
Abstract
Objective
Although DNA-mismatch-repair-deficient (dMMR) status and aberrant expression of miRNAs are both critically implicated in the pathogenesis of resistance to 5-fluorouracil (5-FU) in colorectal cancer (CRC), whether these two factors regulate tumor response to 5-FU in a coordinated manner remains unknown. This study is designed to elucidate whether changes in miR-552 expression levels correlate to 5-FU-based chemoresistance in CRC, and to further identify the putative targets of miR-552 using multiple approaches.
Methods
miR-552 expression was assessed in 5-FU-resistant CRC tissues and cells using real-time PCR. Effects of miR-552 dysregulation on 5-FU resistance in CRC cells were determined by measuring cell viability, apoptosis and in vivo oncogenic capacity. Finally, we studied the posttranscriptional regulation of SMAD2 by miR-552 using multiple approaches including luciferase reporter assay, site-directed mutagenesis and transient/stable transfection, at molecular and functional levels.
Results
Expression of miR-552 was significantly downregulated in 5-FU-resistant CRC tissues and cells, and this downregulation, regulated by dMMR, was associated with poor postchemotherapy prognosis. Functionally, forced expression of miR-552 exhibited a proapoptotic effect and attenuated 5-FU resistance, whereas inhibition of miR-552 expression potentiated 5-FU resistance in CRC cells. Mechanically, miR-552 directly targeted the 3′-UTR of SMAD2, and stable ablation of SMAD2 neutralized the promoting effects of miR-552 deficiency-induced 5-FU resistance.
Conclusions
Overall, our findings have revealed a critical role of miR-552/SMAD2 cascade in modulating cellular response to 5-FU chemotherapy. miR-552 may act as an efficient mechanistic link synchronizing dMMR and 5-FU resistance in CRC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Worldwide, the incidence of colorectal cancer (CRC) is increasing rapidly in both developed and developing counties in recent years. In China, especially in urban regions, the incidence of CRC ranks the third highest and the mortality ranks the fourth among all cancer types [1]. Postoperative chemotherapeutic intervention remains the only backbone of enhanced survival in patients with advanced/metastatic CRC. Among different chemotherapeutic drugs, the antimetabolite 5-fluorouracil (5-FU) is so far the only chemotherapeutic agent available to successfully improve 12 month survival in CRC patients [2]. However, the response rate of 5-FU-based regimens is frustratingly low, with 10–15% in 5-FU alone and < 50% in 5-FU/other cytotoxic drugs combined therapy. This is mainly ascribed to the frequent occurrence of intrinsic and acquired resistance, which remains the major obstacle for the successful treatment [3]. 5-FU is well known to cause DNA damage during S phase due to the misincorporation of FdUTP into DNA. Because the DNA mismatch repair (MMR) system is responsible for the recognition of certain DNA adducts caused by alkylation damage and for the subsequent repair of interstrand nucleotide mismatches and slippage mistakes at microsatellite sequences [4], cells with deficient mismatch repair (dMMR) are expected to be highly resistant to 5-FU-elicited apoptosis. CRC with deficient MMR (dMMR) is such a striking example, and it displays widespread instability at DNA microsatellite sequences (MSI). dMMR is present in 15% of sporadic CRC, but the response in CRC with dMMR to 5-FU-based chemotherapy remains extremely low [5]. There is an unequivocal need to elucidate the molecular mechanisms of 5-FU-based chemoresistance in patients with dMMR CRC.
MicroRNAs (miRNAs), a cluster of small non-protein-coding RNAs, negatively regulate gene expression via translational repression or induction of mRNA decay by partially binding to the three prime untranslated region (3′-UTR) of target genes. Compelling evidence confirms that miRNAs are essentially involved in the initiation, progression, migration, and chemoresistance of cancer [6]. Because DNA MSI occurs at coding repeats, DNA repeats contained in human miRNA genes are preserved from mutations due to MSI in dMMR cancer cells. In this regard, differentially expressed miRNA signatures have been shown to constitute new targets for dMMR CRC [7]. Surprisingly, there is scant information vis-à-vis the functional roles of these miRNAs in the progression of dMMR CRC thereof.
Recent advance in this field has shown that dysregulated expression of miRNAs plays a crucial role in the drug resistance of CRC. For example, miR-34a regulates multidrug resistance via activation of OAZ2 signaling in CRC [8]. The expression of miR-140 is upregulated in 5-FU-resistant CRC cells, and blocking endogenous miR-140 sensitizes resistant colon cancer stem-like cells to 5-FU treatment [9]. Similarly, miR-139-5p [10], miR-22 [11] and miR-1260b [12] are all found to be essential for the pathogenesis of 5-FU-based chemoresistance in CRC.
In the current study, miR-552 was selected and subjected to further investigation based on three criteria: (1) up to now most tumor biology studies regarding miR-552 are focused on its roles in CRC [13,14,15,16], indicating that this unique miRNA may play a conserved role in CRC. (2) The expression profile of miR-552 can help to distinguish CRC metastasis in lung, indicative of its critical involvement in CRC progression [17]. (3) Recent high-throughput analysis has shown that aberrant miR-552 expression is crucially implicated in the undifferentiated proliferative states of dMMR [18]. Moreover, miR-552 is able to promote the cancerous progression in hepatocellular carcinoma by regulating epithelial–mesenchymal transition (EMT) [19]. Given both dMMR [20] and EMT [21] are responsible for the pathogenesis of drug resistance, we hypothesize that miR-552 may function as an oncomiR to modulate chemosensitivity in CRC. In this context, we sought to elucidate whether changes in the expression levels of miR-552 correlate to 5-FU-based chemoresistance in CRC and to further identify the putative targets of miR-552 using in vitro cell-based systems and in vivo animal models.
Materials and methods
Human CRC samples
A total of 97 patients with CRC, who had previously received radical surgical resection and postoperative chemotherapy, were recruited from the Second Affiliated Hospital of Xian Jiaotong University from February 2012 to October 2017 (Supplementary Table 1). The cancer staging, performed by two pathologists, was determined based on pathological findings according to the American Joint Committee on Cancer (AJCC) criteria [22]. Among these 97 patients, 10 pairs of CRC samples that were defined as “5-FU responsive” (n = 10) and “5-FU resistant” (n = 10), based on MRI examination [8], were chosen to study the expression levels of miR-552. The MMR status was determined by assessing expression levels of MLH1, MSH6 and PMS2, and patients with lack of expression of one or more of these genes were considered as “dMMR” [5]. Upon collection from primary tumors resection, CRC samples were snap frozen using liquid nitrogen and stored at − 80 °C until further analysis. Patient consent was waived for all samples, and the human study strictly conformed to the ethical guidelines of Declaration of Helsinki, was approved by the institutional review board of our hospital.
Cell treatment
The human CRC cell lines SW-480, SW-620 and HT-116, along with a normal colon epithelial cell line CCD-18Co, were obtained from ATCC (Rockville, MD, USA). SW-480 and SW-620 are pMMR cell lines, while HT-116 is a dMMR cell line [23]. However, the baseline sensitivity of SW-480 and SW-620 cells to 5-FU is quite similar [24, 25]. Cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum and 2 mM l-glutamine (All reagents from Thermo Scientific, Shanghai, China) in 5% CO2 at 37 °C. 5-FU-resistant sublines (designated as SW-480/5-FU, SW-620/5-FU or HCT-116/5-FU), were established by exposure to gradually elevating concentrations of 5-FU (Sigma-Aldrich, Shanghai, China), at an initial concentration of 1 μM and an ending concentration of 50 μM. The 5-FU-resistant sublines were finally maintained in culture medium containing 1 μM of 5-FU [5]. To manipulate the expression of miR-552, CRC cells were transiently transfected with miR-552 mimics or inhibitors, along with their corresponding negative controls (NC, Thermo Scientific) for 48 h, using GenMute™ Reagent (SignaGen, Gaithersburg, MD, USA). miRNA mimics are small, chemically modified double-stranded RNAs that mimic endogenous miRNAs and enable miRNA functional analysis by up-regulation of miRNA activity, whereas miRNA inhibitors are small, chemically modified single-stranded RNA molecules designed to specifically bind to and inhibit endogenous miRNA molecules and enable miRNA functional analysis by down-regulation of miRNA activity. The mirVana™ miRNA Mimics or Inhibitors obtain from Thermo Scientific have been shown by many studies to be able to alter the expression levels of target miRNAs at both in vitro and in vivo levels very effectively [26,27,28]. SW-620 cells stably deprived of SMAD2 expression (designated as SW-620/SMAD2 shRNA) was established by transfection with SMAD2 shRNA or scramble shRNA (Sigma-Aldrich) using GenMute™ Reagent, followed by selection with 400 μg/ml of G418 (Thermo Scientific). The somatic cell hybrid GM11686 containing human chromosome 3 was obtained from NIGMS Human Genetic Cell Repository (Camden, NJ, USA) and maintained in DMEM containing 400 ng/ml of G418. The HCT-116 containing transferred chromosome 3 from GM11686 was developed according to the published protocol [29] and maintained as HCT-116 + ch3.
Measurement of cell viability and apoptosis
Cells were seeded in a 96-well plate at the concentration of 1.0 × 104 cells/well and cultured overnight. Cells were then treated for 48 h with various doses of 5-FU as indicated, followed by measurement of cell viability using Cell Counting Kit-8 (Engreen, Beijing, China).
Apoptotic cell death in cells and xenografts was evaluated using ApoStrand™ ELISA Apoptosis Detection Kit (ENZO, Farmingdale, NY, USA) according to the manufacturer’s instructions. Final spectrophotometry was developed in triplicate at 405 nm.
Southern blot
Genomic DNA was isolated from CRC cells using a commercial kit from Sigma-Aldrich. After digestion, digested DNA was separated by electrophoresis in 1% agarose gel and transferred to nitrocellulose filters in alkaline conditions. A digoxin-labelled Chromosome 3 probe was obtained from Roche Diagnostics (Shanghai, China), and the hybridization was performed as described elsewhere [29].
In vivo chemosensitivity
In vivo chemosensitivity was determined using xenograft model. Briefly, oligonucleotides-transfected CRC cells (1 × 106 cells/10 μl PBS) were injected subcutaneously into each flank of the 8-week-old male BALB/c nude mice. Two or three weeks later, mice received the intraperitoneal (i.p.) injection of either 5-FU (50 mg/kg, once a week) or saline (Control) for consecutive 28 days. In another experimental setting, 14 days after cell inoculation, LY2109761 (a specific TβRI/TβRII inhibitor, Sigma-Aldrich) was dissolved in oral vehicle (1% sodium carboxymethylcellulose, 0.5% sodium lauryl sulfate and 0.05% antifoam), and mice received cotreatment with LY2109761 administration by oral gavage (100 mg/kg/day) [30] and intraperitoneal injection of 5-FU (50 mg/kg, once a week) [31] for 28 consecutive days. Five mice of each experimental group were sacrificed at the time-points as indicated after 5-FU treatment and tumor volume was recorded and calculated using the formula: V (mm3) = width2 (mm2) × length (mm)/2.
Real-time polymerase chain reaction (real-time PCR)
Total RNA was isolated and purified using High Pure RNA Isolation Kit (Roche Diagnostics) and first-strand cDNA was synthesized using RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific). Quantification of miR-552 expression levels was carried out using TaqMan MicroRNA Assays (Thermo Fisher Scientific), as per the manufacturer’s instructions. Quantitative analysis of expression levels of other target genes was carried out using SYBR Green-based real-time PCR (Roche Diagnostics). Relative expression levels were assessed by the \( 2^{{ - \Delta \Delta C_{\text{t}} }} \) method [32, 33], with U6 small nuclear RNA (snRNA) and 18S serving as internal controls. The primers used were listed in Supplementary Table 2.
Western blot
Western blot analysis was performed according to previously validated protocols [34]. The details of the primary antibodies used in the current study were listed in Supplementary Table 3.
Luciferase reporter assay
The putative binding site of miR-552 in the three prime untranslated region (3′-UTR) of SMAD2 gene, along with its mutated derivative, was cloned into pGL3 Luciferase Reporter Vector (Promega, Shanghai, China), with the aid of CloneEZ® Kit (GenScript, HongKong, China) and QuikChange II Site-Directed Mutagenesis Kit (Agilent, Beijing China). Hela cells were cotransfected with 50 nM of miR-552 mimics or NC and 0.5 μg of pGL3-Luc-SMAD2 3′UTR/WT or pGL3-Luc-SMAD2 3′UTR/Mu using GenMute™ Reagent. 48 h later, cells were harvested and subjected to the dual-luciferase reporter assay using a Promega system.
Statistical analysis
Data were presented as mean ± SEM and were analyzed for statistical difference using Student’s t test or one-way analysis of variance (ANOVA) as appropriate. Survival rates were determined using the Kaplan–Meier method, and the difference between groups was calculated by log-rank tests. The correlation between miR-552 expression and SMAD2 transcripts level was assessed using Pearson’s Chi-square test. A P value of below 0.05 was considered as statistically significant.
Results
Downregulation of miR-552 expression in CRC correlates with poor prognosis after 5-FU treatment
Previous miRNA profiling data show that miR-552 represents one of the most differentially expressed miRNAs in CRC [35, 36]. To validate this, we quantified the relative expression levels of miR-552 using real-time PCR in a cohort of CRC tissue samples (n = 10/group). We observed a robust miR-552 expression in 5-FU-responsive tissues compared to that in adjacent normal and in 5-FU-resistant tissues, with the lowest values being detected in the 5-FU-resistant CRC samples (Fig. 1a). Furthermore, Kaplan–Meier analyses in a cohort of 97 CRC patients demonstrated that down-regulation of miR-552 expression negatively correlated to the overall survival (P < 0.001, Fig. 1b) and disease-free survival (P < 0.001, Fig. 1c) in patients with Stages II–III CRC who had previously received 5-FU-based chemotherapy after radical colectomy.

Downregulation of miR-552 expression correlates with poor prognosis in 97 patients with Stage II-III colorectal cancer (CRC) who had received 5-FU-based chemotherapy. a Real-time PCR analysis of miR-552 expression in human 5-FU-responsive, 5-FU-resistant and adjacent normal colorectal tissues. U6 small nuclear RNA served as the internal control. b, c The patients were classified into two groups according to low or high miR-552 expression levels. Kaplan–Meier survival curves and log-rank tests were then employed to analyze the correlation between miR-552expression and overall survival/disease-free survival rate
Determination of miR-552 expression by dMMR status in CRC
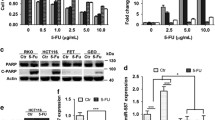
To verify the aforementioned observation at the in vitro level, we generated three 5-FU resistant sublines according to a previously validated protocol (Fig. 2a–c) [5]. Subsequent real-time PCR analysis revealed that miR-552 expression was significantly decreased in the 5-FU resistant sublines compared to that in their parental cells, with the exception of HCT-116 and HCT-116/5-FU cells that both exhibited unanimous low levels of miR-552 expression (Fig. 2d). Importantly, the analysis of the clinicopathological characteristics reflected that reduced miR-552 expression correlated to dMMR status (Supplementary Table 1). To further confirm this, we transfected HCT-116 cells, which is known to have dMMR due to a homozygous mutation in the mismatch repair gene hMLHl on human chromosome 3, with isolated human chromosome 3 from a human monochromosomal hybrid cell line GM11686 [29]. The reintroduction of foreign human chromosome 3 in HCT-116 cells was validated by Southern blot (Fig. 2e). Subsequent real-time PCR analysis confirmed the restoration of miR-552 expression in HCT-116 cells to levels ~ 92.8 to 131.4% of normal colon epithelial cell levels (Fig. 2f).

Regulation of miR-552 expression by DNA-mismatch-repair-deficient (dMMR) status. a–c The 5-FU-resistant SW-480/5-FU, HCT-116/5-FU and SW-620/5-FU cell lines were established as described in “Materials and methods”. CRC cells were then treated for 48 h with various doses of 5-FU as indicated, followed by measurement of cell viability using Cell Counting Kit-8 (*P < 0.05 and **P < 0.01 when compared to the parental CRC cells). d Relative levels of miR-552 expression in different CRC cells were evaluated by real-time PCR analysis (*P < 0.05 and **P < 0.01 when compared to the CCD-18Co cells). e Presence of transferred chromosome 3 in recipient HCT-116 cells was revealed by Southern blot analysis. Arrows denote the transferred chromosome 3. f Real-time PCR analysis of miR-552 expression in HCT-116 and HCT-116 + ch3 cells (*P < 0.05 when compared to the CCD-18Co cells)
Requirement of endogenous miR-552 for the 5-FU chemosensitivity in CRC cells
To provide the direct evidence that miR-552 inhibits 5-FU resistance, we transfected SW-480/5-FU and SW-620/5-FU cells with miR-552 mimics or mimics-NC (Fig. 3a). miR-552 overexpression resulted in decreased viability of SW-480/5-FU and SW-620/5-FU cells upon 5-FU exposure, when compared to that in parental naïve and mimics NC-transfected cells (Fig. 3b). Consistently, cell apoptosis of mimics-miR-552 cells in response to 5-FU was increased compared to that of mimics NC-Ctrl cells (Fig. 3c). To better evaluate the biologic function of miR-552 in vivo, we employed a xenograft model using SW-480/5-FU and SW-620/5-FU cells expressing miR-552 mimics or mimics-NC. 21 days after cell inoculation, 5-FU (50 mg/kg, once a week) or saline (control) was intraperitoneally injected into nude mice for 28 consecutive days. Intriguingly, the tumor volume of the tumors expressing the miR-552 mimics was notably smaller than that of tumors expressing mimics-NC during the late phase of treatment (Fig. 3d). To verify the negative association between miR-552 and 5-FU resistance, we knocked down the expression of endogenous miR-552 by transfecting SW-480 and SW-620 cells with miR-552 inhibitors or inhibitors-NC (Fig. 3e). As expected, cell viability of inhibitors-miR-552 cells was higher than that of inhibitors NC-Ctrl cells in response to 5-FU treatment (Fig. 3f), whereas cell apoptosis of inhibitors-miR-552 cells upon 5-FU treatment was decreased compared to that of inhibitors NC-Ctrl cells (Fig. 3g). Moreover, the application of miR-552 inhibitors promoted the most significant induction of tumor growth in 5-FU-treated nude mice (Fig. 3h). Thus, miR-552 may function as a potent tumor suppressor in 5-FU chemotherapy.

Effects of manipulation of miR-552 expression on 5-FU chemosensitivity. a 5-FU-resistant CRC cells were transfected with miR-552 mimics or negative controls (NC) for 48 h, followed by real-time PCR analysis (*P < 0.05 and **P < 0.01 when compared to NC). b CRC cells were then treated for 48 h with various doses of 5-FU as indicated, followed by measurement of cell viability using Cell Counting Kit-8 (*P < 0.05 and **P < 0.01 when compared to NC). c CRC cells were then treated for 48 h with various doses of 5-FU as indicated, followed by measurement of cell apoptosis using spectrophotometry at 405 nm (*P < 0.05 and **P < 0.01 when compared to NC). d Effects of miR-552 overexpression on in vivo 5-FU sensitivity were evaluated using xenografts model, as described in “Materials and methods” (*P < 0.05 and **P < 0.01 when compared to Mimis-NC + 5-FU). e 5-FU-sensitive CRC cells were transfected with miR-552 inhibitors or negative controls (NC) for 48 h, followed by real-time PCR analysis (*P < 0.05 and **P < 0.01 when compared to NC). f CRC cells were then treated for 48 h with various doses of 5-FU as indicated, followed by measurement of cell viability using Cell Counting Kit-8 (*P < 0.05 and **P < 0.01 when compared to NC). g CRC cells were then treated for 48 h with various doses of 5-FU as indicated, followed by measurement of cell apoptosis using spectrophotometry at 405 nm (*P < 0.05 and **P < 0.01 when compared to NC). h Effects of miR-552 knockdown on in vivo 5-FU sensitivity were evaluated using xenografts model, as described in “Materials and methods” (*P < 0.05 and **P < 0.01 when compared to Inhibitors-NC + 5-FU)
miR-552 directly targets the 3′-UTR of SMAD2
To understand the molecular basis of miR-552 function, we performed in silico analysis using two different algorithms (Targetscan and miRDB). 18 genes were found to be potential candidates by both programs (Fig. 4a). To narrow down the targets range, we analyzed the expression profile of these 18 putative target genes using real-time PCR in 10 sets of 5-FU responsive and 5-FU resistant CRC tissue samples. Only SMAD2 expression was observed to be significantly upregulated, correlated negatively to the miR-552 downregulation (Fig. 4b). Consistently, real-time PCR followed by Pearson’s Chi-square test revealed a negative correlation between miR-552 expression and SMAD2 transcript levels in a cohort of 97 CRC tissue samples (P < 0.0001, Fig. 4c). To confirm that miR-552 could regulate SMAD2, mRNA and protein expression levels were analyzed after miR-552 overexpression. Transfection with miR-552 mimics in SW-480/5-FU and SW-620/5-FU cells significantly suppressed SMAD2 expression, at both transcriptional and translational levels (Fig. 4d). To ask directly whether SMAD2 acts as the downstream target of miR-552, we performed the luciferase reporter assay. Cotransfection with the pGL3-Luc-SMAD2 3′UTR/WT reporter plasmid and miR-552 mimics resulted in a ~ 63.2% reduction in the luciferase reporter activity, and this inhibitory effect was totally abolished by transfection with pGL3-Luc-SMAD2 3′UTR/Mu reporter plasmid (Fig. 4f, g).

Identification of the 3′-UTR of SMAD2 as the direct target of miR-552. a Prediction of putative target genes of miR-552 by two different algorithms (Targetscan and miRDB). b Real-time PCR analysis of the putative target genes in 5-FU-responsive and 5-FU-resistant CRC tissues (n = 10/group, *P < 0.05 and **P < 0.01 when compared to 5-FU-responsive tissues). c Levels of miR-552 and SMAD2 mRNA expression in 97 CRC tissues were assessed using real-time PCR analysis, followed by Pearson’s Chi-square test. SW-480/5-FU and SW-620/5-FU cells were transfected with miR-552 mimics or NC for 48 h, followed by real-time PCR (d) or western blot analysis (e) (*P < 0.05 and **P < 0.01 when compared to NC). e Predicted miR-552 binding sites in the 3ʹ-UTR of SMAD2 gene. f Luciferase reporter assay with co-transfection of wild-type or mutant SMAD2 Luc reporter plasmids and miR-552 mimics in HeLa cells (*P < 0.05 when compared to NC)
Stable ablation of SMAD2 neutralizes the promoting effects of miR-552 deficiency on 5-FU resistance
To unequivocally elucidate the function of the miR-552–SMAD2 axis in 5-FU resistance, we established the SW-620 cells that were stably deprived of endogenous SMAD2 (SW-620/SMAD2 shRNA, Fig. 5a). Cell Counting Kit-8 assays confirmed that upon -5-FU insult, the cell viability in inhibitors-miR-552 cells was markedly enhanced compared to inhibitors NC-Ctrl cells. By contrast, after 5-FU treatment, the cell viability in SW-620 cells simultaneously transfected with miR-552 inhibitors and SMAD2 shRNA was significantly decreased to the levels comparable to naïve SW-620 cells (Fig. 5b). In accordance with these results, stable deprivation of SMAD2 neutralized the promoting effects of miR-552 deficiency on 5-FU resistance and potentiated the inhibitors-transfected SW-620 cells more sensitive to 5-FU-elicited apoptosis (Fig. 5c).

Stable knockdown of SMAD2 ameliorates miR-552 deficiency-induced 5-FU resistance. a Establishment of the SW-620 cells stably deprived of endogenous SMAD2 was verified using western blot analysis. b CRC cells with different transfections were treated for 48 h with 25 μM of 5-FU, followed by measurement of cell viability using Cell Counting Kit-8 (*P < 0.05 and **P < 0.01 when compared to the naïve cells). c CRC cells with different transfections were treated for 48 h with 25 μM of 5-FU, followed by measurement of cell apoptosis using spectrophotometry at 405 nm (*P < 0.05 and **P < 0.01 when compared to the naïve cells)
Synergistic cooperation of TGF-β activation and SMAD2 upregulation in the induction of 5-FU resistance in miR-552-deficient CRC cells
Exhibition of tumor-promoting properties by the canonical SMAD pathway requires the TGF-β receptors (TβRI/TβRII)-mediated phosphorylation of SMAD2, which has been shown, by accumulated data, to be essentially involved in the pathogenesis of drug resistance [37]. In this context, we sought to determined whether SMAD2 upregulation alone can functionally explain for the impaired 5-FU chemosensitivity in miR-552-deficient SW-620 cells. 48 h after transfection, inhibitors-SW-620 cells and inhibitors NC-SW-620 cells were subcutaneously injected into immunodeficient nude mice. After another 14-day of cell growth, mice received cotreatment with LY2109761 administration by oral gavage (100 mg/kg/day) and intraperitoneal injection of 5-FU (50 mg/kg/day) for 28 consecutive days (Fig. 6a). Real-time PCR analysis in SW-620 allografts revealed that either LY2109761 or 5-FU had no effects on miR-552 inhibition by inhibitors, which was substantially repressed at the end of 42 days after cell inoculation (Fig. 6b). Western blot assays showed that the expression levels of SMAD2 and pSMAD2, along with two markers for TGF-β activation (namely E-cadherin and vimentin), were all markedly increased in SW-620 allografts treated with inhibitors alone. By contrast, only SMAD2 expression was significantly induced in SW-620 allografts cotreated with inhibitors and LY2109761 (Fig. 6c). Consequently, administration of LY2109761 successfully abolished miR-552 deficiency-induced resistance to cell apoptosis (Fig. 6d) and restored chemosensitivity in 5-FU-challenged SW-620 allografts (Fig. 6e), at both in vitro and in vivo levels. Together, the available data suggest that in CRC, the 5-FU chemoresistance caused by miR-552 deficiency requires the synergistic action of TGF-β signaling (Fig. 6f).

Synergistic cooperation of TGF-β activation and SMAD2 upregulation in the induction of 5-FU resistance. a Experimental protocol used in our xenografts model. b Expression levels of miR-552 in the SW-620 allografts derived from different mouse models were assayed using real-time PCR analysis. c Expression of SMAD2, pSMAD2, E-cadherin and Vimentin in the SW-620 allografts derived from different mouse models were evaluated using Western blot analysis. d Apoptosis in the SW-620 allografts derived from different mouse models were assayed using ApoStrand™ ELISA. e Effects of cooperation of TGF-β activation and miR-552 inhibition on the in vivo 5-FU sensitivity were evaluated using xenografts model (*P < 0.05 when comparing the values between inhibitors + 5-FU and Inhibitors + 5-FU + LY2109761). f Proposed working model for the current study
Discussion
miR-552 expression has been shown to be significantly increased in primary CRC tissues compared to their adjacent normal tissues [13, 15]. Consistently, our qPCR analysis in a cohort of human samples (n = 10) also revealed a clear-cut elevation of miR-552 expression in CRC tissues (Fig. 1a), suggesting that miR-552 may serve as a potent oncomiR in CRC. Surprisingly, when CRC tissues develop resistance to 5-FU treatment, miR-552 expression was reduced sharply. Levels of miR-552 in 5-FU-less sensitive SW-620 [38] and 5-FU-resistant CRC cells were much lower than those in 5-FU-sensitive SW-480 and parental CRC cells, respectively (Fig. 2), also lending strong support to the notion that miR-552 expression is substantially inhibited along the pathogenesis of 5-FU chemoresistance. Given that acquisition of drug resistance is important for tumor progression, the available data suggest that miR-552 can function as both an oncogenic and a tumor suppressor miRNA in different stages of CRC. A growing number of miRNAs have been shown to be “double-faced” regulators of cancerous behavior depending on the tissue and cellular context. miR-29b negatively regulates proliferation and invasion in multiple myeloma (MM) [39] and acute myeloid leukemia (AML) [40], but potentiates cell growth in bladder cancer [41]. To this end, the current study indicates that miR-552 may exhibit a dual function in response to different environmental stimuli. Considering that miR-552 down-regulation was observed to be negatively correlated to the overall survival and disease-free survival in patients receiving 5-FU-based chemotherapy, as revealed by our Kaplan–Meier analysis in a large cohort of human samples (Fig. 1b, c), we propose the feasibility of using miR-552 as a sensitive biomarker for predicting prognosis of postchemotherapy in CRC.
miR-552 expression appeared to be exquisitely modulated by mismatch repair status in CRC cells. This conclusion is drawn from three observations. (1) Analysis of the clinicopathological data from 97 patients revealed that miR-552 downregulation was associated with dMMR status (Supplementary Table 1). (2) The HCT-116 cell line is known to have homozygous mutations at the hMLHl locus and is defective in mismatch repair [29]. When parental HCT-116 cells were experimentally induced to be resistant to 5-FU, no significant change was found in the miR-552 expression levels between HCT-116 and HCT-116/5-FU cells (Fig. 2d). This line of evidence strongly suggests that dMMR, instead of 5-FU resistance, regulates fundamentally the expression levels of miR-552 in CRC cells. (3) The latter point become more confirmative when reintroduction of a normal copy of hMLHl gene from the human monochromosomal hybrid cell GM11686 have restored miR-552 expression to the levels in normal colon epithelial cells (Fig. 2e, f).
Besides the unique expression profile of miR-552 along the development of 5-FU resistance, our combined analysis allowed us to dissect the major molecular basis underpinning miR-552 action. miR-552 deficiency confers 5-FU resistance by targeting SMAD2 signaling. This is based on the finding that miR-552 inhibited the SMAD2 expression at both transcriptional and translational levels and repressed the activity of a luciferase reporter construct containing the miR-552 binding site (Fig. 4). In good agreement, stable ablation of SMAD2 neutralized the promoting effects of miR-552 deficiency on 5-FU resistance (Fig. 5). SMAD2 is thus a relevant target of miR-552 implicated in these processes. TGF-β/SMADs pathway is emerging as a critical mediator of drug resistance. Actually, the previous study has shown a specific activation of the TGF-β pathway in consequence of 5-FU-based chemotherapeutic treatment in CRC [42]. TGF-β activation stimulates epithelial-to-mesenchymal transition (EMT) and thereby confers acquired resistance in many tumor types [37]. Interestingly, the expression levels of members in TGF-β/SMADs pathway have been shown to be functionally regulated by miRNAs at the posttranscriptional level. For instance, miR-181b regulates cisplatin chemosensitivity by targeting TGFβR1 in NSCLC [43]. miR-34a potentiates oxaliplatin (OXA) sensitivity through inhibition of macroautophagy activation by directly targeting SMAD4 in CRC [44]. In continuance to this understanding, we have identified SMAD2, a core cytoplasmic effector mediating TGF-β activity, as the direct down-stream target of miR-552. Apparently, most of the components of TGF-β/SMADs pathway, such as TGFβR1 [43], SMAD2 [45], SMAD3 [46] and SMAD4 [44], are directly regulated at different layers by miRNAs.
The importance of TGF-β signaling has initiated the expected feasibility of targeting this pathway as an effective antineoplastic strategy. However, the results of the application of TGFBR inhibitors or ligand traps, both preclinically and clinically, turn out to be frustrating [47, 48]. The relevance of this observation is twofold. First, TGF-β possesses a paradoxical activity by acting as a tumor suppressor during early tumor development or as an oncogenic factor in the advanced stages. Therefore, this complexity of TGF-β action, along with the fact that most of the tumors are heterogeneous, is certain to weaken the therapeutic effects of blockade of TGF-β [37]. On the hand, TGF-β may incorporate a synergistic action from other core mechanisms (e.g. miRNAs) to fully exert their biological effects [49, 50]. In favor of this, we have shown that TGF-β inhibition by LY2109761 administration successfully abolished miR-552 deficiency-induced resistance to cell apoptosis (Fig. 6d) and restored chemosensitivity in 5-FU-challenged SW-620 allografts (Fig. 6e). On this basis, we propose that miR-552 replenishment and TGF-β inhibition combination therapy may suppress 5-FU-resistant tumor growth to a greater extent. Overall, our findings have revealed a critical role of miR-552/SMAD2 cascade in modulating cellular response to 5-FU chemotherapy.
References
Zhou Q, Li Y, Liu HZ, Liang YR, Lin GZ (2018) Willingness to pay for colorectal cancer screening in Guangzhou. World J Gastroenterol 24(41):4708–4715. https://doi.org/10.3748/wjg.v24.i41.4708
McQuade RM, Stojanovska V, Bornstein JC, Nurgali K (2017) Colorectal cancer chemotherapy: the evolution of treatment and new approaches. Curr Med Chem 24(15):1537–1557. https://doi.org/10.2174/0929867324666170111152436
Du C, Huang D, Peng Y, Yao Y, Zhao Y, Yang Y, Wang H, Cao L, Zhu WG, Gu J (2017) 5-Fluorouracil targets histone acetyltransferases p300/CBP in the treatment of colorectal cancer. Cancer Lett 400:183–193. https://doi.org/10.1016/j.canlet.2017.04.033
Carethers JM, Chauhan DP, Fink D, Nebel S, Bresalier RS, Howell SB, Boland CR (1999) Mismatch repair proficiency and in vitro response to 5-fluorouracil. Gastroenterology 117(1):123–131
Ye L, Jiang T, Shao H, Zhong L, Wang Z, Liu Y, Tang H, Qin B, Zhang X, Fan J (2017) miR-1290 is a biomarker in dna-mismatch-repair-deficient colon cancer and promotes resistance to 5-fluorouracil by directly targeting hMSH2. Mol Ther Nucleic Acids 7:453–464. https://doi.org/10.1016/j.omtn.2017.05.006
Wen D, Peng Y, Lin F, Singh RK, Mahato RI (2017) Micellar delivery of miR-34a modulator rubone and paclitaxel in resistant prostate cancer. Cancer Res 77(12):3244–3254. https://doi.org/10.1158/0008-5472.CAN-16-2355
El-Murr N, Abidi Z, Wanherdrick K, Svrcek M, Gaub MP, Flejou JF, Hamelin R, Duval A, Lesuffleur T (2012) MiRNA genes constitute new targets for microsatellite instability in colorectal cancer. PLoS One 7(2):e31862. https://doi.org/10.1371/journal.pone.0031862
Li Y, Gong P, Hou JX, Huang W, Ma XP, Wang YL, Li J, Cui XB, Li N (2018) miR-34a regulates multidrug resistance via positively modulating OAZ2 signaling in colon cancer cells. J Immunol Res 2018:7498514. https://doi.org/10.1155/2018/7498514
Song B, Wang Y, Xi Y, Kudo K, Bruheim S, Botchkina GI, Gavin E, Wan Y, Formentini A, Kornmann M, Fodstad O, Ju J (2009) Mechanism of chemoresistance mediated by miR-140 in human osteosarcoma and colon cancer cells. Oncogene 28(46):4065–4074. https://doi.org/10.1038/onc.2009.274
Liu H, Yin Y, Hu Y, Feng Y, Bian Z, Yao S, Li M, You Q, Huang Z (2016) miR-139-5p sensitizes colorectal cancer cells to 5-fluorouracil by targeting NOTCH-1. Pathol Res Pract 212(7):643–649. https://doi.org/10.1016/j.prp.2016.04.011
Zhang H, Tang J, Li C, Kong J, Wang J, Wu Y, Xu E, Lai M (2015) MiR-22 regulates 5-FU sensitivity by inhibiting autophagy and promoting apoptosis in colorectal cancer cells. Cancer Lett 356(2 Pt B):781–790. https://doi.org/10.1016/j.canlet.2014.10.029
Zhao J, Cao J, Zhou L, Du Y, Zhang X, Yang B, Gao Y, Wang Y, Ma N, Yang W (2018) MiR-1260b inhibitor enhances the chemosensitivity of colorectal cancer cells to fluorouracil by targeting PDCD4/IGF1. Oncol Lett 16(4):5131–5139. https://doi.org/10.3892/ol.2018.9307
Wang N, Liu W (2018) Increased expression of miR-552 acts as a potential predictor biomarker for poor prognosis of colorectal cancer. Eur Rev Med Pharmacol Sci 22(2):412–416. https://doi.org/10.26355/eurrev_201801_14189
Cao J, Yan XR, Liu T, Han XB, Yu JJ, Liu SH, Wang LB (2017) MicroRNA-552 promotes tumor cell proliferation and migration by directly targeting DACH1 via the Wnt/beta-catenin signaling pathway in colorectal cancer. Oncol Lett 14(3):3795–3802. https://doi.org/10.3892/ol.2017.6600
Wang J, Li H, Wang Y, Wang L, Yan X, Zhang D, Ma X, Du Y, Liu X, Yang Y (2016) MicroRNA-552 enhances metastatic capacity of colorectal cancer cells by targeting a disintegrin and metalloprotease 28. Oncotarget 7(43):70194–70210. https://doi.org/10.18632/oncotarget.12169
Xia ZS, Wang L, Yu T, Zhong W, Lian GD, Wu D, Zhou HM, Chen GC (2014) MiR-5000-3p, miR-5009-3P and miR-552: potential microRNA biomarkers of side population cells in colon cancer. Oncol Rep 32(2):589–596. https://doi.org/10.3892/or.2014.3232
Kim J, Lim NJ, Jang SG, Kim HK, Lee GK (2014) miR-592 and miR-552 can distinguish between primary lung adenocarcinoma and colorectal cancer metastases in the lung. Anticancer Res 34(5):2297–2302
Sarver AL, French AJ, Borralho PM, Thayanithy V, Oberg AL, Silverstein KA, Morlan BW, Riska SM, Boardman LA, Cunningham JM, Subramanian S, Wang L, Smyrk TC, Rodrigues CM, Thibodeau SN, Steer CJ (2009) Human colon cancer profiles show differential microRNA expression depending on mismatch repair status and are characteristic of undifferentiated proliferative states. BMC Cancer 9:401. https://doi.org/10.1186/1471-2407-9-401
Qu W, Wen X, Su K, Gou W (2019) MiR-552 promotes the proliferation, migration and EMT of hepatocellular carcinoma cells by inhibiting AJAP1 expression. J Cell Mol Med 23(2):1541–1552. https://doi.org/10.1111/jcmm.14062
Etienne-Grimaldi MC, Mahamat A, Chazal M, Laurent-Puig P, Olschwang S, Gaub MP, Formento JL, Formento P, Sudaka A, Boige V, Abderrahim-Ferkoune A, Benchimol D, Andre T, Houry S, Faucheron JL, Letoublon C, Gilly FN, Delpero JR, Lasser P, Pradere B, Pezet D, Penault-Llorca F, Milano G (2014) Molecular patterns in deficient mismatch repair colorectal tumours: results from a French prospective multicentric biological and genetic study. Br J Cancer 110(11):2728–2737. https://doi.org/10.1038/bjc.2014.213
Tanaka S, Hosokawa M, Yonezawa T, Hayashi W, Ueda K, Iwakawa S (2015) Induction of epithelial-mesenchymal transition and down-regulation of miR-200c and miR-141 in oxaliplatin-resistant colorectal cancer cells. Biol Pharm Bull 38(3):435–440. https://doi.org/10.1248/bpb.b14-00695
Park JS, Choi GS, Hasegawa S, Sakai Y, Huh JW, Kim HR, Kwak SG (2012) Validation of the seventh edition of the American Joint Committee on cancer tumor node-staging system in patients with colorectal carcinoma in comparison with sixth classification. J Surg Oncol 106(6):674–679. https://doi.org/10.1002/jso.23117
Peng W, Tan S, Xu Y, Wang L, Qiu D, Cheng C, Lin Y, Liu C, Li Z, Li Y, Zhao Y, Li Q (2018) LCMS/MS metabolome analysis detects the changes in the lipid metabolic profiles of dMMR and pMMR cells. Oncol Rep 40(2):1026–1034. https://doi.org/10.3892/or.2018.6510
Warrington RC, Cheng I, Fang WD (1994) Susceptibility of human colon carcinoma cells to anticancer drugs is enhanced by l-histidinol. Anticancer Res 14(2A):367–372
Zirvi KA, Najjar TA, Slomiany BL (1993) Sensitivity of human colon tumor metastases to anticancer drugs in athymic (nude) mice. Cancer Lett 72(1–2):39–44
Barnes NA, Stephenson S, Cocco M, Tooze RM, Doody GM (2012) BLIMP-1 and STAT3 counterregulate microRNA-21 during plasma cell differentiation. J Immunol 189(1):253–260. https://doi.org/10.4049/jimmunol.1101563
Migliore C, Martin V, Leoni VP, Restivo A, Atzori L, Petrelli A, Isella C, Zorcolo L, Sarotto I, Casula G, Comoglio PM, Columbano A, Giordano S (2012) MiR-1 downregulation cooperates with MACC1 in promoting MET overexpression in human colon cancer. Clin Cancer Res 18(3):737–747. https://doi.org/10.1158/1078-0432.CCR-11-1699
Schaeffer V, Hansen KM, Morris DR, LeBoeuf RC, Abrass CK (2012) RNA-binding protein IGF2BP2/IMP2 is required for laminin-beta2 mRNA translation and is modulated by glucose concentration. Am J Physiol Renal Physiol 303(1):F75–F82. https://doi.org/10.1152/ajprenal.00185.2012
Koi M, Umar A, Chauhan DP, Cherian SP, Carethers JM, Kunkel TA, Boland CR (1994) Human chromosome 3 corrects mismatch repair deficiency and microsatellite instability and reduces N-methyl-N′-nitro-N-nitrosoguanidine tolerance in colon tumor cells with homozygous hMLH1 mutation. Cancer Res 54(16):4308–4312
Connolly EC, Saunier EF, Quigley D, Luu MT, De Sapio A, Hann B, Yingling JM, Akhurst RJ (2011) Outgrowth of drug-resistant carcinomas expressing markers of tumor aggression after long-term TbetaRI/II kinase inhibition with LY2109761. Cancer Res 71(6):2339–2349. https://doi.org/10.1158/0008-5472.CAN-10-2941
Wagner M, Roh V, Strehlen M, Laemmle A, Stroka D, Egger B, Trochsler M, Hunt KK, Candinas D, Vorburger SA (2009) Effective treatment of advanced colorectal cancer by rapamycin and 5-FU/oxaliplatin monitored by TIMP-1. J Gastrointest Surg 13(10):1781–1790. https://doi.org/10.1007/s11605-009-0948-x
Dong YS, Hou WG, Li Y, Liu DB, Hao GZ, Zhang HF, Li JC, Zhao J, Zhang S, Liang GB, Li W (2016) Unexpected requirement for a binding partner of the syntaxin family in phagocytosis by murine testicular Sertoli cells. Cell Death Differ 23(5):787–800. https://doi.org/10.1038/cdd.2015.139
Zhang C, Lai JH, Hu B, Zhang S, Zhao J (1839) Li W (2014) A chromatin modifier regulates Sertoli cell response to mono-(2-ethylhexyl) phthalate (MEHP) via tissue inhibitor of metalloproteinase 2 (TIMP2) signaling. Biochim Biophys Acta 11:1170–1182. https://doi.org/10.1016/j.bbagrm.2014.08.006
Zhang S, Li W, Zhu C, Wang X, Li Z, Zhang J, Zhao J, Hu J, Li T, Zhang Y (2012) Sertoli cell-specific expression of metastasis-associated protein 2 (MTA2) is required for transcriptional regulation of the follicle-stimulating hormone receptor (FSHR) gene during spermatogenesis. J Biol Chem 287(48):40471–40483. https://doi.org/10.1074/jbc.M112.383802
Wu S, Wu F, Jiang Z (2017) Identification of hub genes, key miRNAs and potential molecular mechanisms of colorectal cancer. Oncol Rep 38(4):2043–2050. https://doi.org/10.3892/or.2017.5930
Choi YC, Yoon S, Byun Y, Lee G, Kee H, Jeong Y, Yoon J, Baek K (2015) MicroRNA library screening identifies growth-suppressive microRNAs that regulate genes involved in cell cycle progression and apoptosis. Exp Cell Res 339(2):320–332. https://doi.org/10.1016/j.yexcr.2015.10.012
Brunen D, Willems SM, Kellner U, Midgley R, Simon I, Bernards R (2013) TGF-beta: an emerging player in drug resistance. Cell Cycle 12(18):2960–2968. https://doi.org/10.4161/cc.26034
Bauer KM, Lambert PA, Hummon AB (2012) Comparative label-free LC–MS/MS analysis of colorectal adenocarcinoma and metastatic cells treated with 5-fluorouracil. Proteomics 12(12):1928–1937. https://doi.org/10.1002/pmic.201200041
Wang H, Ding Q, Wang M, Guo M, Zhao Q (2018) miR-29b inhibits the progression of multiple myeloma through downregulating FOXP1. Hematology. https://doi.org/10.1080/10245332.2018.1502961
Huang X, Schwind S, Yu B, Santhanam R, Wang H, Hoellerbauer P, Mims A, Klisovic R, Walker AR, Chan KK, Blum W, Perrotti D, Byrd JC, Bloomfield CD, Caligiuri MA, Lee RJ, Garzon R, Muthusamy N, Lee LJ, Marcucci G (2013) Targeted delivery of microRNA-29b by transferrin-conjugated anionic lipopolyplex nanoparticles: a novel therapeutic strategy in acute myeloid leukemia. Clin Cancer Res 19(9):2355–2367. https://doi.org/10.1158/1078-0432.CCR-12-3191
Xu F, Zhang Q, Cheng W, Zhang Z, Wang J, Ge J (2013) Effect of miR-29b-1* and miR-29c knockdown on cell growth of the bladder cancer cell line T24. J Intern Med Res 41(6):1803–1810. https://doi.org/10.1177/0300060513505266
Romano G, Santi L, Bianco MR, Giuffre MR, Pettinato M, Bugarin C, Garanzini C, Savarese L, Leoni S, Cerrito MG, Leone BE, Gaipa G, Grassilli E, Papa M, Lavitrano M, Giovannoni R (2016) The TGF-beta pathway is activated by 5-fluorouracil treatment in drug resistant colorectal carcinoma cells. Oncotarget 7(16):22077–22091. https://doi.org/10.18632/oncotarget.7895
Wang X, Chen X, Meng Q, Jing H, Lu H, Yang Y, Cai L, Zhao Y (2015) MiR-181b regulates cisplatin chemosensitivity and metastasis by targeting TGFbetaR1/Smad signaling pathway in NSCLC. Sci Rep 5:17618. https://doi.org/10.1038/srep17618
Sun C, Wang FJ, Zhang HG, Xu XZ, Jia RC, Yao L, Qiao PF (2017) miR-34a mediates oxaliplatin resistance of colorectal cancer cells by inhibiting macroautophagy via transforming growth factor-beta/Smad4 pathway. World J Gastroenterol 23(10):1816–1827. https://doi.org/10.3748/wjg.v23.i10.1816
Xu S, Xue C, Li J, Bi Y, Cao Y (2011) Marek’s disease virus type 1 microRNA miR-M3 suppresses cisplatin-induced apoptosis by targeting Smad2 of the transforming growth factor beta signal pathway. J Virol 85(1):276–285. https://doi.org/10.1128/JVI.01392-10
Jiang L, He D, Yang D, Chen Z, Pan Q, Mao A, Cai Y, Li X, Xing H, Shi M, Chen Y, Bruce IC, Wang T, Jin L, Qi X, Hua D, Jin J, Ma X (2014) MiR-489 regulates chemoresistance in breast cancer via epithelial mesenchymal transition pathway. FEBS Lett 588(11):2009–2015. https://doi.org/10.1016/j.febslet.2014.04.024
Connolly EC, Freimuth J, Akhurst RJ (2012) Complexities of TGF-beta targeted cancer therapy. Int J Biol Sci 8(7):964–978. https://doi.org/10.7150/ijbs.4564
Yingling JM, Blanchard KL, Sawyer JS (2004) Development of TGF-beta signalling inhibitors for cancer therapy. Nat Rev Drug Discov 3(12):1011–1022. https://doi.org/10.1038/nrd1580
Smith B, Agarwal P, Bhowmick NA (2017) MicroRNA applications for prostate, ovarian and breast cancer in the era of precision medicine. Endocr Relat Cancer 24(5):R157–R172. https://doi.org/10.1530/ERC-16-0525
Chen W, Zhou S, Mao L, Zhang H, Sun D, Zhang J, Li J, Tang JH (2016) Crosstalk between TGF-beta signaling and miRNAs in breast cancer metastasis. Tumour Biol 37(8):10011–10019. https://doi.org/10.1007/s13277-016-5060-8
Acknowledgements
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhao, P., Ma, Yg., Zhao, Y. et al. MicroRNA-552 deficiency mediates 5-fluorouracil resistance by targeting SMAD2 signaling in DNA-mismatch-repair-deficient colorectal cancer. Cancer Chemother Pharmacol 84, 427–439 (2019). https://doi.org/10.1007/s00280-019-03866-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-019-03866-7