
Abstract
Purpose
Veliparib (V), an oral poly(ADP-ribose) polymerase (PARP) inhibitor, potentiates effects of alkylating agents and topoisomerase inhibitors in preclinical tumor models. We conducted a phase I trial of V with iv cyclophosphamide (C) and V plus iv doxorubicin (A) and C.
Methods
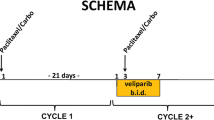
Objectives were to establish the maximum tolerated dose (MTD) of the combinations, characterize V pharmacokinetics (PK) in the presence and absence of C, measure PAR in peripheral blood mononuclear cells (PBMCs) and γH2AX in circulating tumor cells (CTCs). In Group 1, dose escalations of V from 10 to 50 mg every 12 h Days 1–4 plus C 450 to 750 mg/m2 Day 3 in 21-day cycles were evaluated. In Group 2, V doses ranged from 50 to 150 mg every 12 h Days 1–4 with AC (60/600 mg/m2) Day 3 in 21-day cycles. In Group 3, patients received AC Day 1 plus V Days 1–7, and in Group 4, AC Day 1 plus V Days 1–14 was given in 21-day cycles to evaluate effects on γH2AX foci.
Results
Eighty patients were enrolled. MTD was not reached for V and C. MTD for V and AC was V 100 mg every 12 h Days 1–4 with AC (60/600 mg/m2) Day 3 every 21 days. V PK appears to be dose-dependent and has no effect on the PK of C. Overall, neutropenia and anemia were the most common adverse events. Objective response in V and AC treated groups was 22% (11/49). Overall clinical benefit rate was 31% (25/80). PAR decreased in PBMCs. Percentage of γH2AX-positive CTCs increased after treatment with V and AC.
Conclusion
V and AC can be safely combined. Activity was observed in patients with metastatic breast cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Poly(ADP-ribose) polymerases (PARP) are essential nuclear enzymes that play an integral role in the recognition of DNA damage and facilitation of DNA repair [1]. Inhibition of PARP interferes with the repair of DNA damage, resulting in less efficient DNA repair following DNA-damaging insults [2]. Several PARP inhibitors are FDA approved in the treatment of breast and ovarian cancer. Veliparib is a potent, oral inhibitor of PARP-1 and PARP-2 [3]. In preclinical tumor models, veliparib potentiated the effects of several clinically effective DNA-damaging agents, including cyclophosphamide, temozolomide, topotecan, irinotecan, and platinums [4]. In a MX-1 BRCA1 deficient breast cancer model, the combination of cyclophosphamide at 12.5 mg/kg/day on days 20, 14, and 27 and veliparib 25 mg/kg/day resulted in tumor regression whereas single-agent cyclophosphamide, only delayed tumor growth slightly.
This formed the basis of this phase I study which sought to evaluate the role of veliparib as a chemopotentiator when combined with cyclophosphamide. The use of veliparib for chemopotentiation has been evaluated in several clinical trials to date [5,6,7]. Given the efficacy of anthracyclines in the treatment of breast cancer, the safety of combining doxorubicin and cyclophosphamide (AC) with veliparib was undertaken to determine a recommended phase II dose with the goal of potential incorporation into future adjuvant or neoadjuvant regimens. The primary objective of this trial was to establish the maximum tolerated dose (MTD) of veliparib with cyclophosphamide and veliparib combined with AC. Secondary objectives were to characterize the pharmacokinetics (PK) of veliparib and cyclophosphamide, alone and in combination, and to evaluate PARP activity by measurement of PAR levels in peripheral blood mononuclear cells (PBMCs). In the metastatic breast cancer patients treated with AC, we studied modulation of chemotherapy-induced DNA damage and repair by veliparib by measuring γH2AX, (phosphorylated histone protein), a marker of DNA double-strand breaks, in circulating tumor cells (CTCs) and tumor.
Methods
Patient eligibility
Adult patients were eligible with histologically confirmed metastatic malignancy for which no standard therapy was available. They were required to have an Eastern Cooperative Oncology Group Performance Status (ECOG PS) of ≤ 2, adequate organ function, and evaluable or measurable disease by RECIST 1.1. Patients enrolled to treatment with AC required a pre-treatment ejection fraction ≥ 50% and could not have had prior doxorubicin exposure of > 300 mg/m2. Only metastatic breast cancer patients were enrolled onto the schedules of veliparib for 7 days and 14 days with AC (Groups 3 and 4).
Eligible patients had adequate bone marrow (absolute neutrophil count ≥ 1500/mL; platelet count ≥ 100,000/mL; hemoglobin ≥ 9 g/dL), liver function (total bilirubin within normal institutional limits; ALT and AST ≤ 2.5 × ULN or ≤ 5 × with liver metastases, renal function (serum creatinine within normal institutional limits or ≥ 60 mL/min for with creatinine levels above institutional normal), and adequate coagulation status (international normalized ratio or prothrombin time or activated partial thromboplastin time ≤ 1.2 × ULN). Previous anticancer treatment had to be completed at least 4 weeks before study entry. There was no limit on prior cytotoxic chemotherapy regimens. Medications or substances that were strong inhibitors or strong inducers of CYP3A4, CYP2B6, CYP2C9, or CYP2C19 were not allowed. Patients with CNS metastases were also allowed if treated, off steroid treatment for > 3 months, and asymptomatic. Other exclusion criteria included history of active seizures, active systemic infections, symptomatic congestive heart failure, and any impairment to swallow capsules.
Study design and treatment plan
This single-institution phase I study was conducted at Rutgers Cancer Institute of New Jersey. The study received approval of the institutional review board and was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice. Written informed consent was obtained from all patients before enrollment onto the study. In Group 1, a standard 3 + 3 design was used with a starting dose of 10 mg veliparib administered once every 12 h (h) given Days 1–4 and escalating in varying increments to a maximum dose of 200 mg, and the starting dose of iv cyclophosphamide was 450 mg/m2, increasing to 750 mg/m2 given on Day 3 on a 21-day cycle. Group 2 was treated with veliparib doses ranging from 50 to 150 mg every 12 h on Days 1–4 with fixed dosing of AC at 60/600 mg/m2 on day 3 every 21 days. Further doxorubicin was omitted after a cumulative dose of 420 mg/m2 was reached. Alternate schedules of veliparib combined with AC were explored. To maximize the rate of DNA damage, we evaluated giving veliparib on the same day as AC and increased the duration that veliparib was administered. Thus, the schedule for Group 3 was AC on Day 1 with veliparib 100 mg every 12 h Days 1–7 and the schedule for Group 4 was AC on Day 1 with veliparib 100 mg every 12 h Days 1–14. The goal was to obtain a minimum of 10 patients with ≥ 10 CTCs in the sample drawn on Day 1 to perform our biomarker analysis. A DLT was defined as any grade 3 or higher clinically significant non-hematologic toxicity and any grade 4 neutropenia lasting ≥ 7 days, grade 4 neutropenia and fever of ≥ 38.5 °C, ≥ grade 3 neutropenia with ≥ grade 3 infection, or grade 4 thrombocytopenia. MTD was defined as the dose level in which no more than 2/6 or 1/3 patients experience dose-limiting toxicity (DLT) and at least 2/3 or 3/6 patients treated with the next higher dose level will have had DLT.
Study assessments
Physical exams were performed once every 3 weeks. Hematology and chemistry were obtained weekly during the first 9 weeks and then once every 3 weeks thereafter. Laboratory assessments at baseline included a complete blood count with differential (CBC with diff), general chemistry panel, prothrombin time, activated partial thromboplastin time, INR, and urinalysis. A CBC with diff and chemistry panel were collected weekly during the first three cycles. Electrocardiograms were obtained at baseline and Day 1 of each cycle. For patients receiving doxorubicin, an echo or MUGA was obtained every 9 weeks. Adverse events were graded according to the NCI CTCAE (version 4.0). All solid tumor patients underwent CT scanning at baseline and every 9 weeks for evaluation of efficacy based on RECIST version 1.1 [8]. Response in NHL was assessed based on the revised International Working Group Response Criteria for NHL [9].
Pharmacokinetics and pharmacodynamics assessments
Blood samples for PK assessments were collected only in Groups 1 and 2 during cycle 1 and 2 at multiple time points up to 24 h after administration of the C2D1 veliparib dose. Venous blood samples of 4 ml each for analysis of veliparib plasma concentrations, were collected at the following time points: Cycle 1 Day 1, pre-dose, 30 min, 1, 1.5, 2, 3, 4, 6, 7–8, and 24 h after administration (before the third dose of veliparib); Cycle 1 Day 3, pre-dose, 30 min, 1, 1.5, 2, 3, 4, 6,7–8, and 24 h after administration (before the seventh dose of veliparib); and Cycle 2 Day 1, pre-dose, 30 min, 1, 1.5, 2, 3, 4, 6,7–8, and 24 h after administration (before the third dose of veliparib). Venous blood samples of 5 mL each for analysis of cyclophosphamide plasma concentrations, were collected at the following times: Cycle 1 Day 3, pre-dose (before ingestion of veliparib and start of cyclophosphamide infusion), 0.5, 1 (end of infusion), 1.5, 2, 3, 4, 6, 7–8, and 24 h after the start of cyclophosphamide infusion. The effect of cyclophosphamide in Group 1 or cyclophosphamide and doxorubicin in Group 2 on veliparib PK was examined by the comparison of Cmax and AUC and tested by comparing these exposure parameters of Cycle 1 Day 1 with Cycle 1 Day 3 and Cycle 2 Day 1 using Wilcoxon signed-rank tests. The predicted steady-state Cycle 1 Day 1 Cmax was tested against the observed Cycle 1 Day 3 Cmax and predicted steady-state Cycle 2 Day 1 Cmax and the Cycle 1 Day 1 AUC0-inf tested against the Cycle 1 Day 3 AUC0-12 and Cycle 2 Day 1 AUC0-inf. The predicted steady-state Cmax of Cycle 1 Day 1 and Cycle 2 Day 1 were calculated by multiplying the observed Cmax by the theoretical accumulation ratio that was determined using the observed patient and day specific elimination rate and a 12-h dosing interval.
Plasma concentrations of cyclophosphamide and its metabolite, 4-OH cyclophosphamide, were determined by liquid chromatography-mass spectrometry (LC-MS) [10] and were estimated using noncompartmental analysis using WinNonlin 2.1 software (Pharsight Corp, Palo Alto, CA). Veliparib PK was quantitated with a previously validated LC–MS assay [11] and determined non-compartmentally using PK Solutions 2.0 (Summit Research Services, Montrose, CO). Poly(ADP-ribose) (PAR), a product of PARP, was measured in PBMCs in Groups 1 and 2 using a validated immunoassay [12].
Levels of γH2AX were measured in CTCs from Groups 3 and 4 with standard operating procedures previously described for the validated immunofluorescence assay [13]. PBMC samples for PAR analysis were collected in 15 mL tubes of the first two cycles over a 24-h period, at the following time points: Day 1 pre-dose, and Day 3 pre-dose (before start of cyclophosphamide infusion), 2, 4, at 6 or 7, and 24 h after the start of cyclophosphamide infusion. CTC samples for γH2AX analysis were collected in 10 mL CellSave tubes during cycle 1 on days 1, 2, 7, and 14 for Groups 3 and 4 before dosing with veliparib. If there was detectable γH2AX in CTCs by day 7, then the sample on day 14 was not collected. The Cell Search® System (Veridex, LLC) was used to enumerate CTCs. In Groups 3 and 4, patients that had disease that could be safely biopsied, had tumor tissue collected before the start of treatment and then on Cycle 1 Day 2 ± 24 h, after administration of chemotherapy, for γH2AX analysis.
Statistical analysis
Descriptive statistics were used for baseline characteristics, safety assessments, PK parameters, and tumor response. Wilcoxon signed-rank test was used for the comparison of changes from baseline for CTCs, γH2AX positive CTCs, and percentage of γH2AX positive CTCs (%). Wilcoxon two-sample test was used for comparison of group difference on the change from baseline. Logistic regression model was used for analysis of the parameters vs response and BRCA status.
Results
Patient characteristics
A total of 80 patients were enrolled between August 2008 and April 2014. Patient demographics are listed in Table 1. Breast cancer was the most frequent tumor type (74%). Most patients had an ECOG PS of 1 (68%). All patients were pretreated with 68% having received three or more prior lines of systemic treatment.
Safety and tolerability
In Group 1, the starting doses were 10 mg veliparib every 12 h on Days 1–4 and 450 mg/m2 cyclophosphamide on Day 3 in a 21-day cycle. At dose levels 1–3, cyclophosphamide remained constant with veliparib escalating to 10, 20, and 50 mg. No DLTs were observed at the first two dose levels (Table 2). At the 50/450 dosing, a DLT of grade 2 thrombocytopenia that caused a delay of > 2 weeks in starting cycle 2 occurred in a non-Hodgkin’s lymphoma patient. An additional three patients were enrolled, and none had DLTs, so that among the six patients treated at dose level 3 there was one DLT. The dose escalation continued with veliparib remaining constant at 50 mg and cyclophosphamide escalated to 600 and 750 mg/m2 for dose levels 4 and 5, respectively, and no DLTs were observed during the first cycle. In further dose escalations at dose levels 6, 7, and 8, cyclophosphamide remained constant at 750 mg/m2 and veliparib was escalated to 100, 150, and 200 mg. At dose level 6, in which patients were treated with 100 mg veliparib and 750 mg/m2 cyclophosphamide, two patients experienced grade 3 and grade 4 neutropenia requiring a dose reduction of cyclophosphamide, but they did not meet the strict definition for DLT. To gain additional safety data, an additional three patients were intended to enroll for a total of six patients, but one was not evaluable, and had to be replaced which resulted in a total enrollment of seven patients. There were no observed DLTs. Three patients each were enrolled onto 150/750 and 200/750 with no patients experiencing a DLT. While an MTD was not formally defined for this combination, it is notable that two near DLTs of a grade 3 and a grade 4 neutropenia requiring dose reduction of cyclophosphamide were observed at 100 mg veliparib and 750 mg/m2 cyclophosphamide. Moreover, three patients (two at 150/750 level and one at 200/750 level) required growth factor support. Thus, the safety of veliparib given with cyclophosphamide at high doses requires a cautious approach.
Given the interest in using PARP inhibitors in the treatment of breast cancer, and potentially in the adjuvant or neoadjuvant setting, doxorubicin added to veliparib and cyclophosphamide was evaluated (Group 2). Veliparib doses ranged from 50 to 100 mg to 150 mg every 12 h on Days 1–4 with fixed dosing of AC (60/600 mg/m2) on day 3 every 21 days. Two instances of grade 3 febrile neutropenia were considered dose-limiting at the 150 mg veliparib dose level. The MTD or recommended phase II dose of veliparib was declared at the previous dose level 100 mg every 12 h with AC, and this was expanded to enroll up to a total of 12 patients (one was inevaluable for a total of 13). In Group 3, AC was given on Day 1 with veliparib 100 mg every 12 h Days 1–7 in a 21-day cycle, with 14 patients enrolled, and 10 patients had ≥ 10 CTCs in Day 1 samples. In Group 4, AC was given on Day 1 with veliparib 100 mg every 12 h, Days 1–14 (given longer vs Group 3) in a 21-day cycle, with 13 patients enrolled, and only 6 patients had ≥ 10 CTCs in Day 1 samples. The most common treatment related adverse events were hematologic in nature in each group (Table 3). Across the whole trial, 28% (n = 22) patients were given growth factor support after cycle 1. Overall, 23 patients required a dose reduction of veliparib, and 3 patients had a dose reduction of cyclophosphamide. There was higher incidence of grades 3 and 4 hematologic toxicity with increasing the duration of veliparib to 7 days and 14 days.
Pharmacokinetics and pharmacodynamics
Veliparib administered alone exhibited approximately dose-proportional increases in Cmax and AUC0-7 in the explored 50 to 200 mg dose range (Fig. 1A, B for C1D1; online only supplementary information Table S1 and Fig S1 for C2D1). Veliparib Cmax was comparable, but AUC0-7 was significantly higher on C1D3 when veliparib was administered with either combination therapy compared to C1D1 when veliparib was administered alone (Fig. 1C, D). Apparent clearance, volume of distribution and half-life of veliparib in this study were consistent with those reported previously [5]. Cyclophosphamide administered iv at 450 mg/m2 and 750 mg/m2 was not influenced by veliparib orally. The PK parameters of cyclophosphamide following iv infusion were also consistent with previous reports (online only supplementary information Fig S2) [10].

Veliparib dose proportionality in Groups 1 and 2. A Mean (+ SD) veliparib Cmax vs veliparib dose. B Mean AUC0-7 vs veliparib dose Cycle 1 Day 1. C Dose-normalized Cmax Cycle 1 Day 1 vs dose-normalized observed Cmax Cycle 1 Day 3 and Cycle 2 Day 1. D Dose normalized AUC0-∞ Cycle 1 Day 1 vs dose-normalized AUC0-12 Cycle 1 Day 3 and dose-normalized AUC Cycle 2 Day 1
Mean PAR levels in PBMCs over the first 72 h during cycle 1 are shown in Fig. 2A. Of the fifty patients in Groups 1 and 2 with quantifiable PAR, 40 were considered evaluable with PBMCs available at 4 h post-dosing of veliparib on Day 3, and 27 (68%) had over 75% reduction in PAR across all dose levels. Cyclophosphamide did not seem to alter the ability of veliparib to inhibit PARP, because after addition of cyclophosphamide, PAR levels do not return to baseline. Total CTCs and the number of γH2AX-positive CTCs were measured in samples from 27 breast cancer patients at baseline and after treatment with veliparib and AC during cycle 1 (Fig. 2B). There was a significant decrease in CTCs with Groups 3 and 4 combined from Day 1 to Day 7 (p = 0.0007) and from Day 1 to Day 14 (p = 0.0009). Also, there was a significant increase in the percentage of γH2AX-positive CTCs in Groups 3 and 4 combined from Day 1 to Day 7 (p = 0.016) and from Day 1 to Day 14 (p = 0.0006) (Fig. 2C). The percentage of γH2AX-positive CTCs increased to ≥ 50% by Day 7 in 59% (16/27) of patients and persisted to day 14 in 10 patients. There was no association between clinical benefit or BRCA status with the change in CTCs, number of γH2AX-positive CTCs, or percentage of γH2AX-positive CTCs. Paired tumor biopsies from 10 patients were analyzed for γH2AX. Only 6 paired samples yielded enough tissue for evaluating nuclear γH2AX (online only supplementary information Fig S3). In 4 paired samples, the γH2AX nuclear area positive score (%NAP), increased after treatment with veliparib and AC, with increases in NAP scores from < 5% at baseline to 5.2%, 10.96%, 3.23%, (representative γH2AX staining in tumor biopsies shown in Fig. 2D) and 4.5%, with stable disease (SD), progressive disease, partial response (PR) and inevaluable as the responses, respectively.

Pharmacodynamics of veliparib. A PAR levels relative to baseline by dose level in PBMCs before and after veliparib with cyclophosphamide. B Total CTCs and C percentage of CTCs positive for γH2AX, before and after treatment with veliparib and AC. D %NAP was increased after veliparib and AC treatment in a breast cancer patient who achieved a PR. Representative γH2AX signal in green (right)
Treatment efficacy
Complete response was not achieved by any patients (Table 4). In Group 1, a metastatic prostate cancer patient with liver metastases had a PR treated at dose level 4 and 4/31 (10%) had SD lasting ≥ 3 months, for an overall clinical benefit rate (CBR) of 13%. In Group 2, none of the four non-breast cancer patients showed response and of the 18 metastatic breast cancer patients, four had a PR and 5 patients experienced SD ≥ 3 months. The CBR was 41% in Group 2. The four PRs had triple-negative breast cancer and three had a BRCA mutation, with response durations in the range of 5 to 25 months. In Groups 3 and 4, of 27 evaluable patients, seven had PRs and five had SD (range 3 to 21 months). The longest responder was a BRCA2 carrier with a hormone receptor-positive, HER2-negative breast cancer with a PR for 21 months in Group 3. Another BRCA2 carrier with hormone receptor-positive, HER2-negative breast cancer had SD for 21 months treated in Group 4. Among the 59 metastatic breast cancer patients treated, 5 patients with a BRCA1/2 mutation were noted to have PRs. However, BRCA germline status was not known for all subjects.
Discussion
This phase I trial evaluated the tolerability, safety, and PK of veliparib in combination with iv cyclophosphamide in patients with advanced solid tumors and subsequently tested veliparib with AC in metastatic breast cancer. This study was conducted on the basis of preclinical work that veliparib potentiates the activity of cyclophosphamide in a breast cancer xenograft model [4]. Veliparib in combination with iv cyclophosphamide had an acceptable safety profile, which allowed dose escalation to dose level 8, the highest planned dose. MTD was not reached using a 3 + 3 design, but myelotoxicity was observed with this regimen necessitating dose reductions of cyclophosphamide or veliparib and use of growth factor support in some patients.
Variability exists with how veliparib is dosed when partnered with chemotherapy in regard to its administration frequency, duration, and timing relative to chemotherapy as shown in several phase I combination studies. In our trial, for Groups 1 and 2, veliparib was dosed twice daily prior to, during and after chemotherapy to maximize the potential synergy of PARP inhibitor and the DNA-damaging effects from cyclophosphamide. With this schedule, there was rapid and sustained inhibition of PAR in PBMCs. This was one of the earlier trials conducted with veliparib, and at the time the study was written, it was important to demonstrate that veliparib significantly reduced PAR levels, indicating that the drug was hitting its therapeutic target. At the time this study was developed, little was known about any veliparib drug-drug interactions. We were concerned about the potential for co-administration of cyclophosphamide to increase veliparib metabolism due to the potential of CYP450 enzyme induction by cyclophosphamide. Additionally, incubations of veliparib with recombinant cytochrome P450s suggested potential involvement of CYP1A1, 2D6, 2C19, and 3A4. Our study showed that co-administration of cyclophosphamide minimally increased veliparib AUC but did not significantly affect Cmax in Group 1. Similar observations in Group 2 makes it unlikely that doxorubicin independently influenced observed differences in AUC. Veliparib is primarily metabolized by CYP2D6 and only partially by CYP1A2, 2C19, and 3A4 [14]. The overlap in CYP3A4 substrate specificity with cyclophosphamide could explain the observed increase in exposure on C1D3 when co-administered with veliparib [15]. There was a statistical difference between C1D1 and C1D3 veliparib AUC, that may be explained by the accumulation in veliparib exposure after the repeated twice-daily dosing difference and it is not indicative that cyclophosphamide or doxorubicin influenced the PK of veliparib. PKs parameters on all analyzed days were consistent with previous reports of veliparib exposure across the entire dose range of this study [5, 16, 17]. These studies, as well as a single-agent population PK model, were also in agreement with the observed apparent clearance, apparent volume of distribution, and half-life [18]. Drug exposure (AUC) of veliparib showed linearity with dose, which is similar to previous reports of veliparib PK across the doses studied in this trial [5, 7, 19]. Additionally, veliparib did not alter the pharmacokinetics of cyclophosphamide.
Our trial is the only one that evaluated veliparib combined with iv cyclophosphamide, and the highest dose of veliparib reached was 200 mg every 12 h. Other trials have tested once-daily dosing of veliparib with low metronomic-dose oral cyclophosphamide [5, 20, 21]. Anampa et al. conducted a phase I study in metastatic breast cancer patients with twice a day dosing of veliparib, as in our study, and was able to define a recommended phase II dose of veliparib 200 mg BID and cyclophosphamide 125 mg po daily in 21-day cycles, with nausea and headache being DLTs [22]. Similar to our phase I trial, myelosuppression was reported and clinical benefit (19.2%) was observed with oral cyclophosphamide.
To our knowledge, this is the first clinical trial to evaluate the combination of a PARP inhibitor with AC in breast cancer. Febrile neutropenia was dose-limiting. The recommended phase II dose was established at veliparib 100 mg every 12 h Days 1–4 with AC at 60/600 mg/m2 on Day 3 every 21 days. Dose reductions of veliparib occurred in Groups 3 and 4 as well as use of growth factor support.
The analysis of a pharmacodynamic biomarker was an important endpoint in Groups 3 and 4 and the feasibility of performing it in CTCs at our institution was tested. We were able to evaluate modulation of DNA damage by veliparib (when given longer than 4 days) and AC by measuring γH2AX foci, a phosphorylated histone protein and marker of DNA double-strand breaks, in CTCs. We did observe decreases in total CTCs in most patients treated with the combination of veliparib and AC and consistent increases in the percentage of γH2AX-positive CTCs, which indicated the drug effect in the blood. Other phase I studies of veliparib with alkylating agents or topoisomerase inhibitors have reported similar pharmacodynamic results, proving that modulation of DNA damage can be achieved with the combination [5, 6].
In the clinical safety evaluation, drug-related adverse events were surprisingly limited. The results of a favorable safety profile may be related to infrequent clinical examinations which occurred every 3 weeks. Incorporation of a weekly targeted physical exam during the initial cycles may have captured a more comprehensive picture of the toxicity profile of these combinations.
In summary, the combination of veliparib with iv cyclophosphamide and veliparib plus AC is associated with enhanced myelosuppression and reductions of treatment doses, like other phase I studies combining PARP inhibitors and DNA-damaging agents. This regimen also showed some anti-tumor activity in metastatic breast cancer patients. We provided proof of concept of pharmacodynamic modulation of PARP by veliparib via measurement of PAR levels and γH2AX-positive CTCs. Though the combination of veliparib and AC seemed tolerable, it is likely challenging to incorporate this three-drug regimen as treatment for early-stage breast cancer given the hematologic toxicities. These data add to the already growing body of literature of the potential therapeutic synergy of PARP inhibitors and DNA-damaging chemotherapy.
References
Ame JC, Spenlehauer C, de Murcia G (2004) The PARP superfamily. BioEssays 26(8):882–893. https://doi.org/10.1002/bies.20085
Jagtap P, Szabo C (2005) Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat Rev Drug Discov 4(5):421–440. https://doi.org/10.1038/nrd1718
Penning TD, Zhu GD, Gandhi VB, Gong J, Liu X, Shi Y, Klinghofer V, Johnson EF, Donawho CK, Frost DJ, Bontcheva-Diaz V, Bouska JJ, Osterling DJ, Olson AM, Marsh KC, Luo Y, Giranda VL (2009) Discovery of the Poly(ADP-ribose) polymerase (PARP) inhibitor 2-[(R)-2-methylpyrrolidin-2-yl]-1H-benzimidazole-4-carboxamide (ABT-888) for the treatment of cancer. J Med Chem 52(2):514–523. https://doi.org/10.1021/jm801171j
Donawho CK, Luo Y, Luo Y, Penning TD, Bauch JL, Bouska JJ, Bontcheva-Diaz VD, Cox BF, DeWeese TL, Dillehay LE, Ferguson DC, Ghoreishi-Haack NS, Grimm DR, Guan R, Han EK, Holley-Shanks RR, Hristov B, Idler KB, Jarvis K, Johnson EF, Kleinberg LR, Klinghofer V, Lasko LM, Liu X, Marsh KC, McGonigal TP, Meulbroek JA, Olson AM, Palma JP, Rodriguez LE, Shi Y, Stavropoulos JA, Tsurutani AC, Zhu GD, Rosenberg SH, Giranda VL, Frost DJ (2007) ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin Cancer Res 13(9):2728–2737. https://doi.org/10.1158/1078-0432.CCR-06-3039
Kummar S, Ji J, Morgan R, Lenz HJ, Puhalla SL, Belani CP, Gandara DR, Allen D, Kiesel B, Beumer JH, Newman EM, Rubinstein L, Chen A, Zhang Y, Wang L, Kinders RJ, Parchment RE, Tomaszewski JE, Doroshow JH (2012) A phase I study of veliparib in combination with metronomic cyclophosphamide in adults with refractory solid tumors and lymphomas. Clin Cancer Res 18(6):1726–1734. https://doi.org/10.1158/1078-0432.CCR-11-2821
LoRusso PM, Li J, Burger A, Heilbrun LK, Sausville EA, Boerner SA, Smith D, Pilat MJ, Zhang J, Tolaney SM, Cleary JM, Chen AP, Rubinstein L, Boerner JL, Bowditch A, Cai D, Bell T, Wolanski A, Marrero AM, Zhang Y, Ji J, Ferry-Galow K, Kinders RJ, Parchment RE, Shapiro GI (2016) Phase I safety, pharmacokinetic, and pharmacodynamic study of the poly(ADP-ribose) polymerase (PARP) inhibitor veliparib (ABT-888) in combination with irinotecan in patients with advanced solid tumors. Clin Cancer Res 22(13):3227–3237. https://doi.org/10.1158/1078-0432.CCR-15-0652
Atrafi F, Groen HJM, Byers LA, Garralda E, Lolkema MP, Sangha RS, Viteri S, Chae YK, Camidge DR, Gabrail NY, Hu B, Tian T, Nuthalapati S, Hoening E, He L, Komarnitsky P, Calles A (2019) A phase I dose-escalation study of veliparib combined with carboplatin and etoposide in patients with extensive-stage small cell lung cancer and other solid tumors. Clin Cancer Res 25(2):496–505. https://doi.org/10.1158/1078-0432.CCR-18-2014
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45(2):228–247. https://doi.org/10.1016/j.ejca.2008.10.026
Cheson BD, Pfistner B, Juweid ME, Gascoyne RD, Specht L, Horning SJ, Coiffier B, Fisher RI, Hagenbeek A, Zucca E, Rosen ST, Stroobants S, Lister TA, Hoppe RT, Dreyling M, Tobinai K, Vose JM, Connors JM, Federico M, Diehl V, International Harmonization Project on Lymphoma (2007) Revised response criteria for malignant lymphoma. J Clin Oncol 25(5):579–586. https://doi.org/10.1200/JCO.2006.09.2403
Emmenegger U, Shaked Y, Man S, Bocci G, Spasojevic I, Francia G, Kouri A, Coke R, Cruz-Munoz W, Ludeman SM, Colvin OM, Kerbel RS (2007) Pharmacodynamic and pharmacokinetic study of chronic low-dose metronomic cyclophosphamide therapy in mice. Mol Cancer Ther 6(8):2280–2289. https://doi.org/10.1158/1535-7163.MCT-07-0181
Parise RA, Shawaqfeh M, Egorin MJ, Beumer JH (2008) Liquid chromatography-mass spectrometric assay for the quantitation in human plasma of ABT-888, an orally available, small molecule inhibitor of poly(ADP-ribose) polymerase. J Chromatogr B Analyt Technol Biomed Life Sci 872(1–2):141–147. https://doi.org/10.1016/j.jchromb.2008.07.032
Ji J, Kinders RJ, Zhang Y, Rubinstein L, Kummar S, Parchment RE, Tomaszewski JE, Doroshow JH (2011) Modeling pharmacodynamic response to the poly(ADP-Ribose) polymerase inhibitor ABT-888 in human peripheral blood mononuclear cells. PLoS ONE 6(10):e26152. https://doi.org/10.1371/journal.pone.0026152
Wang LH, Pfister TD, Parchment RE, Kummar S, Rubinstein L, Evrard YA, Gutierrez ME, Murgo AJ, Tomaszewski JE, Doroshow JH, Kinders RJ (2010) Monitoring drug-induced gammaH2AX as a pharmacodynamic biomarker in individual circulating tumor cells. Clin Cancer Res 16(3):1073–1084. https://doi.org/10.1158/1078-0432.CCR-09-2799
Li X, Delzer J, Voorman R, de Morais SM, Lao Y (2011) Disposition and drug-drug interaction potential of veliparib (ABT-888), a novel and potent inhibitor of poly(ADP-ribose) polymerase. Drug Metab Dispos 39(7):1161–1169. https://doi.org/10.1124/dmd.110.037820
Beijnen JH, Schellens JH (2004) Drug interactions in oncology. Lancet Oncol 5(8):489–496. https://doi.org/10.1016/S1470-2045(04)01528-1
Rodler ET, Kurland BF, Griffin M, Gralow JR, Porter P, Yeh RF, Gadi VK, Guenthoer J, Beumer JH, Korde L, Strychor S, Kiesel BF, Linden HM, Thompson JA, Swisher E, Chai X, Shepherd S, Giranda V, Specht JM (2016) Phase I study of veliparib (ABT-888) combined with cisplatin and vinorelbine in advanced triple-negative breast cancer and/or BRCA mutation-associated breast cancer. Clin Cancer Res 22(12):2855–2864. https://doi.org/10.1158/1078-0432.CCR-15-2137
Stoller R, Schmitz JC, Ding F, Puhalla S, Belani CP, Appleman L, Lin Y, Jiang Y, Almokadem S, Petro D, Holleran J, Kiesel BF, Ken Czambel R, Carneiro BA, Kontopodis E, Hershberger PA, Rachid M, Chen A, Chu E, Beumer JH (2017) Phase I study of veliparib in combination with gemcitabine. Cancer Chemother Pharmacol 80(3):631–643. https://doi.org/10.1007/s00280-017-3409-3
Niu J, Scheuerell C, Mehrotra S, Karan S, Puhalla S, Kiesel BF, Ji J, Chu E, Gopalakrishnan M, Ivaturi V, Gobburu J, Beumer JH (2017) Parent-metabolite pharmacokinetic modeling and pharmacodynamics of veliparib (ABT-888), a PARP inhibitor, in patients with BRCA 1/2-mutated cancer or PARP-sensitive tumor types. J Clin Pharmacol 57(8):977–987. https://doi.org/10.1002/jcph.892
Berlin J, Ramanathan RK, Strickler JH, Subramaniam DS, Marshall J, Kang YK, Hetman R, Dudley MW, Zeng J, Nickner C, Xiong H, Komarnitsky P, Shepherd SP, Hurwitz H, Lenz HJ (2018) A phase 1 dose-escalation study of veliparib with bimonthly FOLFIRI in patients with advanced solid tumours. Br J Cancer 118(7):938–946. https://doi.org/10.1038/s41416-018-0003-3
Kummar S, Oza AM, Fleming GF, Sullivan DM, Gandara DR, Naughton MJ, Villalona-Calero MA, Morgan RJ Jr, Szabo PM, Youn A, Chen AP, Ji J, Allen DE, Lih CJ, Mehaffey MG, Walsh WD, McGregor PM 3rd, Steinberg SM, Williams PM, Kinders RJ, Conley BA, Simon RM, Doroshow JH (2015) Randomized trial of oral cyclophosphamide and veliparib in high-grade serous ovarian, primary peritoneal, or fallopian tube cancers, or BRCA-mutant ovarian cancer. Clin Cancer Res 21(7):1574–1582. https://doi.org/10.1158/1078-0432.CCR-14-2565
Kummar S, Wade JL, Oza AM, Sullivan D, Chen AP, Gandara DR, Ji J, Kinders RJ, Wang L, Allen D, Coyne GO, Steinberg SM, Doroshow JH (2016) Randomized phase II trial of cyclophosphamide and the oral poly (ADP-ribose) polymerase inhibitor veliparib in patients with recurrent, advanced triple-negative breast cancer. Invest New Drugs 34(3):355–363. https://doi.org/10.1007/s10637-016-0335-x
Anampa J, Chen A, Wright J, Patel M, Pellegrino C, Fehn K, Sparano JA, Andreopoulou E (2018) Phase I trial of veliparib, a poly ADP ribose polymerase inhibitor, plus metronomic cyclophosphamide in metastatic HER2-negative breast cancer. Clin Breast Cancer 18(1):e135–e142. https://doi.org/10.1016/j.clbc.2017.08.013
Acknowledgements
We acknowledge Dr. Ulf Niemeyer, of Niomech -IIT GmbH, Bielefeld, Germany for the generous gift of PBOX-d4 (internal standard) for analysis of 4-OH-CPA.
Funding
Supported by Rutgers Cancer Institute of New Jersey Cancer Center Support Grant/Core Grant and NCI grants (U01-CA-132194-01, UM1-CA-186716, UM1-CA-186690, U01-CA099168, and R50 CA211241). This project used the UPCI Cancer Pharmacokinetics and Pharmacodynamics Facility and was supported in part by award P30-CA47904.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
A. Tan has received research grants from Arvinas, Deciphera, Daiichi-Sankyo, Genentech, Merck, and Pfizer; advisory board member/paid consultant for Athenex, AstraZeneca, Eisai, G1 Therapeutics, Novartis, and Immunomedics. M. Stein has received research grants from Merck, Exelixis, Oncoceutics, Janssen, Medivation/Astellas, Advaxis, Suzhou Kintor, Harpoon, Bristol-Meyers Squibb, Genocea, Eli Lilly, Seattle Genetics and Xencor. R. Moss is an employee for Bristol-Myers Squibb. J. Malhotra has received commercial research grants from AstraZeneca, Beyond Spring, Bristol-Myers Squibb, Biohaven and Pfizer. J. Aisner is on the DMC for EMD Serono. J. Mehnert has received research grants from Merck, EMD Serono, Pfizer, Genentech, Amgen, Boehringer Ingelheim, Array BioPharma, Immunocore, AstraZeneca, Incyte, Macrogenics, Bristol-Myers Squibb, Novartis, and Polynoma. No potential conflicts of interest were disclosed by the other authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Tan, A.R., Chan, N., Kiesel, B.F. et al. A phase I study of veliparib with cyclophosphamide and veliparib combined with doxorubicin and cyclophosphamide in advanced malignancies. Cancer Chemother Pharmacol 89, 49–58 (2022). https://doi.org/10.1007/s00280-021-04350-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-021-04350-x