liposomal vincristine (Marqibo®) for acute lymphoblastic leukemia in August 2012, omacetaxine mepesuccinate (Synribo®) for chronic myeloid leukemia in October 2012, and lurbinectedin (Zepzelca®) for metastatic small cell lung cancer in June 2020. In addition, three marketed CTs received approval for new tumor types: trabectedin (Yondelis®) for metastatic liposarcoma or leiomyosarcoma in October 2015, mitomycin (Jelmyto®) for upper tract urothelial cancer in April 2020, and azacitidine tablets (Onureg®) for continued treatment of acute myeloid leukemia in September 2020.
IO has been one of the undoubted landmark breakthroughs in the fight against cancer of the past decade. Nevertheless, IO alone, and in particular anti programmed cell death protein-1 (PD-1) or anti programmed death-ligand 1 (PD-L1) mAbs used as single agents, generally provides insufficient responses or nonsignificant therapeutic advantages [28]. A major benefit is being attained from these agents combined with other therapeutic classes and, in particular, with CT as it boosts anti-tumor immunogenicity, sensitizing cancer cells to IO.
For hematologic malignancies, the most common combination approvals consisted of two non-KI targeted agents or one non-KI targeted agent plus one mAb. This finding is definitely related to the high proportion of approvals for MM, nearly half for hematologic malignancies, while that disease only accounts for 29.5% of them [29]. Indeed, most combinations for MM consisted of three types of non-KI targeted agents: a proteasome inhibitor (bortezomib or carfilzomib), an immunosuppressant (pomalidomide or lenalidomide), and a corticosteroid (dexamethasone), with or without a mAb (daratumumab or elotuzumab). The longer survival as compared to other hematologic malignances likely explain the investigations using so many similar combinations of agents being characterized by a favorable tolerability.
On the one hand, dose regimens of agents when given in combination and as monotherapy usually matched, although to a lesser extent that of CTs. Targeted therapies, although far from being toxicity free, usually display a non-linear relationship between dose and toxicity, so efficacy may occur at doses that do not induce clinically significant toxicity, at least during earlier cycles of treatment. In fact, model-based (D–R or E–R) approaches are deemed more appropriate than conventional rule-based (3 + 3) approaches for dose selection. As a result, the recommended doses in the labels of half of SM-targeted oncology drugs are lower than the MTD defined in the first in-human (FIH) study [30]. The recommended dose of these agents as monotherapy, which is based on optimal target engagement rather than on MTD, will more likely be tolerated also when given in combination.
On the other hand, CTs are usually given at their MTD defined in FIH studies by conventional 3 + 3 design, assuming that both efficacy and toxicity increase directly with dose. When given in combination, concomitant toxicities, either overlapping or not, may require dose reductions. This does not necessary entail a loss of efficacy, as demonstrated by those approved combinations with lower doses than when agents are given as monotherapy, in which the additive effect could overcome the dose reduction.
Furthermore, dose-finding studies were more intensive when the innovative agent was an SM. For these combinations, escalation studies were more abundant and thorough in their design, aiming at identifying the optimum dose regimens to be used thereafter. By contrast, early phase studies when the innovative agent was an LM rarely explored more than one dose level. This may be explained by the fact that doses of mAbs selected in early phases coincide less frequently with the MTD, and poorly predict the dose used in pivotal trials [31]. Indeed, conventional phase I studies with mAbs cannot capture their medium- and long-term toxicity, so alternative designs including a longer time frame for endpoint assessment at selected doses are deemed more appropriate for LMs.
This new dose-finding paradigm is currently being addressed by FDA’s Project Optimus [32], aimed at setting up expectations, enhance communication with developers, and develop new strategies for dose optimization for targeted agents.
Regardless of therapeutic class, an upward trend in the use of more innovative dose-escalation (e.g., adaptive) designs in early phase combination studies was not detected, despite efforts to promote adaptive approaches among statisticians and clinicians. The underlying reasons should be explored, since these designs claim to reach the MTD faster and treat most patients at or near the MTD in phase I single agent [33], and combination studies [34, 35].
Nevertheless, the design of early phase studies has gained complexity over this decade; in particular, phase I combination studies restricted to the definition of MTD are now infrequent, whereas expansion cohorts involving several hundred patients, building on an initial dose-escalation study that may have no more than 20 or 30 patients, are becoming more frequent.
A PoC study was not conducted for the majority of approved combinations. Despite the wide criteria used in this review that considered both efficacy extensions of phase I studies and stand-alone phase II studies, PoC studies were the exception rather than the rule. The use of futility analyses in pivotal studies was likely the preferred option to obtain substantial evidence of effectiveness instead of PoC studies, to speed up clinical development while paving the way toward new drug application (NDA) submission. This review could not capture which type of approved combinations were more likely to include a PoC study, either randomized controlled or single arm using historical data to estimate the SOC response rate.
Most approved combinations for hemato-oncology consisted of an add-on to SOC. Either NMEs/NBEs or already approved agents added to a backbone therapy were, by far, the most efficient approaches to reaching positive outcomes in pivotal studies over the comparator arm. Consequently, combination therapies have become the SOC for many cancer types and line settings, and more innovative approaches will be needed to add benefit to the SOC. In this sense, in 2013, the FDA released guidance to assist sponsors in the co-development of NMEs for use in combination, aimed at affecting multiple therapeutic targets to improve treatment response, minimize development of resistance, or minimize adverse events [3]. Although only a marginal proportion of the approved hemato-oncology combinations have followed this path, since then, it may become in the future the most promising approach to achieving superior outcomes over SOC, when certain criteria are met (i.e., serious disease, strong biological rationale, preclinical characterization suggesting a therapeutic advance, and reduction of activity if one or both agents are given as monotherapy). In terms of clinical pharmacology studies (e.g., assessment of bioavailability, characterization of PKs, mass balance, etc.), the guidance states that they should be done for each individual drug as if they were being developed separately, while studies to evaluate effects of intrinsic and extrinsic factors on PK and PD, and E–R, could be conducted either with the individual drugs or the combination [3].
Most studies involved PK assessments, although more restricted to the innovative agent the later the phase. Since a drug combination constitutes a unique therapy on its own, sponsors should carefully consider whether or not to include PK assessment of the secondary agent in the experimental and the control arm of late phase studies. Specifically, pivotal randomized studies with an A + B vs. B design, by far the most frequent, provide the ideal scenario to evaluate potential effects of the innovative agent over the PK of the secondary agent. But most importantly, they allow the development of innovative E–R models aimed at exploring the contribution of each agent to the overall effect of the combination therapy, especially when the dose regimen of any of the agents varies between arms. A paradigmatic case occurred in the area of immunosuppression, where an E–R model was successfully applied to derive the effect size of a noninferiority margin of the everolimus–tacrolimus combination in an otherwise negative pivotal study [36]. FDA members stated that they were not aware of any examples where E–R analysis was utilized to derive the effect size in the case of oncology drug combination approvals, although that example in transplant medicine can shed light on the utility of E–R analysis in other therapeutic areas [37]. Indeed, E–R analyses identified in this review were all limited to the innovative agent, and were dedicated to provide supportive evidence about the effectiveness of the innovative agent and to justify the adequacy of the selected dose (either by detecting an E–R relationship where most patients treated at the selected dose achieve active exposures, or by showing a the lack of meaningful relationship between a wide range of exposures and efficacy and/or safety).
Finally, performing PK assessments in all patients can be invaluable, since, as documented in numerous FDA reviews, these analyses are often leveraged to answer questions and support labeling when clinical pharmacology studies are not available [30].
Evaluations of PK interactions between combined agents were not routinely performed, especially when an SM and an LM were combined. This finding aligns with a review regarding the utility of PK studies in dose-finding studies of two oncology drugs over a previous 5-year period (2007–2011), which disregarded dedicated PK evaluations when a mechanistic basis was absent, in spite of the narrow therapeutic window, steep dose-toxicity profile and high interindividual variability in PK and pharmacodynamic (PD) on antineoplastics [38]. Nevertheless, evaluations of PK interactions, based on within-study, historically controlled, or PopPK analyses, were still reported for a large number of combinations, reflecting the awareness of drug developers and regulators to the risk of overexposure. Within-study evaluations, either as a crossover or parallel study, are the type of PK evaluation that provides more reliable information about potential interactions, since patients are randomized to sequence and/or treatment, thus ensuring the absence of selection bias. In any of the identified PK evaluations in this review, either within-study, historical or PopPK-based, no PK drug–drug interactions that could be ultimately used to guide dosing recommendations, were reported.
Conclusions
This review outlines the main clinical development features of drug combinations that have become part of the current armamentarium for hemato-oncologists during the past decade, based on a substantial amount of evidence of effectiveness over the SOC, according to FDA criteria.
CT-containing combinations were the most prevalent approvals, highlighting the role of these agents as the backbone therapy in cancer. Dose regimens of agents when given in combination and as monotherapy usually matched, although to a lesser extent for CTs.
When the innovative agent was an SM, dose-finding studies were more intensive. However, classical escalation is still the preferred option for phase I, despite claims of superiority of adaptive designs to find the recommended doses of combined agents. PoC studies were not conducted for the majority of combinations, which was likely to shorten development time. Most pivotal studies consisted of an add-on to the SOC versus the SOC, enabling the demonstration of a substantial gain in efficacy and no detrimental effect in tolerability.
Inclusion and design of PK assessments were largely determined by the specificities of each combination, even though their utility is deemed greater than for monotherapies, as they can inform about PK interactions. Most importantly, they can provide evidence about the contribution of each agent to the overall anti-tumor effect.
Looking toward the next decade, more effective and tolerable treatments combining either existing or next-generation drug types and cells (e.g., bispecific antibodies, CAR-T cells, etc.), will hopefully be meriting regulatory approval, as a result of dose optimization, and streamlined and flexible studies supported by E–R analyses, enabling both the definition of the best dosing regimens, and the accurate description of each agent’s effect size.
References
Dameshek W, Necheles T, Finkle H, Allen D (1965) Therapy of Acute Leukemia, 1965. Blood 26(2):220
Sitki Copur M, Harrold L, Chu E (2021) Common Chemotherapy Regimens in Clinical Practice. In: Chu E, DeVita VT Physicians' Cancer Chemotherapy Drug Manual 2020. In: Jones and Bartlett Publishers I (ed). Boston, USA,
Department of Health and Human Services FDA (2013) Codevelopment of two or more new investigational drugs for use in combination. https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM236669.pdf. Accessed 8 July 2018
Administration USFaD (2016–2019) Hematology/Oncology (Cancer) Approvals & Safety Notifications. https://www.fda.gov/drugs/resources-information-approved-drugs/hematologyoncology-cancer-approvals-safety-notifications. Accessed January 5, 2020
Centerwatch FDA approved drugs for oncology. http://www.centerwatch.com/drug-information/fda-approved-drugs/therapeutic-area/12/oncology. Accessed 2 January, 2020
Administration USFaD Drugs@FDA: FDA approved drug products. . http://www.accessdata.fda.gov/scripts/cder/daf/index.cfm. Accessed 2 January, 2020
Department of Health and Human Services FaDA Drugs@FDA: FDA Approved Drug Products. https://www.accessdata.fda.gov/scripts/cder/daf/. Accessed July 18, 2018
Nottage M, Siu LL (2002) Principles of clinical trial design. J Clin Oncol 20(18 Suppl):42S-46S
Lexchin J, Graham J, Herder M, Jefferson T, Lemmens T (2021) Regulators, pivotal clinical trials, and drug regulation in the age of COVID-19. Int J Health Serv 51(1):5–13. https://doi.org/10.1177/0020731420979824
Lu D, Lu T, Stroh M, Graham RA, Agarwal P, Musib L, Li CC, Lum BL, Joshi A (2016) A survey of new oncology drug approvals in the USA from 2010 to 2015: a focus on optimal dose and related postmarketing activities. Cancer Chemother Pharmacol 77(3):459–476. https://doi.org/10.1007/s00280-015-2931-4
Mirza M, Avall-Lundqvist E, Birrer M (2019) Combination of niraparib and bevacizumab versus niraparib alone as treatment of recurrent platinum-sensitive ovarian cancer: a randomized controlled chemotherapy-free study — NSGOAVANOVA2/ENGOT-OV24. J Clin Oncol 37:Suppl: 5505
Vogl DT, Dingli D, Cornell RF, Huff CA, Jagannath S, Bhutani D, Zonder J, Baz R, Nooka A, Richter J, Cole C, Vij R, Jakubowiak A, Abonour R, Schiller G, Parker TL, Costa LJ, Kaminetzky D, Hoffman JE, Yee AJ, Chari A, Siegel D, Fonseca R, Van Wier S, Ahmann G, Lopez I, Kauffman M, Shacham S, Saint-Martin JR, Picklesimer CD, Choe-Juliak C, Stewart AK (2018) Selective inhibition of nuclear export with oral selinexor for treatment of relapsed or refractory multiple myeloma. J Clin Oncol 36(9):859–866. https://doi.org/10.1200/JCO.2017.75.5207
Leonard JP, Jung SH, Johnson J, Pitcher BN, Bartlett NL, Blum KA, Czuczman M, Giguere JK, Cheson BD (2015) Randomized trial of lenalidomide alone versus lenalidomide plus rituximab in patients with recurrent follicular lymphoma: CALGB 50401 (Alliance). J Clin Oncol 33(31):3635–3640. https://doi.org/10.1200/JCO.2014.59.9258
Baselga J, Semiglazov V, van Dam P, Manikhas A, Bellet M, Mayordomo J, Campone M, Kubista E, Greil R, Bianchi G, Steinseifer J, Molloy B, Tokaji E, Gardner H, Phillips P, Stumm M, Lane HA, Dixon JM, Jonat W, Rugo HS (2009) Phase II randomized study of neoadjuvant everolimus plus letrozole compared with placebo plus letrozole in patients with estrogen receptor-positive breast cancer. J Clin Oncol 27(16):2630–2637. https://doi.org/10.1200/JCO.2008.18.8391
Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, Ettl J, Patel R, Pinter T, Schmidt M, Shparyk Y, Thummala AR, Voytko NL, Fowst C, Huang X, Kim ST, Randolph S, Slamon DJ (2015) The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol 16(1):25–35. https://doi.org/10.1016/S1470-2045(14)71159-3
Skolnik JM, Barrett JS, Jayaraman B, Patel D, Adamson PC (2008) Shortening the timeline of pediatric phase I trials: the rolling six design. J Clin Oncol 26(2):190–195. https://doi.org/10.1200/JCO.2007.12.7712
Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, Hamid O, Schuchter L, Cebon J, Ibrahim N, Kudchadkar R, Burris HA 3rd, Falchook G, Algazi A, Lewis K, Long GV, Puzanov I, Lebowitz P, Singh A, Little S, Sun P, Allred A, Ouellet D, Kim KB, Patel K, Weber J (2012) Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med 367(18):1694–1703. https://doi.org/10.1056/NEJMoa1210093
Tabernero J, Geel RV, Guren TK, Yaeger RD, Spreafico A, Faris JE, Yoshino T, Yamada Y, Kim TW, Bendell JC, Schuler MH, Lenz H, Eskens F, Desai J, Hochster HS, Avsar E, Demuth T, Sandor V, Elez E, Schellens JHM (2016) Phase 2 results: Encorafenib (ENCO) and cetuximab (CETUX) with or without alpelisib (ALP) in patients with advanced BRAF-mutant colorectal cancer (BRAFm CRC). J Clin Oncol 34(15_suppl):3544–3544. https://doi.org/10.1200/JCO.2016.34.15_suppl.3544
Lancet JE, Uy GL, Cortes JE, Newell LF, Lin TL, Ritchie EK, Stuart RK, Strickland SA, Hogge D, Solomon SR, Stone RM, Bixby DL, Kolitz JE, Schiller GJ, Wieduwilt MJ, Ryan DH, Hoering A, Banerjee K, Chiarella M, Louie AC, Medeiros BC (2018) CPX-351 (cytarabine and daunorubicin) liposome for injection versus conventional cytarabine plus daunorubicin in older patients with newly diagnosed secondary acute myeloid leukemia. J Clin Oncol 36(26):2684–2692. https://doi.org/10.1200/JCO.2017.77.6112
Garcia-Manero J, McCloskey J, Griffiths EA (2019) Pharmacokinetic Exposure Equivalence and Preliminary Efficacy and Safety from a Randomized Cross over Phase 3 Study (ASCERTAIN study) of an Oral Hypomethylating Agent ASTX727 (cedazuridine/decitabine) Compared to IV Decitabine. Blood 134(Supplement_1):846. https://doi.org/10.1182/blood-2019-122980
Dummer R, Ascierto PA, Gogas HJ, Arance A, Mandala M, Liszkay G, Garbe C, Schadendorf D, Krajsova I, Gutzmer R, Chiarion-Sileni V, Dutriaux C, de Groot JWB, Yamazaki N, Loquai C, Moutouh-de Parseval LA, Pickard MD, Sandor V, Robert C, Flaherty KT (2018) Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 19(5):603–615. https://doi.org/10.1016/S1470-2045(18)30142-6
Chari A, Vogl DT, Gavriatopoulou M, Nooka AK, Yee AJ, Huff CA, Moreau P, Dingli D, Cole C, Lonial S, Dimopoulos M, Stewart AK, Richter J, Vij R, Tuchman S, Raab MS, Weisel KC, Delforge M, Cornell RF, Kaminetzky D, Hoffman JE, Costa LJ, Parker TL, Levy M, Schreder M, Meuleman N, Frenzel L, Mohty M, Choquet S, Schiller G, Comenzo RL, Engelhardt M, Illmer T, Vlummens P, Doyen C, Facon T, Karlin L, Perrot A, Podar K, Kauffman MG, Shacham S, Li L, Tang S, Picklesimer C, Saint-Martin JR, Crochiere M, Chang H, Parekh S, Landesman Y, Shah J, Richardson PG, Jagannath S (2019) Oral selinexor-dexamethasone for triple-class refractory multiple myeloma. N Engl J Med 381(8):727–738. https://doi.org/10.1056/NEJMoa1903455
Salles G, Duell J, Gonzalez Barca E, Tournilhac O, Jurczak W, Liberati AM, Nagy Z, Obr A, Gaidano G, Andre M, Kalakonda N, Dreyling M, Weirather J, Dirnberger-Hertweck M, Ambarkhane S, Fingerle-Rowson G, Maddocks K (2020) Tafasitamab plus lenalidomide in relapsed or refractory diffuse large B-cell lymphoma (L-MIND): a multicentre, prospective, single-arm, phase 2 study. Lancet Oncol 21(7):978–988. https://doi.org/10.1016/S1470-2045(20)30225-4
Chari A, Suvannasankha A, Fay JW, Arnulf B, Kaufman JL, Ifthikharuddin JJ, Weiss BM, Krishnan A, Lentzsch S, Comenzo R, Wang J, Nottage K, Chiu C, Khokhar NZ, Ahmadi T, Lonial S (2017) Daratumumab plus pomalidomide and dexamethasone in relapsed and/or refractory multiple myeloma. Blood 130(8):974–981. https://doi.org/10.1182/blood-2017-05-785246
Makker V, Taylor MH, Aghajanian C, Oaknin A, Mier J, Cohn AL, Romeo M, Bratos R, Brose MS, DiSimone C, Messing M, Stepan DE, Dutcus CE, Wu J, Schmidt EV, Orlowski R, Sachdev P, Shumaker R, Casado Herraez A (2020) Lenvatinib plus pembrolizumab in patients with advanced endometrial cancer. J Clin Oncol 38(26):2981–2992. https://doi.org/10.1200/JCO.19.02627
Administration USFaD (2015) FARYDAK® (panobinostat) clinical pharmacology and biopharmaceutics review. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/205353Orig1s000TOC.cfm. Accessed October 16, 2021
(CHMP) CfMPfHU (2019) Tecentriq: EPAR - assessment report - variation
Bailly C, Thuru X, Quesnel B (2020) Combined cytotoxic chemotherapy and immunotherapy of cancer: modern times. NAR Cancer. https://doi.org/10.1093/narcan/zcaa002
Hungria VTM, Chiattone C, Pavlovsky M, Abenoza LM, Agreda GP, Armenta J, Arrais C, Avendaño-Flores O, Barroso F, Basquiera AL, Cao C, Cugliari MS, Enrico A, Foggliatto LM, Galvez KM, Gomez D, Gomez A, de Iracema D, Farias D, Lopez L, Mantilla WA, Martínez D, Mela MJ, Miguel CE, Ovilla R, Palmer L, Pavlovsky C, Ramos C, Remaggi G, Santucci R, Schusterschitz S, Sossa CL, Tuna-Aguilar E, Vela J, Santos T, de la Mora O, Machnicki G, Fernandez M, Barreyro P (2019) Epidemiology of hematologic malignancies in real-world settings: findings from the hemato-oncology latin america observational registry study. J Glob Oncol 5:1–19. https://doi.org/10.1200/jgo.19.00025
Bullock JM, Rahman A, Liu Q (2016) Lessons learned: dose selection of small molecule-targeted oncology drugs. Clin Cancer Res 22(11):2630–2638. https://doi.org/10.1158/1078-0432.CCR-15-2646
Jardim DL, Hess KR, Lorusso P, Kurzrock R, Hong DS (2014) Predictive value of phase I trials for safety in later trials and final approved dose: analysis of 61 approved cancer drugs. Clin Cancer Res 20(2):281–288. https://doi.org/10.1158/1078-0432.CCR-13-2103
Oncology Center of Excellence, Food and Drug Administration (2022) Project Optimus: Reforming the dose optimization and dose selection paradigm in oncology. https://www.fda.gov/about-fda/oncology-center-excellence/project-optimus. Accessed July 21, 2022
Iasonos A, O’Quigley J (2014) Adaptive dose-finding studies: a review of model-guided phase I clinical trials. J Clin Oncol 32(23):2505–2511. https://doi.org/10.1200/JCO.2013.54.6051
Riviere MK, Le Tourneau C, Paoletti X, Dubois F, Zohar S (2015) Designs of drug-combination phase I trials in oncology: a systematic review of the literature. Annals Oncol 26(4):669–674. https://doi.org/10.1093/annonc/mdu516
Wages NA, Ivanova A, Marchenko O (2016) Practical designs for Phase I combination studies in oncology. J Biopharm Stat 26(1):150–166. https://doi.org/10.1080/10543406.2015.1092029
Dumortier T, Looby M, Luttringer O, Heimann G, Klupp J, Junge G, Witte S, VanValen R, Stanski D (2015) Estimating the contribution of everolimus to immunosuppressive efficacy when combined with tacrolimus in liver transplantation: a model-based approach. Clin Pharmacol Ther 97(4):411–418. https://doi.org/10.1002/cpt.63
Zhao L, Hongshan L, Marathe A, Yu J, Rekić D, Mehrotra N, Sinha V, Wang Y (2016) New advancements in exposure-response analysis to inform regulatory decision making. In: Bonate PL, Howrad DR (eds) Pharmacokinetics in drug development. Springer International, Cham. https://doi.org/10.1007/978-3-319-39053-6_13
Wu K, House L, Ramirez J, Seminerio MJ, Ratain MJ (2013) Evaluation of utility of pharmacokinetic studies in phase I trials of two oncology drugs. Clin Cancer Res 19(21):6039–6043. https://doi.org/10.1158/1078-0432.CCR-13-0597
Author information
Authors and Affiliations
Contributions
SF and RL contributed to conception and design of the review. AS helped with the literature search and organization. AS, MU, and AC contributed to data extraction. SF wrote the manuscript. LPR drew the figures. BGA contributed with a critical assessment, and editorial review of the manuscript. AZ contributed with a critical assessment and expert review. All authors contributed to manuscript revision. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Fudio, S., Sellers, A., Pérez Ramos, L. et al. Anti-cancer drug combinations approved by US FDA from 2011 to 2021: main design features of clinical trials and role of pharmacokinetics. Cancer Chemother Pharmacol 90, 285–299 (2022). https://doi.org/10.1007/s00280-022-04467-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-022-04467-7