
Abstract
Despite significant advances in our ability to treat cancer, cytotoxic chemotherapy continues to be the mainstay treatment for many solid tumours. Chemotherapy is commonly associated with a raft of largely manageable adverse events; however, gastrointestinal (GI) toxicity (also termed mucositis) remains a significant challenge with little in the way of preventative and therapeutic options. The inability to manage GI complications likely reflects our incomplete understanding of its aetiology and the idiosyncrasies of each chemotherapeutic agent. This review highlights aims to provide a narrative for the involvement of Toll-like receptor (TLR4) in the development of chemotherapy-induced GI mucositis, an already emerging theme within this field. Particular focus will be placed upon the signalling interaction between TLR4 and interleukin (IL)-6. This parallels recent preclinical findings showing that TLR4 knockout mice, which are protected from developing severe GI mucositis, completely lack an IL-6 response. As such, we suggest that this signalling pathway presents as a novel mechanism with potential for therapeutic intervention.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Chemotherapy is a commonly prescribed treatment for cancer, however it is associated with serious adverse effects that influence patient well-being and clinical outcomes [1]. Affecting the entire alimentary tract (AT) from mouth to anus [2], mucositis presents with symptoms such as mouth ulcers, intractable vomiting, diarrhoea and pain, which are complicated by weight loss and infectious events such as sepsis [2]. Mucositis affects almost half of all patients receiving standard dose chemotherapy and 100% of those receiving high-dose chemotherapy or total body irradiation [3]. These patients are at a greater risk of treatment complications requiring significant supportive care methods to be adopted, thus adding to the growing economic burden of cancer therapy [4]. Although occurring in response to most anti-cancer agents, including newer targeted agents, mucositis is most problematic following 5-fluorouracil (5-FU) and irinotecan (CPT-11), with rates of severe mucositis exceeding 15% [2], thus limiting their widespread clinical implementation.
Although the mechanisms underpinning mucositis remains poorly understood [5], it is thought to involve interactions between the resident microflora, epithelial barrier function and the mucosal immune system [6]. This physiological balance requires a basal activation state of pattern recognition receptors, known as Toll-like receptors (TLRs), which limit inflammatory responses in intestinal epithelial cells (IEC) [7]. Conversely, TLRs are essential for triggering an appropriate immune response to infection [8], which manifests as inflammation initially. This is currently proposed to underpin a number of gastrointestinal (GI) diseases including inflammatory bowel disease (IBD), and as such is a working hypothesis for the damage that occurs in the GIT following chemotherapy. Damage to the cells by chemotherapy releases danger signals that result in multiple inflammatory events [5] causing activation of TLRs, especially TLR4 [1]. This elicits upregulation and release of inflammatory cytokines such as interleukin-1β, interleukin-6 (IL-6) and tumour necrosis factor-α (TNF-α) via the transcription factor, nuclear factor-κB (NF-κB) [9]. IL-6 is considered a hallmark of mucositis [10] and a variety of other inflammatory disorders [11], due to its pro-inflammatory properties [12]. Although association between TLR4-mediated IL-6 production and mucositis has been established [13] it is unclear if the protective effects of TLR4 deletion act through the blockade of IL-6 production directly. This review will therefore outline current evidence for the potential involvement of the TLR4/IL-6 signalling pathway in GI mucositis development.
Mucositis
Clinical and economic impact
Although able to affect any part of the AT, mucositis is often separated into oral mucositis (OM) and gastrointestinal mucositis (GIM) [14]. Both OM and GIM are thought to stem from common underlying mechanisms, underpinned by irreversible DNA damage, both histopathological and symptomatic differences do exists owing to the unique specialities of these distinctly different parts of the AT. OM often presents as erythematous and ulcerative lesions of the oral mucosa observed in patients with cancer being treated with chemotherapy or radiotherapy of the oral region [15]. These impact daily activities such as speaking, eating and drinking, and predispose to oral infections. In the large majority of cases, however, GIM is considered more impactful to the patient given the overriding symptom of diarrhoea +/− rectal bleeding, with not only impacts on patient comfort but also predisposition to infection, dehydration, weight loss, pain and kidney failure. Even in its most mild form, diarrhoea renders people housebound due to embarrassment, pain, fatigue, dehydration and simply the fear of needing to defecate suddenly. This can exacerbate social pressures, preventing active participation in the workforce (or school for younger patients), and lead to psychological distress and personal economic burden. In severe cases, diarrhoea is not adequately managed in the outpatient setting, requiring hospitalisation and extensive supportive care measure to prevent infectious complications and severe dehydration. The persistence of these symptoms often requires dose reductions, delays or complete cessation of treatment, thus impacting on the oncological outcomes for the patient [2, 13].
Economically, the management of mucositis remains a significant burden at the personal, societal and governmental level. Although the true impact of diarrhoea is difficult to quantify, conservative estimates suggest that for each diarrhoea-related episode there are an additional AUD1300 Medicare-related costs [16]. This estimate does not consider lost income due to time off work, lost productivity of carers and the use of complementary medicines; thus, the associated financial toxicity is likely much greater [17, 18].
Pathogenesis
The pathogenesis of mucositis (both OM and GIM) comprises a complex five-phase model proposed by Sonis [9, 19]. Direct DNA damage and subsequent cell death initiates a cascade of events, resulting in the production of reactive oxygen species (ROS), damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs), including secretory products, high mobility group box 1 (HMGB1) protein, DNA and RNA from bacteria, virus and fungi, and lipopolysaccharides (LPS) from Gram-negative bacteria [20]. DAMPs and PAMPs serve to activate TLRs [9], leading to primary damage response characterised by activation of transcription factor, NF-κB [10]. NF-κB activation is a key driver of mucositis development [9]. This is followed by a signal amplification phase, with a pro-inflammatory ‘storm’ of cytokines such as IL-6, TNF-α and IL-1β [10], which cause enhanced tissue damage, leading to the phase of ulceration that is the clinically relevant stage, as it is paralleled by major symptoms. This is supported by evidence of an increase in tissue level of NF-κB and inflammatory cytokines prior to histological changes in the mucosa of chemotherapy-treated rats [21]. Healing is the final phase which occurs spontaneously upon cessation of the chemotherapy treatment; however, residual changes within the mucosa remain [9]. The whole process results in marked histological changes in the GI mucosa. Histological findings of mucositis involve characteristic apoptotic bodies, villous blunting, and crypt degeneration throughout the GI mucosa; as seen in the jejunum and colon of rats treated with irinotecan [22]. Restoration of the mucosa in the GIT of the rats was evident 120 h after irinotecan administration, indicated by the return of architectural integrity [22].
Intestinal barrier dysfunction and bacterial translocation
Although not directly addressed in the current model of mucositis, a growing body of evidence now indicates that the epithelial barrier properties are critical in the severity of mucositis [23, 24]. This is particularly important for the development of GIM, given the critical role of the epithelial barrier in maintaining ion/water movement and preventing bacterial translocation. Marked abnormalities in intestinal permeability have been observed clinically in patients receiving myeloablative treatment [25] and high-dose chemotherapy [26]. This leads to altered fluid movement through leak-flux mechanisms, thus contributing to diarrhoea development [27]. This breakdown of the mucosal barrier also enables pathogens to penetrate the epithelium [22]; resulting in bacterial translocation which can predispose to systemic infection and secondary complications [28]. Guabiraba et al. [29] reported that irinotecan induces direct epithelial cell damage by modulating the release of IL-33. IL-33, in turn, stimulates chemokine production leading to tissue damage and bacterial translocation. This concept has been studied further in animal models, with bacteria found in the mesenteric lymph nodes of chemotherapy-treated rats [22]. Further evidence supporting the role of bacterial translocation in the pathogenesis of GIM is provided by an iatrogenic cohort study, in which elevated levels of LPS were found in the peripheral circulation of patients with haematological malignancies, signifying bacterial invasion [30]. Escherichia coli, a Gram-negative organism commonly resident in the lower intestine, was most frequently isolated from the blood of bacteraemic patients [31].
Gut microbiome and mucositis
About 100 trillion microorganisms [32] inhabit the GIT, existing in a finely tuned balance of commensal, pathobiont and pathogenic species. The microbiome has been increasingly considered for its role in the development of GI mucositis due to its ability to modify the host’s local and systemic immune system [33] and metabolise certain chemotherapeutic agents [5, 34]. As such, significant research efforts have been directed towards characterising the changes that occur in the microbiome following chemotherapy.
It is now understood that chemotherapy causes dysbiotic changes in the microbiome with a shift from β-glucuronidase inhibiting Bifidobacterium spp. towards β-glucuronidase producing Salmonella spp. and E. coli, corresponding with diarrhoea [35]. Studies have shown that lactobacillus spp. decreases 12–48 h after treatment with irinotecan, while E. coli increases as measured by PCR [36]; these changes in commensal bacteria correlate with irinotecan-induced diarrhoea [12]. Although the specific species involved in symptom generation is unclear, there is uniformity in the overall shift in the microbiome that occurs following chemotherapy, dominated by Gram-negative pathogenic species in both preclinical [37] and clinical settings [38, 39]. This is also accompanied by an overall loss in species diversity, enabling the proliferation and colonisation of opportunistic pathogens. Mechanistically, however, the causal relationship between gut dysbiosis and symptom generation remains difficult to achieve. Research using germ-free mice, or antibiotic-induced depletion of the microbiome, has produced conflicting results. In the setting of irinotecan-induced diarrhoea, germ-free mice have shown significantly improved diarrhoea compared to their wild-type counterparts [34]. Importantly, this protection was reversed following re-colonisation of the germ-free mice. However, clinically, the use of prophylactic antibiotics is typically associated with worsened mucositis severity [13, 40].
Further research on the key role of intestinal dysbiosis in chemotherapy-induced mucositis led to the concept that normalisation of intestinal homeostasis could be an appropriate strategy to reduce chemo-toxicity [41], with the obvious investigation of probiotics [41]. Evidence in human [42, 43] and animal [44] studies suggests that probiotics have a role in the prevention of mucositis and they rarely cause sepsis [45]. A randomised clinical study by Valta and co-workers [42] showed that colorectal cancer patients treated with 5-fluorouracil-based regimens who received Lactobacillus preparations presented with diarrhoea less often. A small placebo-controlled trial of B. breve strain Yakult in children undergoing chemotherapy also demonstrated that the treatment group experienced fewer episodes of fever compared with controls [43]. Bowen and colleagues found that VSL#3, which contains a mixture of Streptococcus thermophilus, Bifidobacterium breve, B. longum, B. infantis, Lactobacillus paracasei, L. delbrueckii subsp. bulgaricus, L. acidophilus and L. plantarum, reduced diarrhoea and weight loss in irinotecan-treated rats and inhibited intestinal apoptosis [44]. However, two recent meta-analyses only reported beneficial results for prophylactic probiotics for radiation-induced GIM, with unclear support in the setting of chemotherapy [46, 47]. This likely reflects the heterogeneity that exists in various chemotherapy regimens, agents and doses; all of which are likely to involve different microbial phenotypes. Nonetheless, there is clear evidence for the correlative involvement of the microbiome in the development of chemotherapy-induced GIM, and as such TLRs are considered critical in its symptomology [23, 48].
Toll-like receptors in the GI tract
Recent research has acknowledged an emerging role for pattern recognition receptors (PRR) [8, 49] such as TLRs, in the setting of chemotherapy-induced mucositis [13]. Typically expressed on the plasma and intracellular membranes of innate immune cells, epithelial cells, endothelial cells and cancer cells [20], TLRs play a key role in a number of inflammatory disorders affecting the GIT [50]. This is also the case for mucositis [51], with increasing evidence implicating various members of the TLR family in its pathogenesis. Among 13 known members of the TLR family, 10 are known to be present in humans [32] of which TLR4 is of particular importance in the setting of chemotherapy-induced mucositis, specifically irinotecan-induced mucositis [48].
Activation of TLR4 and its co-receptors CD-14 and MD-2 [5] by the toxic metabolite of irinotecan, SN-38, and LPS [1], involves recruitment and activation of adapter protein myeloid differentiation protein (MyD88) [49, 50]; leading to initiation of an inflammatory cascade involving NF-κB and IL-6 [9, 10]. Excessive activation of TLR4 has been suggested to exacerbate the production of inflammatory cytokines leading to a vicious cycle of tissue destruction through increased inflammation [5]. It is this process that is thought to underpin mucositis [30]; however, this hypothesis remains predominantly based on preclinical research with only anecdotal clinical evidence supporting a role for TLR4.
TLR4 and mucositis
Tight control of basal TLR4 signalling in the healthy gut is essential to maintain intestinal homeostasis and physiological state [5]. Extensive data exist that imply paradoxical roles for TLR4 in different inflammatory-based pathologies including inflammatory bowel disease. Expression of TLR4 is found to increase in the gut during peak injury and undetectable during healing [13]. However, intact TLR4 signalling through MyD88 is important in limiting bacterial translocation, with increased sepsis seen in TLR4 knockout mice compared to wild-type mice [50].
In the setting of chemotherapy-induced mucositis, substantial anecdotal evidence exists suggesting a role for TLR4, yet few studies have directly assessed TLR4-dependent mechanisms. [52] reported a significant increase in TLR4 mRNA expression in the small intestine of methotrexate-treated rats during the acute phase of gut toxicity. These changes coincided with a pro-inflammatory setting and increased permeability [52]. In addition, studies indicate upregulated TLR4 mRNA, NF kB, IL-1β and IL-6 [10] levels in the colon of dark agouti rats following irinotecan [4, 53]. Some studies suggest that TLR4 deletion has a protective role in the setting of chemotherapy-induced mucositis [13]. This concept is supported by an experiment where TLR4 knockout mice had significant reductions in diarrhoea, weight loss [54] and intestinal apoptosis as compared to wild-type mice after irinotecan administration [13]. Conversely, pre-treatment with the TLR4 agonist, LPS, has shown to be protective against radiotherapy-induced intestinal injury in mice [55].
TLR4-mediated protective effects seem to be limited to acute insults, with TLR4 deficiency exacerbating chronic inflammatory diseases [13]. Furthermore, previous studies have shown that a TLR4 knockout mouse model was associated with increased breast tumour growth during treatment with oxaliplatin and doxorubicin [54]. In addition, patients with a TLR4 genetic deficiency had an increased incidence of metastases 5 years after surgery for breast cancer [54]. Pre-clinical trials have also been conducted that for the first time indicate that a non-specific TLR4 antagonist, naloxone, is not effective in reducing irinotecan-induced mucositis [54]. In another study involving a model of dextran sulphate sodium- (DSS) induced colitis, mice deficient in TLR4 or MyD88 exhibited diminished tissue repair [51]. Hence, these contradictory studies strongly suggest that activation of TLRs could be a double-edged sword that can be either beneficial or detrimental to intestinal integrity [28], with the efficacy of TLR targeted interventions depending on the setting (e.g. health vs cancer) and chronicity of symptoms. It also suggests that pharmacological inhibition of TLR4 may not be the best approach in the prevention of GI toxicity. Instead, it may be more beneficial to target a product of TLR4 signalling that confers intestinal protection and resistance to toxicity. Based on previous findings in the field of GIM, we suggest that this may be achieved through targeting IL-6.
TLR4/IL-6 interactions and their relevance to mucositis
IL-6 is an important cytokine in the TLR4-mediated inflammatory pathway of mucositis, particularly affecting the gut [49]. Being a member of a trio of cytokines of acute inflammation (IL-1β, TNF-α, IL-6) [56]; it is unique in having both pro-inflammatory and anti-inflammatory properties [56]. The primary source of IL-6 is monocytes, tissue macrophages at the site of acute inflammation and T cells at the site of chronic inflammation [56]. Although IL-1β and TNF-α also constitute stimulus to produce IL-6 through activation of NF-κB [56], TLR4 activation is one of the earliest events leading to IL-6 production [57].
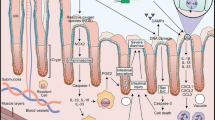
The anti-inflammatory action of IL-6 is mediated via a classical (cis) signalling pathway and pro-inflammatory property is mediated via a trans signalling pathway [56]. In cis-signalling, binding of IL-6 to IL-6R recruits a membrane-bound glycoprotein 130 (gp130) co-receptor that initiates downstream signalling, including Janus kinases (JAKs) and finally phosphorylation and activation of STAT3 [49]. STAT3 further releases IL-6 directly and indirectly via a TLR4-mediated pathway (Fig. 1). Hepatocytes and some immune cells have IL-6R and thus respond to cis-signalling [56]. This pathway promotes growth differentiation, regeneration and induction of apoptosis in neutrophils that resolves acute inflammation [56]. Alternatively, IL-6 can function through a pathway known as trans-signalling by binding to the soluble form of IL-6R (sIL-6R). This complex also interacts with gp130 and activates the same intracellular pathways as cis-signalling [49]. This pathway is responsible for the pro-inflammatory property of IL-6 as gp130 is present on all body cells; it causes accumulation of T cells resistant to apoptosis, migration and accumulation of inflammatory cells and production of TNF-α and IL-1β [56].

Pathways for IL-6 production. Direct IL-6 production occurs through the activation of a receptor formed by two distinct subunits, an alpha subunit for ligand specificity and glycoprotein (GP) 130. Binding of IL-6 to its receptor (IL-6R) initiates cellular events including JAK kinases, which when phosphorylated, activates signal transducers and activators of transcription-3 (STAT3). This then translocates to the nucleus resulting in the production of IL-6. Indirect IL-6 production is mediated through innate immune receptor, Toll-like receptor 4 (TLR4). TLR4 is expressed on glial, immune and intestinal epithelial cells with bacterial lipopolysaccharide (LPS) as its primary ligand. Upon LPS binding, TLR4 undergoes a conformational change resulting in the recruitment of TIR domains containing adaptor molecules. In particular, myeloid differentiation primary response (MyD)88-dependent signalling results in nuclear translocation of nuclear factor kappa B (NF-κB) and the subsequent production of IL-6 [57]
The same mechanism underlies a variety of inflammatory disorders like inflammatory bowel disease (IBD) and rheumatoid arthritis [49], where IL-6 trans-signalling is critically involved in the maintenance of a disease state [56]. This concept is further supported by findings where high expression of IL-6 mRNA was seen in inflamed mucosa of IBD patients as compared to healthy controls and even in normal mucosa adjacent to inflamed mucosa [11]. The same trans-signalling is believed to be involved in the pathogenesis of chemotherapy-induced mucositis [56]. Logan et al. [10, 53] found elevated levels of NF-κB and inflammatory cytokines, TNF-α, IL-1β and IL-6 in serum and AT (both buccal mucosa and intestinal tissue) of rodents after administration of irinotecan, 5-fluorouracil and methotrexate. Similar findings were seen in adults receiving chemotherapy [21] and children having treatment for acute lymphoblastic leukemia, where an increase in blood levels of IL-6 and TNF-α was seen compared to healthy controls, 72 h after administration of methotrexate [58].
Keeping in view these findings, one can suggest that polymorphonuclear inflammatory cells recruited by IL-6 might be a relevant component of chemotherapy-induced GIM. Also, this inflammatory reaction of IL-6 seems to be mediated through TLR4 activation [57]. This concept is further supported by a study in which expression of intestinal IL-6 was significantly reduced in TLR4 knockout mice as compared to wild type, 6 h after irinotecan administration [13]. In fact, TLR4 null mice completely lacked an IL-6 response, supporting the hypothesis that TLR4-dependent NF-κB activation is required for the release of IL-6 from macrophages [59]. Stunted IL-6 production has also been seen in MyD88-deficient mice [60] and TLR4-null macrophages [61], implicating TLR4 in the regulation of IL-6. Hence, IL-6 is recognised as a pivotal component of GIM through its pro-inflammatory properties and TLR4 constitutes a major pathway for its production.
Important considerations for systemic IL-6 blockade
We have outlined a strong body of evidence supporting investigation into IL-6 blockade for the prevention of GIM. However, this approach must be considered in the larger setting of supportive oncology. IL-6 is not exclusively linked to mucosal injury in the gastrointestinal tract. Certainly, there are roles for IL-6 in immunological cell death, critical to the efficacy of some newer targeted agents and immunotherapies. However, more importantly, IL-6 is also considered critical in the response to potentially life-threatening infections that underpin febrile neutropaenia. On one hand, this presents as a promising avenue in which to explore, with the opportunity to simultaneously manage febrile neutropaenia associated with GIM (also termed febrile mucositis [62]). However systemic IL-7 blockade may also mask this febrile response to infection, thus preventing rapid detection of infection in neutropaenia patients [30]. As such, targeting systemic production of IL-6 to alleviate mucositis must be approached with caution, and preclinical models must adequately incorporate measures of infection and neutropaenia in their analyses.
Conclusion
Chemotherapy-induced mucositis, affecting the GIT, is a debilitating side effect of chemotherapy which limits the therapeutic efficiency of cancer treatment. The pathobiology of GIM is complex involving innate immune responses, pharmacokinetics modulation and the gut microbiome. Data increasingly points towards TLR4 as a key regulator of GIM, particularly induced by irinotecan. However, pharmacologically targeting TLR4 and genetic inhibition have presented challenges regarding tumour growth and the inability to overcome chronic insults. Given that TLR4-null mice lack an IL-6 response, it is plausible that protection from GIM is mediated through this pathway. As such, we suggest that IL-6 may be a more promising therapeutic target to prevent or reduce the severity of GIM caused by chemotherapy and should form the basis of new research models.
References
Gibson RJ, Bowen JM, Coller JK (2015) What are the predictive factors in the risk and severity of chemotherapy-induced gastrointestinal toxicity? Future Oncol 11(17):2367–2370
Elting LS et al (2003) The burdens of cancer therapy. Clinical and economic outcomes of chemotherapy-induced mucositis. Cancer 98(7):1531–1539
Keefe DM et al (1997) Effect of high-dose chemotherapy on intestinal permeability in humans. Clin Sci (Lond) 92(4):385–389
Wardill HR et al. (2014) TLR4/PKC-mediated tight junction modulation: a clinical marker of chemotherapy-induced gut toxicity? Int J Cancer 135(11):2483–2492
Cario E (2016) Toll-like receptors in the pathogenesis of chemotherapy-induced gastrointestinal toxicity. Curr Opin Support Palliat Care 10(2):157–164
Kelly D et al (2004) Commensal anaerobic gut bacteria attenuate inflammation by regulating nuclear-cytoplasmic shuttling of PPAR-gamma and RelA. Nat Immunol 5(1):104–112
Eyking A et al (2011) Toll-like receptor 4 variant D299G induces features of neoplastic progression in Caco-2 intestinal cells and is associated with advanced human colon cancer. Gastroenterology 141(6):2154–2165
Spiller S et al (2008) TLR4-induced IFN-gamma production increases TLR2 sensitivity and drives Gram-negative sepsis in mice. J Exp Med 205(8):1747–1754
Sonis ST (2007) Pathobiology of oral mucositis: novel insights and opportunities. J Support Oncol 5(9 Suppl 4):3–11
Logan RM et al (2008) Characterisation of mucosal changes in the alimentary tract following administration of irinotecan: implications for the pathobiology of mucositis. Cancer Chemother Pharmacol 62(1):33–41
Velikova T et al (2017) Alterations in cytokine gene expression profile in colon mucosa of Inflammatory Bowel Disease patients on different therapeutic regimens. Cytokine 92:12–19
Logan RM et al (2007) The role of pro-inflammatory cytokines in cancer treatment-induced alimentary tract mucositis: pathobiology, animal models and cytotoxic drugs. Cancer Treat Rev 33(5):448–460
Wardill HR et al (2016) Irinotecan-induced gastrointestinal dysfunction and pain are mediated by common TLR4-dependent mechanisms. Mol Cancer Ther 15(6):1376–1386
Peterson DE et al (2011) Management of oral and gastrointestinal mucositis: ESMO clinical practice guidelines. Ann Oncol 22(Suppl 6):vi78–vi84
Lalla RV et al (2014) MASCC/ISOO clinical practice guidelines for the management of mucositis secondary to cancer therapy. Cancer 120(10):1453–1461
Vouk K et al (2016) Cost and economic burden of adverse events associated with metastatic melanoma treatments in five countries. J Med Econ 19(9):900–912
Carlotto A et al (2013) The economic burden of toxicities associated with cancer treatment: review of the literature and analysis of nausea and vomiting, diarrhoea, oral mucositis and fatigue. Pharmacoeconomics 31(9):753–766
White T, De Abreu Lourenco R, Kenny P, Lehane L, D'Abrew N (2017) Abstracts of the MASCC/ISOO 2017 annual meeting. Support Care Cancer. https://doi.org/10.1007/s00520-017-3704-x
Sonis ST (2004) The pathobiology of mucositis. Nat Rev Cancer 4(4):277–84
Yu J (2013) Intestinal stem cell injury and protection during cancer therapy. Transl Cancer Res 2(5):384–396
Logan RM et al (2007) Nuclear factor-kappaB (NF-kappaB) and cyclooxygenase-2 (COX-2) expression in the oral mucosa following cancer chemotherapy. Oral Oncol 43(4):395–401
Wardill HR et al (2014) Irinotecan disrupts tight junction proteins within the gut: implications for chemotherapy-induced gut toxicity. Cancer Biol Ther 15(2):236–244
Wardill HR et al (2016) TLR4-dependent claudin-1 internalization and secretagogue-mediated chloride secretion regulate irinotecan-induced diarrhea. Mol Cancer Ther 15(11):2767–2779
Wardill HR et al (2016) Tight junction defects are seen in the buccal mucosa of patients receiving standard dose chemotherapy for cancer. Support Care Cancer 24(4):1779–1788
Blijlevens NM, Donnelly JP, de Pauw BE (2005) Prospective evaluation of gut mucosal barrier injury following various myeloablative regimens for haematopoietic stem cell transplant. Bone Marrow Transplant 35(7):707–711
Keefe D et al (2000) Chemotherapy for cancer causes apoptosis that precedes hypoplasia in crypts of the small intestine in humans. Gut 47(5):632–637
Pereira VB et al (2016) A new animal model of intestinal mucositis induced by the combination of irinotecan and 5-fluorouracil in mice. Cancer Chemother Pharmacol 77(2):323–332
Kaczmarek A et al (2012) Severity of doxorubicin-induced small intestinal mucositis is regulated by the TLR-2 and TLR-9 pathways. J Pathol 226(4):598–608
Guabiraba R et al (2014) IL-33 targeting attenuates intestinal mucositis and enhances effective tumor chemotherapy in mice. Mucosal Immunol 7(5):1079–1093
Wong M et al (2013) Microbial translocation contribute to febrile episodes in adults with chemotherapy-induced neutropenia. PLoS One 8(7):e68056
Wisplinghoff H et al (2004) Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin Infect Dis 39(3):309–317
Thorpe DW, Stringer AM, Gibson RJ (2013) Chemotherapy-induced mucositis: the role of the gastrointestinal microbiome and toll-like receptors. Exp Biol Med (Maywood) 238(1):1–6
Thaiss CA et al (2016) The microbiome and innate immunity. Nature 535(7610):65–74
Pedroso SH et al (2015) Evaluation of mucositis induced by irinotecan after microbial colonization in germ-free mice. Microbiology 161(10):1950–1960
Stringer AM et al (2009) Gastrointestinal microflora and mucins may play a critical role in the development of 5-fluorouracil-induced gastrointestinal mucositis. Exp Biol Med (Maywood) 234(4):430–441
Stringer AM et al (2008) Faecal microflora and beta-glucuronidase expression are altered in an irinotecan-induced diarrhea model in rats. Cancer Biol Ther 7(12):1919–1925
Stringer AM et al (2009) Irinotecan-induced mucositis manifesting as diarrhoea corresponds with an amended intestinal flora and mucin profile. Int J Exp Pathol 90(5):489–499
Montassier E et al (2014) 16S rRNA gene pyrosequencing reveals shift in patient faecal microbiota during high-dose chemotherapy as conditioning regimen for bone marrow transplantation. Microb Ecol 67(3):690–699
Montassier E et al (2015) Chemotherapy-driven dysbiosis in the intestinal microbiome. Aliment Pharmacol Ther 42(5):515–528
Brandi G et al (2006) Intestinal microflora and digestive toxicity of irinotecan in mice. Clin Cancer Res 12(4):1299–1307
Tang Y et al (2017) Administration of probiotic mixture DM#1 ameliorated 5-fluorouracil-induced intestinal mucositis and dysbiosis in rats. Nutrition 33:96–104
Osterlund P et al (2007) Lactobacillus supplementation for diarrhoea related to chemotherapy of colorectal cancer: a randomised study. Br J Cancer 97(8):1028–1034
Motoori M et al (2017) Randomized study of the effect of synbiotics during neoadjuvant chemotherapy on adverse events in esophageal cancer patients. Clin Nutr 36(1):93–99
Bowen JM et al (2007) VSL#3 probiotic treatment reduces chemotherapy-induced diarrhea and weight loss. Cancer Biol Ther 6(9):1449–1454
Redman MG, Ward EJ, Phillips RS (2014) The efficacy and safety of probiotics in people with cancer: a systematic review. Ann Oncol 25(10):1919–1929
Wang YH et al (2016) The efficacy and safety of probiotics for prevention of chemoradiotherapy-induced diarrhea in people with abdominal and pelvic cancer: a systematic review and meta-analysis. Eur J Clin Nutr 70(11):1246–1253
Wardill HR et al (2018) Prophylactic probiotics for cancer therapy-induced diarrhoea: a meta-analysis. Curr Opin Support Palliat Care 12(2):187–197
Wardill H et al (2016) Toll-like receptor 4 (tlr4)-mediated tight junction disruption and dysregulated ion secretion are key drivers of irinotecan-induced diarrhoea. Support Care Cancer 24(1):S60
Greenhill CJ et al (2011) IL-6 trans-signaling modulates TLR4-dependent inflammatory responses via STAT3. J Immunol 186(2):1199–1208
Fukata M et al (2005) Toll-like receptor-4 is required for intestinal response to epithelial injury and limiting bacterial translocation in a murine model of acute colitis. Am J Physiol Gastrointest Liver Physiol 288(5):G1055–G1065
Frank M et al (2015) TLR signaling modulates side effects of anticancer therapy in the small intestine. J Immunol 194(4):1983–1995
Hamada K, Kakigawa N, Sekine S, Shitara Y, Horie T (2013) Disruption of ZO-1/claudin-4 interaction in relation to inflammatory responses in methotrexate-induced intestinal mucositis. Cancer Chemother Pharmacol 72(4):757–765. https://doi.org/10.1007/s00280-013-2238-2
Logan RM et al (2008) Serum levels of NFkappaB and pro-inflammatory cytokines following administration of mucotoxic drugs. Cancer Biol Ther 7(7):1139–1145
Coller JK et al (2017) Potential safety concerns of TLR4 antagonism with irinotecan: a preclinical observational report. Cancer Chemother Pharmacol 79(2):431–434
Riehl T et al (2000) Lipopolysaccharide is radioprotective in the mouse intestine through a prostaglandin-mediated mechanism. Gastroenterology 118(6):1106–1116
Naugler WE, Karin M (2008) The wolf in sheep’s clothing: the role of interleukin-6 in immunity, inflammation and cancer. Trends Mol Med 14(3):109–119
Karin M, Lawrence T, Nizet V (2006) Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell 124(4):823–835
Morales-Rojas T et al (2012) Proinflammatory cytokines during the initial phase of oral mucositis in patients with acute lymphoblastic leukaemia. Int J Paediatr Dent 22(3):191–196
Pathak SK et al (2013) Helicobacter pylori protein JHP0290 binds to multiple cell types and induces macrophage apoptosis via tumor necrosis factor (TNF)-dependent and independent pathways. PLoS One 8(11):e77872. https://doi.org/10.1371/journal.pone.0077872
Hayashi F et al (2001) The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 410(6832):1099–1103
Shoenfelt J et al (2009) Involvement of TLR2 and TLR4 in inflammatory immune responses induced by fine and coarse ambient air particulate matter. J Leukoc Biol 86(2):303–312
Demacker PN et al (2009) Plasma citrulline measurement using UPLC tandem mass-spectrometry to determine small intestinal enterocyte pathology. J Chromatogr B Anal Technol Biomed Life Sci 877(4):387–392
Acknowledgements
Dr. Hannah Wardill is the recipient of an NHMRC CJ Martin Postdoctoral Fellowship (2018–2022).
Funding
No funding was sought for this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare they have no conflicts of interest.
Ethical approval
This article does not contain any studies with animals or humans performed by any of the authors.
Rights and permissions
About this article

Cite this article
Khan, S., Wardill, H.R. & Bowen, J.M. Role of toll-like receptor 4 (TLR4)-mediated interleukin-6 (IL-6) production in chemotherapy-induced mucositis. Cancer Chemother Pharmacol 82, 31–37 (2018). https://doi.org/10.1007/s00280-018-3605-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-018-3605-9