
Abstract
Purpose
Anticancer-drug efficacy seems to involve the direct interaction with host immune cells. Although topoisomerase I (Top I) inhibitors have been suggested to block LPS-evoked inflammation, the interaction between these drugs and toll-like receptor 4 (TLR4) is unaddressed.
Methods
SN-38, the active metabolite of the Top I inhibitor irinotecan, and TLR4 interaction was assessed using the in vitro luciferase nuclear factor-κB reporter assay, neutrophil migration to murine air-pouch, in silico simulation, and the thermal shift assay (TSA). Topotecan was used as a positive anti-inflammatory control.
Results
Non-cytotoxic concentrations of SN-38 attenuated LPS (a TLR4 agonist)-driven cell activation without affecting peptidoglycan (a TLR2 agonist)-activating response. Similarly, topotecan also prevented LPS-induced inflammation. Conversely, increasing concentrations of LPS reversed the SN-38 inhibitory effect. In addition, SN-38 abrogated LPS-dependent neutrophil migration and reduced TNF-α, IL-6, and keratinocyte chemoattractant levels in the air-pouch model, but failed to inhibit zymosan (a TLR2 agonist)-induced cell migration. A two-step molecular docking analysis indicated two potential binding sites for the SN-38 in the MD-2/TLR4 complex, the hydrophobic MD-2 pocket (binding energy of − 8.1 kcal/mol) and the rim of the same molecule (− 6.9 kcal/mol). The topotecan also bound to the MD-2 pocket. In addition, not only the lactone forms, but also the carboxylate conformations of both Top I inhibitors interacted with the MD-2 molecule. Furthermore, the TSA suggested the interaction of SN-38 with MD-2.
Conclusions
Therefore, SN-38 inhibits acute inflammation by blocking LPS-driven TLR4 signaling. This mechanism seems to be shared by other Top I inhibitors.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Toll-like receptors (TLRs) are a family of proteins that recognize pathogen- and danger-associated molecular patterns (PAMPs and DAMPs, respectively) to drive immune responses [1]. Ligands bind to their respective TLRs either in the cell surface (for instance, TLR4, TLR5, and TLR11) or in endosomes (TLR3, TLR9, and TLR13) to trigger pro-inflammatory signaling [2, 3]. Toll-like receptor 4 (TLR4), a receptor for the Gram-negative bacterial lipopolysaccharide (LPS), orchestrates transduction signaling that culminates with the activation of transcription factors, such as nuclear factor-κB (NF-κB) and activator protein 1 (AP-1). Once in the nucleus, these transcription factors stimulate the over-expression of pro-inflammatory enzymes and cytokines, including cyclooxygenase-2 (COX-2), inducible nitric oxide synthase (iNOS), tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), IL-6, and IL-8 [2, 4]. Notably, the dual association of TLR4 with inflammation and cancer indicates the therapeutic potential of TLR4 modulators [5].
Interestingly, anticancer drugs, such as 5-fluorouracil and gemcitabine, can activate immune cells by the regulation of molecular and cellular pathways [6]. In addition, paclitaxel, an anticancer drug, is described to mimic the agonistic action of LPS [7] by targeting TLR4 together with its co-receptor myeloid differentiation-2 (MD-2) complex [8, 9]. Conversely, paclitaxel fails to reproduce LPS-mimetic responses in macrophages with TLR4 point mutations [8]. In contrast to paclitaxel, topoisomerase 1 (Top I) inhibitors, such as camptothecin and topotecan, another class of anticancer agents, demonstrate a strong capacity to suppresses inflammatory genes and protect mice from death by inflammation in LPS-driven sepsis-like models [10]. In that context, Top I is suggested to exert a positive regulation of RNA polymerase II transcriptional activity at pathogen-induced genes [10]. Such a mechanism might be dependent on the low pH, commonly observed in the tumor microenvironment and in inflammatory conditions, which increases intracellular concentration of the drug [11].
Notably, the direct interaction between Top I inhibitors and TLR4 could also explain their capacity to negatively modulating LPS-dependent inflammation, which has never been addressed. Here, SN-38, a Top I inhibitor, is demonstrated to present and anti-inflammatory effect by targeting TLR4 receptor, which is a mechanism shared by other class members of Top I inhibitors.
Materials and methods
Animals
C57BL/6 male mice, weighing 20–22 g, were randomly divided into experimental groups (n = 8 animals/group) and were maintained in a temperature-controlled room under a dark–light cycle, with water and food provided ad libitum. This paper adheres to the principles for transparent reporting and scientific rigor of preclinical research as set out in the UK Concordat on Openness on Animal Research, the USA NIH Guidelines on reporting preclinical research, the ARRIVE Guidelines, the Instructional Animal Care and Use Committee and the Guidelines for the Care and Use of Laboratory Animals [12]. All efforts were made to minimize animal suffering. The Committee on the Ethics of Animal Experiments of the Federal University of Ceará approved the experimental protocol (number: 100/2014).
Drugs
Lipopolysaccharides from Escherichia coli O111:B4 (LPS, a TLR4 agonist), peptidoglycan (PGN, a TLR2 agonist), phorbol 12-myristate 13-acetate (PMA, which activates NF-κB-dependent transcription in a receptor-independent manner) and zymosan (a TLR2 agonist) were purchased from Sigma-Aldrich (São Paulo, Brazil). 7-Ethyl-10-hydroxycamptothecin (SN-38) was obtained from Pensabio Biotecnologia (Tocris Bioscience, São Paulo, Brazil), and topotecan chloridrate was obtained from Accord (São Paulo, Brazil; 4-mg ampoule). MD-2 protein was obtained from Lgcbio (MyBiosource, San Diego, USA). SN-38 stock solution was prepared in 100% DMSO solvent. DMSO concentration in work solution (sterile saline as vehicle) was lower than 0.04%. All the drugs were dissolved in sterile saline.
Luciferase nuclear factor-κB (NF-κB) reporter assay
RAW 264.7.Luc murine macrophages (Ingenex, USA) that stably bear the luciferase reporter gene controlled by the NF-κB sensitive promoter (pNF-κB-Luc; passage number of cell line 25) were used. RAW 264.7.Luc cells maintained in supplemented RPMI-1640 medium were seeded in a 24-well plate at a concentration of 2 × 106 cells/well. Supernatants were replaced by RPMI-1640 supplemented with 2% fetal bovine serum and were incubated for 12 h under 5% CO2 at 37 °C. Next, cells were incubated with different protocols to test the selectivity to TLR4: (1) SN-38 (0.2, 2, or 20 μM, concentrations based on IC50 from [13] and adapted for our 6h-incubation time protocol) alone or in combination with LPS (100 ng/ml); (2) SN-38 (2 μM) alone or in combination with LPS (10, 100, or 1000 ng/ml, [9]); and (3) SN-38 (10 μM, high concentration to evaluate the potential lack of selectivity to TLR4) alone or in combination with PGN (1 μg/ml, [14]) or PMA (10 ng/ml, [15]) under 5% CO2 atmosphere at 37 °C. After a 6-h incubation time, cells were lysed with TNT lysis buffer (0.1 M Tris–HCl [pH 7.5], 0.15 M NaCl, 0.05% Tween 20), and the luciferase activity in cell lysates was determined using a luminometer (Victor X5, PerkinElmer, Waltham, MA) and the Dual Luciferase Reporter assay system (Promega, Wisconsin, USA). The data are expressed as the fold-increase of the relative luciferase activity of four independent biological replicates, each one consisting of three technical measurements. A schematic protocol of cell incubation with the compounds is presented in Fig. 1a. Since TLR4 must be activated to release pro-inflammatory cytokines, a 30-min interval between each injection was a mean to demonstrate that SN-38 and topotecan would be capable of preventing the release of inflammatory mediators under stimuli exposure.

SN-38 competitively inhibits LPS-dependent signaling. a Experimental protocol used in the luciferase NF-κB reporter assay in RAW 264.7 cells. b SN-38 significantly attenuated LPS-induced luciferase activity. c Increasing concentrations of LPS reversed the inhibitory effect of SN-38. d Peptidoglycan (PGN, a TLR2 agonist) and e PMA (a NF-κB stimulator) increased luciferase activity, which was not inhibited by SN-38. The data are expressed as the mean ± SD and were analyzed using one-way ANOVA followed by Bonferroni’s test. The results indicate the fold-increase of the relative luciferase activity of four independent biological replicates, each consisting of three technical measurements. **P < 0.01 and ***P < 0.001
In vitro antiproliferative activity by Sulforhodamine B (SRB) assay
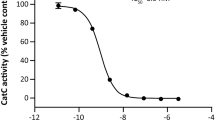
The macrophage cell line RAW 264.7 (from Banco de Células do Rio de Janeiro, Rio de Janeiro) or RAW 264.7.Luc was cultured in DMEM medium (LONZA®) supplemented with 10% heat inactivated fetal bovine serum (GIBCO®), penicillin (10,000 U/ml), and streptomycin (10,000 μg/ml) (GIBCO®) at 37 °C under 5% CO2 atmosphere. Cell cultures were regularly split to keep them in a logarithmic growth phase. The sulforhodamine B (SRB) assay was used for evaluation of antiproliferative effect, based on the measurement of total cellular protein content. The assay was performed according to Skehan et al. [16]. Briefly, the macrophages cell lines were plated 24 h prior to the addition of the test sample in 96-well plates at the density of 4 × 104 cells/ml. Then, cells were treated with SN-38 or topotecan with concentration ranging from 0.2 to 20 μM [13] alone or in combination with LPS 100 ng/ml for 6 h [9]. Cell density was indirectly quantified colorimetrically by sulforhodamine B (SRB) staining immediately after the 6 h of cell incubation. Absorbances were read on a plate reader (Fisher Scientific, model Multiskan FC) at 570 nm (adapted from [17]). The absorbance values were normalized to percentage of cell growth. The mean growth inhibition (GI50), the total growth inhibition (TGI), and the mean lethal concentration (LC50) values were investigated by interpolation of non-linear regression of normalized absorbance data to percentage of cell growth [16, 18].
In vivo neutrophil migration to murine air pouch
Mouse skin 6-day-old air pouches were produced as described by Sin and co-workers [19]. The backs of the mice were shaved, and 5 ml of sterile air were injected subcutaneously. After 3 days, 2.5 ml of sterile air were again injected to maintain pouch patency. Six days after the first air injection, saline (sterile PBS, 1 ml/cavity), SN-38 (100 ng/cavity), or topotecan (100 ng/cavity) alone or in combination with LPS (1 μg/cavity) or Zymosan (1 mg/cavity) was instilled [20]. A 30-min interval between each injection was a mean to demonstrate that SN-38 would be capable of preventing the release of inflammatory mediators by LPS or zymosan (Fig. 2a). Six hours after injection of the chemotactic stimuli into air pouches, the animals were euthanized, and the air pouches were washed with 1 ml of PBS containing 5 U/ml of heparin (Fig. 2a). The lavage fluid was cooled on ice, and 200 μl was used for the myeloperoxidase activity. The remaining fluids were centrifuged at 800×g for 5 min at 4 °C, and supernatants were stored at − 80 °C for cytokine and chemokine analyses.

SN-38 inhibits acute inflammation by selectively targeting LPS-driven response. a Experimental protocol performed in the murine (C57BL/6) air-pouch model of inflammation. b LPS (a TLR4 agonist) induced significant accumulation of neutrophils in the air pouch that was prevented by SN-38. c Zymosan (a TLR2 activator)-driven neutrophil migration was not prevented by SN-38. d–f SN-38 abrogated LPS-driven inflammation, as detected by the reduced levels of d TNF-α, e IL-6 and f KC. The data are shown as the mean ± SD of 8 mice per experimental group and were analyzed using one-way ANOVA followed by Bonferroni’s test. ***P < 0.001
Determination of myeloperoxidase activity (MPO)
Briefly, a lavage fluid sample (200 μl) was harvested and centrifuged at 800×g for 15 min at 4 °C. The pellet was then subjected to hypotonic lysis (0.2% NaCl solution) followed 30 s later by a further centrifugation step at 800×g for 15 min at 4 °C. The pellet was resuspended in 300 µl of 0.05 M NaPO4 buffer, pH 5.4, containing 0.5% hexadecyltrimethyl-ammonium bromide (HTAB, Sigma-Aldrich, São Paulo, Brazil). The MPO activity was developed with the color reagent tetramethylbenzidine (1.6 mM) and H2O2 (0.5 mM). The reaction was stopped with a 2 M H2SO4 solution, and the absorbance at 450 nm was determined using a spectrophotometer. The readings were compared with those of a standard curve of mouse peritoneal neutrophils processed in the same way. Briefly, carrageenan-induced peritonitis assay was used for isolation of neutrophils (aliquots of 1 × 105 neutrophils/ml of PBS) and construction of a standard curve as described by Dornelas-Filho et al. [21]. The data obtained were expressed as cells × 103/ml of exudate.
Cytokine dosage
The TNF-α, IL-6, and KC (the murine IL-8 homologue) concentrations in fluid lavage or cell supernatant were measured using an ELISA kit (R&D Systems, USA). Briefly, primary anti-TNF-α, anti-IL-6, or anti-KC antibody (2 μg/ml) was incubated overnight at 4 °C in a 96-well microtiter plate. After the plate was blocked, the samples and standard curves were added and incubated at 4 °C for 24 h. The plate was washed with buffer, after which anti-TNF-α, anti-IL-6, or anti-KC biotinylated antibody (1:1000 dilution with 1% BSA) was added to the wells. After further incubation at room temperature for 1 h, the plate was washed, and streptavidin-HRP (100 μl) at 1:200 dilutions was added, followed by a 100 μl substrate solution (1:1 mixture of H2O2 and tetramethylbenzidine; R&D Systems, USA). The plate was then incubated in the dark at room temperature for 20 min. The enzyme reaction was stopped with 2 N H2SO4, and the absorbance was measured at 450 nm. The results were expressed as pg/ml of exudate.
Molecular docking
Molecular docking was performed using the software Autodock 4.2 [22] to predict the interaction site between SN-38 and the TLR4/MD-2 complex. In addition, binding sites for topotecan, which has a similar chemical structure to SN-38, were also predicted. Since SN-38 and topotecan lactone rings tend to be hydrolyzed at physiological pH, we carried out docking experiments using two ligand chemical forms: carboxylate and lactone form. Protonation state of SN-38 and topotecan as well as isomers species were verified using the software MarvinSketch 18.8 (ChemAxon). In the ligand preparation step, lactone and carboxylate 3D structure conformations of SN-38 and topotecan were minimized using AMBER FF14SB force field and Gasteiger partial charges was computed using the software UCSF Chimera [23]. The TLR4/MD-2 structure was obtained from Protein Data Bank (PDB: 2Z64) and hydrogens were added as well as Gasteiger partial charges that were computed using AutoDockTools-1.5.6 (ADT). For the ligand, the maximum number of torsions was allowed, while the protein was considered rigid. For all docking experiments, the genetic algorithm was used. The first step of the docking experiments was performed using a grid that covered the entire TLR4/MD-2 complex. The number of energy evaluations and docking runs were set to 2,500,000 and 1000, respectively. The second step of the docking experiments was performed restricting the grid to the sites, where the best scored ligand poses obtained in the first step interacted. For this step, the numbers of energy evaluations and docking runs were set to 25,000,000 and 50, respectively. To compare the binding site in MD-2 between SN-38 or topotecan and LPS, the co-crystal structure of MD-2-TLR4 and LPS (PDB: 3VQ2) were aligned with the SN-38 or topotecan docking model using the software Chimera. The final images of the models were created also using the software Maestro (Schrödinger).
Thermal shift assay (TSA)
TSA, a commonly used biophysical technique to study protein–ligand interaction [24], was performed herein to investigate the direct binding of SN-38 to MD-2. TSA experiments have been successfully used for instance to screen compounds that bind to nuclear receptors [25, 26] and were performed as previously described [25]. The thermal stability of MD-2, in the presence or absence of LPS (positive control) or SN-38, was accessed by monitoring the SYPRO® Orange probe (Sigma-Aldrich, Brazil) fluorescence as a function of temperature. MD-2 recombinant protein (aminoacid sequence 2–160, molecular weight 22 kDa, product name MD-2 (LY96), and recombinant protein) was expressed in Escherichia coli, diluted in 1X PBS containing 0.1% SDS and purchased from MyBioSource (California, San Diego, USA) with ~ 95% purity. MD-2 (5 µM) was incubated with threefold ligand excess (15 µM of ligands) or 2% DMSO at 4 °C for 1 h (compound concentrations adapted from [25]). SYPRO® Orange was added to the reaction mixture, composed by physiological buffer saline at pH 7.4, to a fivefold final dilution and final volume of 20 μL in 96-well PCR microplates (Applied Biosystems-Life technologies). TSA was performed using a RT-PCR device (7500 Real Time PCR System) and fluorescence measurements were obtained during the heating of samples from 15 to 90 °C (heating rate 1 °C/min). Fluorescence intensities were used to obtain the melting temperature (ΔTm) of the protein unfolding transition through a Boltzmann model in Origin Pro 8.1.
Statistical analysis
The data were expressed as mean ± standard deviation (SD) and analyzed using one-way ANOVA followed by Bonferroni’s test. Statistical significance was accepted when P < 0.05. Statistical analysis was performed using GraphPad Prism software version 6.1 (Intuitive Software for Science, La Jolla, CA, USA). Investigators responsible for data analysis were blinded to which group samples were obtained from.
Results
SN-38 Antagonizes LPS-driven inflammation
To investigate the potential anti-inflammatory effect of SN-38, an in vitro NF-κB reporter assay was employed. First, RAW 264.7.Luc and RAW 264.7 cells were incubated with SN-38 alone or combined with LPS (Fig. 1a). Interestingly, SN-38 did not affect RAW 264.7.Luc cell growth at low (0.2 μM) or medium (2 μM) concentrations, but induced cytostasis at a high (20 μM) concentration (Supplementary Fig. 1a). Conversely, SN-38 depicted a cytotoxic effect on non-transfected RAW 264.7 cells in all concentrations tested (Supplementary Fig. 1b), as confirmed by the SRB assay. Considering the lower cytotoxic potential of SN-38 in RAW 264.7.Luc cells, a second set of experiments was designed using the luciferase reporter assay using these cells.
As observed in Fig. 1b, cells incubated with SN-38 alone showed no variation in luciferase activity compared with untreated cells (P > 0.05). However, LPS-induced significant (P < 0.001) cell activation, which was partially attenuated by SN-38 in all the concentrations tested (Fig. 1b). In a third experimental approach, cells were incubated with a single concentration of SN-38 alone or in combination with different LPS concentrations (Fig. 1c). Accordingly, a 2 µM SN-38 concentration abrogated the LPS (10 ng/ml)-evoked luciferase activity. Conversely, LPS (100 and 1000 ng/ml) fully reversed the SN-38 inhibitory effect in a concentration-dependent manner (Fig. 1c), indicating that SN-38 antagonized LPS-activating response.
Next, SN-38 selectivity for TLR4 was investigated using PGN, a TLR2 agonist, and PMA, a compound that directly activates NF-κB-dependent transcription in a receptor-independent manner [27]. Remarkably, PGN and PMA were found to increase luciferase activity (Fig. 1d, e, respectively), but SN-38 failed to block that response, confirming that SN-38 selectively reduces TLR4-dependent NF-κB activation. Notably, topotecan, which was used as a positive anti-inflammatory control, also inhibited LPS (100 ng/ml)-driven inflammation in a concentration-dependent manner (0.2 μM versus 2 μM), as detected by the reduced levels of pro-inflammatory cytokines TNF-α, IL-6, and KC (Supplementary Fig. 2a, b, c, respectively). However, topotecan induced a false-positive anti-inflammatory response at the highest concentration (20 μM), since it was cytotoxic on RAW 264.7 cells (Supplementary Fig. 1c).
To further confirm the in vitro findings, leukocyte accumulation into mouse air pouches was studied. Figure 2a illustrates the experimental protocol used. Consistently, LPS and zymosan injection induced a pronounced neutrophil infiltration [28, 29]. Notably, SN-38 effectively blocked the LPS-dependent inflammatory response, but failed to prevent zymosan-induced cell migration (Fig. 2b, c), indicating that SN-38 preferentially targets TLR4-related inflammation, but not TLR2. TLR agonists regulate neutrophil migration by orchestrating the release of several inflammatory and chemotactic mediators [30]. To verify whether SN-38 could inhibit the production of pro-inflammatory mediators downstream of TLR4, SN-38 was injected into the air-pouch before LPS. Accordingly, SN-38 significantly abrogated LPS-driven inflammation, as detected by reduced levels of TNF-α (Fig. 2d), IL-6 (Fig. 2e) and KC (Fig. 2f), confirming the SN-38 anti-inflammatory effect. To further confirm that the anti-inflammatory effect of SN-38 was not drug specific, topotecan was used as a positive control. Interestingly, topotecan also inhibited LPS-induced cell accumulation into the mouse air-pouch (Supplementary Fig. 3).
Top I inhibitors have potential binding sites in MD-2
Considering that SN-38 and topotecan blocked LPS inflammatory response, potential binding sites for SN-38 in TLR4/MD-2 were further evaluated by a two-step molecular docking. The first step comprised a search for binding sites in the whole TLR4/MD-2 complex and indicated a preference of both SN-38 forms for the MD-2 hydrophobic pocket, with the higher number of SN-38 conformations (32.2% for lactone and 41.8% for carboxylate form) and with favorable binding energy (Fig. 3a). The conformations of SN-38 in MD-2 pocket were refined at the second docking step and two possible SN-38 poses were identified for each SN-38 form (Fig. 3a, b). In carboxylate form, which is more abundant in physiological pH [11], the first pose (pose 1) was the best scored (− 6.7 kcal/mol) one. In this pose, SN-38 is buried inside the MD-2 pocket making extensive hydrophobic contacts mainly with phenylalanine and isoleucine residues (Fig. 3c). At the bottom of the MD-2 pocket, the SN-38 carboxyl group, produced by hydrolysis of the lactone ring, interacted with two serine residues (S47 and S48). An inverted orientation (pose 2) with the SN-38 carboxyl group pointing away from the bottom of MD-2 pocket is also allowed, but in this case the binding score is worse (− 6.1 kcal/mol).

SN-38 interacts with the MD-2. Molecular docking and the thermal shift assay were used to verify the interaction between SN-38 and TLR4. Best scored poses of SN-38 carboxylate form (a) (green stick, pose one; cyan stick, pose two) or SN-38 lactone form (b) (green stick, pose one; cyan stick, pose two) in the hydrophobic pocked of the MD-2 molecule and comparison among the predicted binding sites of SN-38, topotecan (orange sticks) and LPS (yellow sticks). Autodock score is demonstrated below the pose names. The main contacts of the best scored pose of SN-38 carboxylate and lactone forms are represented in c and d, respectively. e Direct interaction between SN-38 (light green) or LPS (dark green) and MD-2 is demonstrated by the thermal shift assay performed in physiological pH. The interaction was detected by the right shift of the unfolded transition curve of MD-2 (pink). Melting temperatures (ΔTm) are also depicted
The hydrophobic MD-2 pocket seems also to be able to accommodate SN-38 molecules that have their lactone ring intact (Fig. 3b). In general, docking scores predicted for SN-38 lactone form were better, but quite similar to SN-38 carboxylate form, and two SN-38 poses were also suggested. The best scored one (− 8.1 kcal/mol) occupied a region in MD-2 pocket similar to the pose 1 of SN-38 carboxylate form, with its lactone ring buried inside the MD-2 pocket and contacting S47 and S48 residues (Fig. 3d). The second possible pose identified for SN-38 (pose 2) presented a binding energy of -6.9 kcal/mol. In this pose, SN-38 is not buried inside the MD-2 pocket, but arranged in parallel to its F126 loop (Fig. 3b).
Despite particular differences in SN-38 orientation and position predicted for its lactone and carboxylate form, both interact with binding sites of LPS in MD-2, since this lipopolysaccharide occupy a large region in MD-2 pocket (Fig. 3a, b). All SN-38 predicted poses present large overlap with LPS, as also expected to the pose 2 of SN-38 lactone form. As SN-38 is not buried in MD-2 pocket in pose 2, the only structural overlap observed is with the hydroxyl group from one lipid chain of LPS (Fig. 3b).
The other Top I inhibitor, topotecan, has a chemical structure quite similar to SN-38 and the only difference is the presence of an amine group instead of a methyl group and its position in the aromatic ring of these molecules. However, this difference seems not to be enough to affect topotecan binding to the MD-2 pocket. Thus, docking experiments suggest that the large MD-2 pocket also can accommodate topotecan in both lactone and carboxylate forms (Fig. 3a, b). Moreover, the best topotecan poses had similar scores in comparison with the respective SN-38 forms.
To further investigate whether SN-38 binds directly to MD-2, we performed a thermal shift assay. In this assay, modifications in thermal stability of MD-2 in the absence or in the presence of SN-38 or LPS was determined. As expected, the MD-2 agonist LPS right shifted the unfolded transition curve of MD-2, increasing its Tm by 7 °C (Fig. 3e). A similar pattern was observed for SN-38 that increased the Tm of MD-2 by 8.5 °C, indicating a direct interaction between SN-38 and MD-2.
Discussion
In the present study, we described that the Top I inhibitors, topotecan and SN-38, the active metabolite of the anticancer-drug irinotecan, antagonize the LPS-dependent pro-inflammatory effect, by competing for the same LPS-binding site in the toll-like receptor 4 (TLR4).
The efficacy of chemotherapeutic drugs seems to be partially due to augmentation of the host immune cell-mediated inhibition of the antitumor response [6]. Conversely, anticancer agents also orchestrate pro-inflammatory and nociceptive responses that contribute to side effects [31,32,33,34,35]. For instance, paclitaxel regulates the immune response by targeting TLR4 [9] and activating the downstream MyD88-dependent signaling cascade [36], which contributes to the establishment of peripheral neuropathy [37]. Considering the well-known involvement of MyD88 in irinotecan-induced mucositis [33], we questioned whether SN-38 and topotecan could interact with TLR4.
The expression of genes induced by microbial stimuli activates the host innate immune response, which initiates an exacerbated inflammation [10]. Top I inhibitors have already been demonstrated to suppress inflammatory genes and to protect animals from LPS-induced death by regulating RNA polymerase II activity [10]. One potential explanation to the protective effect of Top I inhibitors on LPS-dependent inflammation might also involve the direct targeting of LPS. To verify whether SN-38 could be a TLR4 agonist or an antagonist, we performed in vitro and in vivo assays. Interestingly, RAW 264.7.Luc cells showed a negligible response to LPS in the presence of SN-38, but full cellular activity was reestablished when incubated with increasing concentrations of LPS, strongly indicating a reversible competitive antagonism. The SN-38-modulating response was on a receptor level and specific to TLR4, since the compound did not affect the effect of PMA, an NF-κB activator, or PGN, a TLR2 agonist. Accordingly, topotecan prevented LPS-related production of pro-inflammatory cytokines, suggesting the same SN-38 antagonism on TLR4. It is very surprising that most of the concentrations of SN-38 and topotecan caused no or mild percentage of cell growth inhibition. Noteworthy, the in vitro assays and cytostatic/cytotoxic effects were performed immediately after 6 h of cell incubation, whose time was chosen due to the high cytotoxicity verified at a longer incubation time. One plausible explanation to the lack of drug-related cytotoxicity might involve the neutral pH of vehicle (PBS) used for drug dilution. Top I inhibitors are predominantly in the therapeutically active lactone conformation (lipophilic form that crosses cell membranes) in acidic conditions and undergo reversible, physiological pH-sensitive ring-opening hydrolysis to give origin to their less active carboxylate forms (hydrophilic conformation that barely crosses cell membranes) [11]. In our opinion, the carboxylate forms have scarce access to the intracellular compartment, which might justify the anti-inflammatory action of these compounds on the membrane/receptor level allied with low cytotoxicity. To inhibit cell transcription, a drug must be lipophilic to properly cross cell membranes. It is also reasonable to infer that the low cytotoxicity reduced the likely contribution of DAMPs to further cell activation. Therefore, NF-kB luciferase activity measured in in vitro assays is solely due to the stimuli (LPS and PGN) that were applied to the cell culture.
Accordingly, SN-38 and topotecan also promoted an in vivo anti-inflammatory response in the LPS-induced leukocyte-migration air-pouch animal model. Conversely, SN-38 failed to prevent the pro-inflammatory effect of zymosan, a TLR2 agonist [38], confirming the selective antagonism on TLR4. Interestingly, cell incubation with camptothecin and topotecan, other topoisomerase I inhibitors, attenuates LPS-driven IL-6 and -8 expression and improves animal survival under sepsis-like conditions [10], which was also confirmed in in vitro and in vivo conditions in our study, suggesting the anti-inflammatory effect is shared by the class of topoisomerase I inhibitors.
To search for putative binding sites of SN-38 in TLR4, molecular docking experiments were performed, which have been successfully used to identify potential TLR4 ligands [39, 40]. Notably, lactone and carboxylate conformations of SN-38 and topotecan were predicted to have an energetically preference for binding to MD-2 sites. MD-2 forms a complex with TLR4 and allows the recognition of LPS, which bind to the MD-2 hydrophobic pocket [41]. This pocket is the target of several small molecules, such as berberine, the chalcone derivative L6H21 and curcumin [42,43,44], that compete with LPS and antagonize its effects. Similarly, SN-38 and topotecan have a hydrophobic moiety that allows a proper interaction with the MD-2 pocket. Two possible binding poses to MD-2 were predicted for the SN-38 by the docking experiments. Interestingly, the proposed poses were located in different MD-2 sites and their atoms are not superposed. As the MD-2 pocket is large and can accommodate more than one small molecule, as previously suggested for the MD-2 ligand berberine [42], it is sterically possible that the two SN-38 poses coexist.
In the first pose, SN-38 is buried inside MD-2 pocket, interacting mainly using hydrophobic interactions and sharing part of the same LPS binding site. As SN-38 and topotecan position in MD-2 overlap two lipid chains of LPS, it is possible that the interaction of LPS to MD-2 is impaired in presence of Top I inhibitors, corroborating with the competition between these compounds and LPS observed in our in vitro assays. Similarly, the second proposed pose for SN-38 also overlapped part of LPS-binding site, though less extensively. In addition to that structural overlap, SN-38 made a direct interaction with F126 in the second pose, which is described as an important residue that contributes to TLR4 activation and responsiveness to LPS, leading to the downstream protein–protein interactions needed for signal transduction [45]. In fact, lipid chains of LPS directly interact with F126 and mutation of this residue blocks LPS-induced MD-2/TLR4 dimerization [45]. Thus, besides the competition of SN-38 for the LPS-binding site, the interaction of SN-38 with F126 could impair LPS binding and would contribute to turn off TLR4 sensitivity to LPS, leading to an anti-inflammatory response. Consistently, topotecan also bound MD-2 with very low binding energy, reinforcing the idea that Top I inhibitors are TLR4/MD-2 antagonists.
Considering the neutral pH used in cell-based assays, TSA experiments were also performed at pH 7.4. The thermal shift assay results could have been altered by the pH of solutions used. To test that hypothesis the molecular docking was performed with the lactone and the carboxylate forms of SN-38 and topotecan. Notably, in contrast to topoisomerase inhibition, the binding of SN-38 and topotecan to MD-2 and the impairment of TLR4/MD-2 signaling seemed not to be dependent on the lactone ring, since the carboxylate form also interacted with the target protein. It reinforces our hypothesis that in our experimental conditions SN-38 and topotecan anti-inflammatory effects involve TLR4/MD-2 complex and does not involve the modulation of Top I dependent mechanisms.
It is well established that proteins bound to ligands have improved structural stability, since ligand binding increases the number of protein interactions [46]. In that context, more energy (temperature) is required to disrupt the overall protein structure upon ligand binding. Consistently with the in vitro, in vivo, and the molecular docking assays, the thermo shift analysis showed that SN-38 increased the melting temperature of the protein unfolding transition in a similar profile as LPS. Thus, the thermal stabilization induced by SN-38 indicates a direct SN-38/MD-2 interaction. Other techniques, such as isothermal titration calorimetry (ITC), could also be used to confirm MD-2/SN-38 interaction [47]. However, thermal shift assay was preferred in the present study due to the technique reliability, like ITC. In addition, the biophysical technique requires a very small amount of the target protein for the assay when compared with ITC, for instance.
The translation between these findings and the clinical setting is yet to be validated. Top I inhibitors are broadly used as anticancer agents and are also associated with severe gastrointestinal toxicities. Considering that (1) mucositis is an inflammatory toxicity of anticancer drugs on the alimentary tract and also that (2) TLR4 is important to activate the innate immune response to limit Gram-negative bacterial translocation, TLR4 blockade by Top I inhibitors could contribute to aggravate mucositis. Conversely, the literature is conflicting. TLR4 gene deletion is suggested to protect mice from irinotecan-induced intestinal mucositis [48]. On the other hand, LPS is also described to prevent radiotherapy-related gut damage [49]. These apparently opposing findings might indeed be due to different underlying mechanisms (disease specific), stimulus (dose intensity) and several other factors. Then, TLR4 might contribute to the pathogenesis of mucositis but at the same time be protective depending on the disease stage [50]. Further studies are required as a proof of concept.
Therefore, these findings collectively show that topotecan and SN-38 show effective acute anti-inflammatory effect by binding the MD-2 hydrophobic pocket and blocking LPS-driven TLR4 signaling.
Abbreviations
- AP-1:
-
Activator protein 1
- COX-2:
-
Cyclooxygenase-2
- DAMP:
-
Danger-associated molecular patterns
- DMSO:
-
Dimethyl sulfoxide
- HTAB:
-
Hexadecyltrimethyl-ammonium bromide
- IL-1β:
-
Interleukin-1 beta
- IL-6:
-
Interleukin-6
- IL-8:
-
Interleukin-8
- iNOS:
-
Inducible nitric oxide synthase
- KC:
-
Keratinocyte chemoattractant
- LPS:
-
Lipopolysaccharide
- MD-2:
-
Myeloid differentiation-2
- MPO:
-
Myeloperoxidase
- MyD88:
-
Myeloid differentiation primary response 88
- NF-κB:
-
Nuclear factor-κappa B
- PAMP:
-
Pathogen-associated molecular patterns
- PBS:
-
Phosphate buffered saline
- PGN:
-
Peptidoglycan
- PMA:
-
Phorbol 12-myristate 13-acetate
- SN-38:
-
7-Ethyl-10-hydroxycamptothecin
- TLR2:
-
Toll-like receptor 4
- TLR4:
-
Toll-like receptor 2
- TLRs:
-
Toll-like receptors
- TNF-α:
-
Tumor necrosis factor-alpha
- Top I:
-
Topoisomerase I
References
Liston A, Masters SL (2017) Homeostasis-altering molecular processes as mechanisms of inflammasome activation. Nat Rev Immunol 17:208–214
O’Neill LAJ, Golenbock D, Bowie AG (2013) The history of Toll-like receptors—redefining innate immunity. Nat Rev Immunol 13:453–460. https://doi.org/10.1038/nri3446
Rosadini CV, Kagan JC (2017) Early innate immune responses to bacterial LPS. Curr Opin Immunol 44:14–19
Wang JQ, Jeelall YS, Ferguson LL, Horikawa K (2014) Toll-like receptors and cancer: MYD88 mutation and inflammation. Front Immunol 5:1–10
Awasthi S (2014) Toll-like receptor-4 modulation for cancer immunotherapy. Front Immunol 5:1–5. https://doi.org/10.3389/fimmu.2014.00328
Shurin MR (2013) Dual role of immunomodulation by anticancer chemotherapy. Nat Med 19:20–22. https://doi.org/10.1038/nm.3045
Ding AH, Porteu F, Sanchez E, Nathan CF (1990) Shared actions of endotoxin and taxol on TNF receptors and TNF release. Science 248:370–372. https://doi.org/10.1126/Science.1970196
Kawasaki K, Akashi S, Shimazu R et al (2000) Mouse toll-like receptor 4.MD-2 complex mediates lipopolysaccharide-mimetic signal transduction by Taxol. J Biol Chem 275:2251–2254. https://doi.org/10.1074/jbc.275.4.2251
Wanderley CW, Colon DF, Luiz JPM et al (2018) Paclitaxel reduces tumor growth by reprogramming tumor-associated macrophages to an M1-profile in a TLR4-dependent manner. Cancer Res 78:5891–5900. https://doi.org/10.1158/0008-5472.CAN-17-3480
Rialdi A, Campisi L, Zhao N et al (2016) Topoisomerase 1 inhibition suppresses inflammatory genes and protects from death by inflammation. Science 352:7993. https://doi.org/10.1126/science.aad7993
Adams D (2005) The impact of tumor physiology on camptothecin-based drug development. Curr Med Chem Agents 5:1–13. https://doi.org/10.2174/1568011053352596
Danos O, Davies K, Lehn P, Mulligan R (2010) The ARRIVE guidelines, a welcome improvement to standards for reporting animal research. J Gene Med 12:559–560. https://doi.org/10.1002/jgm.1472
Gao H, Zhang X, Chen Y et al (2005) Synthesis and antitumor activity of 7-ethyl-9-alkyl derivatives of camptothecin. Bioorganic Med Chem Lett 15:2003–2006. https://doi.org/10.1016/j.bmcl.2005.02.072
Wang M, Wang L, Guo Y et al (2015) The broad pattern recognition spectrum of the Toll-like receptor in mollusk Zhikong scallop Chlamys farreri. Dev Comp Immunol 52:192–201. https://doi.org/10.1016/j.dci.2015.05.011
Busuttil V, Bottero V, Frelin C et al (2002) Blocking NF-κB activation in Jurkat leukemic T cells converts the survival agent and tumor promoter PMA into an apoptotic effector. Oncogene 21:3213–3224. https://doi.org/10.1038/sj/onc/1205433
Skehan P, Storeng R, Scudiero D et al (1990) New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst 82:1107–1112. https://doi.org/10.1093/jnci/82.13.1107
Wittmann M, Penzkofer A (1993) Concentration-dependent absorption and emission behaviour of sulforhodamine B in ethylene glycol. Chem Phys 172:339–348. https://doi.org/10.1016/0301-0104(93)80128-V
Boyd MR, Paull KD (1995) Some practical considerations and applications of the national cancer institute in vitro anticancer drug discovery screen. Drug Dev Res 34:91–109. https://doi.org/10.1002/ddr.430340203
Sin Y, Sedgwick A, Chea EP, Willoughby D (1986) Mast cells in newly formed lining tissue during acute inflammation: a six day air pouch model in the mouse. Ann Rheum Dis 45:873–877. https://doi.org/10.1136/ard.45.10.873
Leite ACRM, Cunha FQ, Dal-Secco D et al (2009) Effects of nitric oxide on neutrophil influx depends on the tissue: role of leukotriene B4 and adhesion molecules. Br J Pharmacol 156:818–825. https://doi.org/10.1111/j.1476-5381.2008.00094.x
Dornelas-Filho AF, Pereira VBM et al (2018) Neutrophils contribute to the pathogenesis of hemorrhagic cystitis induced by ifosfamide. Int Immunopharmacol 62:96–108. https://doi.org/10.1016/j.intimp.2018.06.031
Morris G, Huey R, Lindstrom W et al (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 30:2785–2791. https://doi.org/10.1002/jcc.21256.AutoDock4
Pettersen EF, Goddard TD, Huang CC et al (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. https://doi.org/10.1002/jcc.20084
Andreotti G, Monticelli M, Cubellis MV (2015) Looking for protein stabilizing drugs with thermal shift assay. Drug Test Anal 7:831–834. https://doi.org/10.1002/dta.1798
Ribeiro-Filho HV, Videira NB, Bridi AV et al (2018) Screening for PPAR non-agonist ligands followed by characterization of a hit, AM-879, with additional no-adipogenic and cdk5-mediated phosphorylation inhibition properties. Front Endocrinol (Lausanne) 9:1–12. https://doi.org/10.3389/fendo.2018.00011
Videira NB, Batista FAH, Torres Cordeiro A, Figueira ACM (2018) Cellular and biophysical pipeline for the screening of peroxisome proliferator-activated receptor beta/delta agonists: avoiding false positives. PPAR Res 2018:3681590. https://doi.org/10.1155/2018/3681590
Holden NS, Squires PE, Kaur M et al (2008) Phorbol ester-stimulated NF-kappaB-dependent transcription: roles for isoforms of novel protein kinase C. Cell Signal 20:1338–1348. https://doi.org/10.1016/j.cellsig.2008.03.001
Huang Z, Zhao C, Chen Y et al (2014) Recombinant human hyaluronidase PH20 does not stimulate an acute inflammatory response and inhibits lipopolysaccharide-induced neutrophil recruitment in the air pouch model of inflammation. J Immunol 192:5285–5295. https://doi.org/10.4049/jimmunol.1303060
Mancuso F, Calignano A, Cozzolino A et al (1996) Inhibition of zymosan-induced air-pouch inflammation by rat seminal vesicle protein and by its spermidine derivative. Eur J Pharmacol 312:327–332. https://doi.org/10.1016/0014-2999(96)00394-9
Aomatsu K, Kato T, Fujita H et al (2008) Toll-like receptor agonists stimulate human neutrophil migration via activation of mitogen-activated protein kinases. Immunology 123:171–180. https://doi.org/10.1111/j.1365-2567.2007.02684.x
Lima-Júnior RCP, Freitas HC, Wong DVT et al (2014) Targeted inhibition of IL-18 attenuates irinotecan-induced intestinal mucositis in mice. Br J Pharmacol 171:2335–2350. https://doi.org/10.1111/bph.12584
Leite CAVG, Alencar VTL, Melo DLR et al (2015) Target inhibition of IL-1 receptor prevents ifosfamide induced hemorrhagic cystitis in mice. J Urol 194:1777–1786. https://doi.org/10.1016/j.juro.2015.07.088
Wong DVT, Lima-júnior RCP, Carvalho CBM (2015) The Adaptor Protein Myd88 Is a Key Signaling Molecule in the Pathogenesis of Irinotecan-Induced Intestinal Mucositis 10:e0139985. https://doi.org/10.1371/journal.pone.0139985
Pachman DR, Qin R, Seisler D et al (2016) Comparison of oxaliplatin and paclitaxel-induced neuropathy (Alliance A151505). Support Care Cancer 24:5059–5068. https://doi.org/10.1007/s00520-016-3373-1
Bao T, Basal C, Seluzicki C et al (2016) Long-term chemotherapy-induced peripheral neuropathy among breast cancer survivors: prevalence, risk factors, and fall risk. Breast Cancer Res Treat 159:327–333. https://doi.org/10.1007/s10549-016-3939-0
Byrd-Leifer CA, Block EF, Takeda K et al (2001) The role of MyD88 and TLR4 in the LPS-mimetic activity of Taxol. Eur J Immunol 31:2448–2457. https://doi.org/10.1002/1521-4141(200108)31:8%3c2448:AID-IMMU2448%3e3.0.CO;2-N
Li Y, Zhang H, Zhang H et al (2014) Toll-like receptor 4 signaling contributes to Paclitaxel-induced peripheral neuropathy. J Pain 15:712–725. https://doi.org/10.1016/j.jpain.2014.04.001
Underhill DM, Ozinsky A, Hajjar AM et al (1999) The Toll-like receptor 2 is recruited to macrophage phagosomes and discriminates between pathogens. Nature 401:811–815. https://doi.org/10.1038/44605
Billod JM, Lacetera A, Guzmán-Caldentey J, Martín-Santamaría S (2016) Computational approaches to Toll-like receptor 4 modulation. Molecules. https://doi.org/10.3390/molecules21080994
Kuzmich N, Sivak K, Chubarev V, et al (2017) TLR4 Signaling Pathway Modulators as Potential Therapeutics in Inflammation and Sepsis. Vaccines. https://doi.org/10.3390/vaccines5040034
Shimazu R, Akashi S, Ogata H et al (1999) MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J Exp Med 189:1777–1782. https://doi.org/10.1084/jem.189.11.1777
Chu M, Ding R, Chu Z et al (2014) Role of berberine in anti-bacterial as a high-affinity LPS antagonist binding to TLR4/MD-2 receptor. BMC Complement Altern Med 14:1–9. https://doi.org/10.1186/1472-6882-14-89
Wang Y, Shan X, Chen G et al (2015) MD-2 as the target of a novel small molecule, L6H21, in the attenuation of LPS-induced inflammatory response and sepsis. Br J Pharmacol 172:4391–4405. https://doi.org/10.1111/bph.13221
Gradisar H, Keber MM, Pristovsek P, Jerala R (2007) MD-2 as the target of curcumin in the inhibition of response to LPS. J Leukoc Biol 82:968–974. https://doi.org/10.1189/jlb.1206727
Teghanemt A, Re F, Prohinar P et al (2008) Novel roles in human MD-2 of phenylalanines 121 and 126 and tyrosine 131 in activation of Toll-like receptor 4 by endotoxin. J Biol Chem 283:1257–1266. https://doi.org/10.1074/jbc.M705994200
Grøftehauge MK, Hajizadeh NR, Swann MJ, Pohl E (2015) Protein-ligand interactions investigated by thermal shift assays (TSA) and dual polarization interferometry (DPI). Acta Crystallogr Sect D Biol Crystallogr 71:36–44. https://doi.org/10.1107/S1399004714016617
Damian L (2013) Isothermal titration calorimetry for studying protein-ligand interactions. Methods Mol Biol 1008:103–118. https://doi.org/10.1007/978-1-62703-398-5_4
Wardill HR, Gibson RJ, Van Sebille YZA et al (2016) Irinotecan-induced gastrointestinal dysfunction and pain are mediated by common TLR4-dependent mechanisms. Mol Cancer Ther 15:1376–1386. https://doi.org/10.1158/1535-7163.MCT-15-0990
Riehl T, Cohn S, Tessner T et al (2000) Lipopolysaccharide is radioprotective in the mouse intestine through a prostaglandin-mediated mechanism. Gastroenterology 118:1106–1116. https://doi.org/10.1016/S0016-5085(00)70363-5
Khan S, Wardill HR, Bowen JM (2018) Role of toll-like receptor 4 (TLR4)-mediated interleukin-6 (IL-6) production in chemotherapy-induced mucositis. Cancer Chemother Pharmacol 82:31–37. https://doi.org/10.1007/s00280-018-3605-9
Acknowledgements
We are grateful to Giuliana Bertozi, Diva Amabile, Ana Katia dos Santos, Sergio Roberto Rosa, Ieda Regina dos Santos, Maria Silvandira Freire Socorro França, and José Olavo Morais for technical assistance.
Funding
This study was supported by CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico, Grant numbers: 307143/2014–7 and 428354/2016–5), FUNCAP (Fundação Cearense de Apoio ao Desenvolvimento Científico, Grant number: PR2-0101-00054.01.00/15) and CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior). This manuscript is dedicated to the loving memory of Prof. Dr. Ronaldo Albuquerque Ribeiro (in memoriam).
Author information
Authors and Affiliations
Contributions
Study design: DVTW, CWSW, HVRF, NMNA, FQC, and RCPLJ. Performed the experiments: DVTW, CWSW, HVRF, CAVGL, JBL, ANBA, AGC, GLPB, RHG, KOS, and LPCB. Data analysis: DVTW, CWSW, HVEF, DVW, and TMC. Interpretation of the results: DVTW, NMNA, DVW, TMC, FQC, and RCPLJ. Wrote the paper: DVTW, HVRF, and RCPLJ. All the authors revised and approved the paper.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare they have no financial, personal, or professional interests to disclose.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wong, D.V.T., Ribeiro-Filho, H.V., Wanderley, C.W.S. et al. SN-38, the active metabolite of irinotecan, inhibits the acute inflammatory response by targeting toll-like receptor 4. Cancer Chemother Pharmacol 84, 287–298 (2019). https://doi.org/10.1007/s00280-019-03844-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-019-03844-z