
Abstract
Purpose
To characterize cobimetinib pharmacokinetics and evaluate impact of clinically relevant covariates on cobimetinib pharmacokinetics.
Methods
Plasma samples (N = 4886) were collected from 487 patients with various solid tumors (mainly melanoma) in three clinical studies (MEK4592g, NO25395, GO28141). Cobimetinib was administered orally, once daily on either a 21-day-on/7-day-off, 14-day-on/14-day-off or 28-day-on schedule in a 28-day dosing cycle as single agent or in combination with vemurafenib. Cobimetinib doses ranged from 2.1 to 125 mg. NONMEM was used for pharmacokinetic analysis.
Results
A linear two-compartment model with first-order absorption, lag time and first-order elimination described cobimetinib pharmacokinetics. The typical estimates (inter-individual variability) of apparent clearance (CL/F), central volume of distribution (V2/F) and terminal half-life were 322 L/day (58 %), 511 L (49 %) and 2.2 days, respectively. Inter-occasion variability on relative bioavailability was estimated at 46 %. CL/F decreased with age. V2/F increased with body weight (BWT). However, the impact of age and BWT on cobimetinib steady-state exposure (peak and trough concentrations and AUC following the recommended daily dose of 60 mg 21-day-on/7-day-off) was limited (<25 % changes across the distribution of age and BWT). No significant difference in cobimetinib pharmacokinetics or steady-state exposure was observed between patient subgroups based on sex, renal function, ECOG score, hepatic function tests, race, region, cancer type, and co-administration of moderate and weak CYP3A inducers or inhibitors and vemurafenib.
Conclusion
A population pharmacokinetic model was developed for cobimetinib in cancer patients. Covariates had minimal impact on steady-state exposure, suggesting no need for dose adjustments and supporting the recommended dose for all patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cobimetinib (also known as GDC-0973, XL518 and RO5514041) is a potent and highly selective small molecule inhibitor of mitogen-activated protein kinases (MEK) including MEK1/2, central components of the RAS/RAF/MEK/ERK signal transduction pathway. This signaling pathway is highly conserved and plays an important role in cell proliferation, survival, migration, cell cycle regulation and angiogenesis [1]. In the phase III double-blind placebo-controlled randomized clinical trial [2] in 495 previously untreated patients with BRAF V600 mutation-positive unresectable locally advanced or metastatic melanoma (detected by the cobas® 4800 BRAF Mutation Test), cobimetinib in combination with vemurafenib (Zelboraf®) significantly improved progression-free survival (PFS) as compared to vemurafenib alone with a hazard ratio of 0.51 (p < 0.001). Median PFS was improved from 6.2 months in the vemurafenib arm to 9.9 months in the cobimetinib plus vemurafenib arm. The interim analysis of overall survival in the intention-to-treat population showed a significant hazard ratio of 0.65 (p = 0.046).
In vitro data indicated that cobimetinib is metabolized via CYP3A and UGT2B7 [3]. Cobimetinib pharmacokinetics (PK) was previously reported in healthy volunteers [3, 4], but little information has been published about cobimetinib PK in cancer patients [3–5]. Cobimetinib PK samples have been collected in three clinical studies investigating cobimetinib in cancer patients: a single-agent dose escalation and expansion study of cobimetinib in patients with solid tumors (MEK4592g, NCT00467779), a combination dose escalation and expansion study of cobimetinib and vemurafenib in BRAF V600 mutation-positive advanced melanoma patients (NO25395, NCT01271803 [6]), and a pivotal phase III study of cobimetinib and vemurafenib combination in BRAF V600 mutation-positive advanced melanoma patients (GO28141, NCT01689519 [2]). A population PK analysis was performed to analyze these data and reported here. The objectives of this analysis were to estimate typical values and inter-individual variability of cobimetinib PK parameters and the effects of clinically relevant covariates on cobimetinib PK parameters and exposure in cancer patients in order to inform dosing.
Methods
Patients, samples and bioanalysis
Patients were defined as evaluable for population pharmacokinetic (PK) analysis if at least one adequately documented cobimetinib administration and a documented corresponding PK sample collection after the dose were available. Dosing information is summarized in Table 1. Each dosing cycle was 28 days in all three studies. Serial and sparse plasma samples were collected at the pre-specified time points to capture PK after the first dose and at steady state in all three studies.
Cobimetinib plasma concentrations were determined by a validated liquid chromatography/tandem mass spectrometry (LC–MS/MS) method [4] by Advion (Ithaca, NY) and Covance Laboratories, Inc. (Madison, WI). The lower limit of the quantification (LLOQ) of the assay was 0.2 ng/mL except for some patients in MEK4592g (LLOQ = 0.183 ng/mL).
Population pharmacokinetic modeling
Population PK analysis was performed using a nonlinear mixed-effects modeling approach with NONMEM, version 7.2 (ICON Development Solutions, Ellicott City, MD, USA), Perl-Speak-NONMEM (version 3.7.6; Uppsala University, Uppsala, Sweden) and R version 3.0.2 [7]. The FOCE method with interaction was used in NONMEM. Concentrations below the LLOQ were omitted. The clinically relevant covariates tested included those related to demographics, hepatic function, renal function, co-administered medications and pathophysiological factors. For each study, if the covariate value was available for ≥85 % of the patients, the missing values were imputed. The imputed value was the median for continuous covariates or the most frequent value for categorical covariates derived with available data for each sex.
Several models were fit to the PK data to select the best base model. Concentrations were log-transformed for model fitting. Various OMEGA matrix models were evaluated. Nonlinearity of PK was assessed using different dose-dependency models on relative bioavailability (F1). The quality of fit was evaluated using a standard model discrimination process including statistical criteria [i.e., minimum of objective function value (OFV)] as well as graphical representations of goodness of fit. The final model was established in a stepwise manner by forward addition followed by backward elimination with a significance level of p < 0.05 and p < 0.001, respectively.
The effect of n covariates on apparent clearance (CL/F), apparent central volume of distribution (V2/F) and F1 was coded using a multiplicative model:
where θ i is the typical value of the parameter for the individual patient i with a set of covariates 1, …, n, θ TV is the typical value of the PK parameter for patients having the covariate values equal to the reference, and Effect1,i through Effect n,i were multiplicative factors of the effects for covariate 1 through n, for the individual patient i.
The multiplicative factor was defined using the power function for continuous covariates:
and defined as follows for categorical covariates with m (m > 1) categories:
-
If this categorical covariate is in the reference category, then Effect i = 1
-
If this categorical covariate is in the category k (k = 1, …, m), then Effect i = eθeff(k)
where Effect i is the multiplicative factor of the covariate effect of the particular covariate Cov in the individual patient i, Cov i is the covariate value, Covreference value is the median of this covariate for all patients and θ eff(k) is the exponent of the power function to be estimated for the category k.
After all the other covariates were evaluated by forward addition and backward elimination, drug–drug interaction and vemurafenib co-administration effect were evaluated on final model with a significance level of p < 0.001, including the association between cobimetinib PK parameters and co-administration of weak-to-moderate CYP3A inducers and inhibitors and vemurafenib. Vemurafenib co-administration was assessed both as categorical variable (yes/no) and as continuous variable (vemurafenib steady-state AUC).
Model evaluation and sensitivity analysis
The population PK models were evaluated using diagnostic plots [8, 9], visual predictive check (VPC) [9, 10], prediction-corrected VPC (pcVPC) [11], bootstrapping [12] and shrinkage [13] assessments. VPC, pcVPC and bootstrapping were all performed using 1000 replicates based on the final model. The relative impact of significant covariates on cobimetinib exposure metrics (AUCss, C max,ss, C min,ss) was assessed by simulating PK using final PK model with keeping other covariates at the reference value. Exposure metrics were simulated at steady state using the final population PK model under the dosing regimen of 60 mg daily with 21 days on followed by 7 days off, and included the maximum concentration (C max,ss), trough concentration (C min,ss) and area under the plasma concentration versus time curve (AUCss) of cobimetinib.
Results
Patients
A total of 4886 cobimetinib plasma concentrations from 487 patients including 277 female and 210 male patients were used for the population PK analysis. The number of patients who received a dose of 40 mg or lower, 60, 80, 100 and 125 mg was 22 (4.5 %), 407 (83.6 %), 19 (3.9 %), 33 (6.8 %) and 6 (1.2 %), respectively. The number of cobimetinib concentrations available for analysis in these dose groups were 401 (8.2 %), 3534 (72.3 %), 310 (6.3 %), 538 (11.0 %) and 103 (2.1 %), respectively.
Patient characteristics (continuous covariates) are summarized in Table 2. Age in this patient population ranged from 19 to 88 years with an average of 55.5 years, and body weight (BWT) ranged from 42.7 to 185 kg with an average of 81 kg. All the categorical variables are summarized as follows. Majority of the patients (92 %) are Caucasian. Fifty-six percent of the patients had normal renal function. The percentages of patients with mild, moderate and severe renal impairment are 29, 10 and <1 %, respectively. The degree of renal impairment was defined and classified based on estimated creatinine clearance (CRCL) as: Normal (CRCL ≥ 90 mL/min), Mild (CRCL ≥ 60 and < 90 mL/min), Moderate (CRCL ≥ 30 and <60 mL/min) and Severe (CRCL < 30 mL/min) [14]. This classification of renal impairment categories was performed to show the impact on exposure (sensitivity analysis); the effect of renal impairment on PK was tested using creatinine clearance as a continuous covariate in the model. The percentages of patients with baseline ECOG performance score of 0, 1 or 2 are 55, 42 and 2 %, respectively. Approximately, half of the patients were enrolled in the USA. Majority of the patients (80 %) enrolled had melanoma. The percentages of patients who received concomitant medications that were classified as CYP3A inducers or inhibitors are 7 and 25 %, respectively. Most of the co-administered CYP3A inducers and inhibitors are weak to moderate. BRAF V600 mutation subtype (V600E vs. V600K) information was available only in Study GO28141. All the variables listed above and in Table 2 were tested in the model.
Population pharmacokinetic modeling
Concentrations below the LLOQ were omitted (59 of 4886, 1.2 %). A linear two-compartment model with first-order absorption, lag time and first-order elimination was the best base model to describe cobimetinib PK. Inter-individual variability (IIV) was assessed on CL/F, V2/F, Q/F (apparent inter-compartmental clearance) and V3/F (apparent peripheral volume of distribution). Inter-occasion variability (IOV) was assessed on F1 with three occasions defined as follows: Occasion 1: up to 24 h after the first dose; Occasion 2: steady state (from day 14); Occasion 3: all other time points. Residual unexplained variability was best described using an additive residual error model on log-transformed concentration data. Due to the strong correlations between body size covariates (body surface area, body mass index and body weight), only one of these covariates, body weight, was retained in the full population PK model.
Covariate analysis identified an age effect on CL/F and body weight effect on V2/F. In the final model, CL/F decreased with age and V2/F increased with body weight (BWT) (Supplementary Figure 1). CL/F and V2/F for patient i in the final model were as follows:
For a typical patient who is 57 years old with a BWT of 80 kg, the estimated CL/F was 322 L/day and V2/F was 511 L. The estimated population distribution and terminal half-lives for a typical patient were 11.5 h and 2.2 days, respectively. Parameter estimates of the final model are summarized in Table 3. No other covariates or patient factors, e.g., renal function, disease severity, hepatic function, race (Caucasian vs. non-Caucasian), region (USA vs. non-USA), cancer type (melanoma vs. non-melanoma), BRAF V600 mutation subtype (V600E vs. V600K), co-administration of weak-to-moderate CYP3A inducers or inhibitors, and co-administration of vemurafenib, were identified as statistically significant on cobimetinib CL/F or V2/F (Supplementary Figures 2 & 3) in this analysis.
Model evaluation and sensitivity analysis
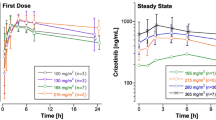
Goodness-of-fit plots showed good agreement between predicted and observed cobimetinib concentrations with no apparent bias in residual plots over time or across population-predicted concentrations (Supplementary Figure 4). Graphical representation of the prediction-corrected visual predictive check for the final population PK model is presented in Fig. 1. Overall, the 2.5th, 50th and 97.5th percentiles of observed concentrations are within the predicted 95 % confidence interval (CI) of these percentiles, suggesting accurate model fitting across a wide range of dosing regimens and time course. Bootstrapping resulted in median parameter estimates and 95 % CI similar to the estimates from the original dataset, indicating that the final model provided good precision for parameter estimation.

Prediction-corrected visual predictive check for the cobimetinib final population PK model. Note 324 out of the 4886 (6.6 %) prediction-corrected observations with time after dose beyond 30 h were not displayed. Pred: population prediction
The impact of the variation for a single covariate included in the final model on key cobimetinib PK parameters was low: no more than 14 % for CL/F and no more than 33 % for V2/F. Covariate-normalized CL/F (Fig. 2) and V2/F (not shown) were similar between patient subgroups by all other clinically relevant covariates including degrees of renal impairment, race (Caucasian vs. non-Caucasian), cancer type (melanoma vs. non-melanoma), CYP3A inducer or inhibitor co-administration and vemurafenib co-administration.

Comparison of cobimetinib CL/F from the final model between patient subgroups by clinically relevant covariates: a degrees of renal impairment, b race (Caucasian vs. non-Caucasian), c cancer type (melanoma vs. non-melanoma), d CYP3A inducer co-administration, e CYP3A inhibitor co-administration and f vemurafenib co-administration. Circles represent the Bayesian post hoc PK parameter estimates from the final population PK model (covariate-normalized CL/F and/or V2/F). The blue lines represent the typical population value. The red squares represent the geometric means of the individual estimates. N is the number of patients in each group
For the final model, the effects of covariates on cobimetinib steady-state exposure (C max,ss, C min,ss and AUSss) are presented in Fig. 3. Using the predicted steady-state exposure of cobimetinib at 60 mg (21 days on followed by 7 days off) in a typical patient with age of 57 years and BWT of 80 kg as the base, variation in age and BWT caused no more than 16 % of decrease and no more than 9 % of increase in exposure from “the base” (see Fig. 3 for definition). All exposure metrics were comparable among patient subgroups by all other covariates (Supplementary Table 1).

Impact of covariates in the final model (age and BWT) on cobimetinib steady-state exposure: a maximum concentration (C max,ss), b trough concentration (C min,ss) and c area under the plasma concentration versus time curve (AUCss). BWT denotes baseline body weight. The black vertical line presents the base, defined as the predicted steady-state exposure of cobimetinib at 60 mg in a typical patient with age of 57 years and BWT of 80 kg. The gray shaded bar shows the fifth and 95th percentile (p5–p95) of the simulated steady-state exposure range across the entire population. Each green shaded bar represents the influence of a single covariate on the steady-state exposure. The label at left end of the bar represents age and BWT with the boundary values of the fifth and 95th percentiles (p5–p95). The length of each green bar describes the impact of age and BWT on cobimetinib steady-state exposure. The percentage value at each end of the green bar represents the percent change from the base
Discussion
The objective of this analysis was to inform the prescribing and dosing practice of cobimetinib based on the impact of patient variables such as age, body weight, renal function, disease severity, hepatic function, race, region, cancer type (melanoma vs. non-melanoma), BRAF V600 mutation subtype and co-administered medications on cobimetinib PK. A population PK model was developed for cobimetinib using a total of 4886 plasma concentrations from 487 patients with solid tumors across three clinical studies. A two-compartment model with first-order absorption, lag time and first-order elimination best described cobimetinib PK in patients. The model-derived terminal elimination half-life was 2.2 days. The PK data after single dose and at steady state allowed characterizing the dug accumulation and estimation of elimination half-life of cobimetinib.
Cobimetinib PK was linear in the tested dose range. Inclusion of dose on relative bioavailability (F1) in the model resulted in a drop in OFV of <10. However, the relative standard error of the estimated dose effect parameter was high (>40 %), the covariance step often aborted and the 95 % CI included zero. As a result, the effect of dose on F1 was not retained in the model.
Inter-individual variability (IIV) for CL/F and V2/F was estimated to be 58 and 49 %, respectively. The relative standard error for IIV on V3/F and Q/F was higher; however, the models without IIV on these parameters had high objective function values. Higher variability estimates were observed for the other PK parameters and particularly for Ka (166 %) as commonly observed with orally administered drugs. A relatively small η-shrinkage of <25 % for Ka, CL/F and V2/F was observed both in the base model and in the final model, suggesting that PK data collected were sufficient to characterize the IIV for these key PK parameters.
The covariates, age and body weight were found to be significant on CL/F and V2/F, respectively. Although CL/F decreased with age and V2/F increased with BWT according to the final model, their impact on steady-state exposure was minimal (Fig. 3). All effects were well within the IIV represented by the fifth and 95th percentiles of prediction intervals. In addition, vemurafenib co-administration (a weak CYP3A inducer [6]) had no impact on cobimetinib PK (Fig. 2). These results indicated that dose adjustment based on these covariates is not needed.
Cobimetinib is extensively metabolized, and there appears to be a greater contribution of intestinal metabolism than hepatic metabolism to the overall oral clearance [15]. Although in vitro data indicated that cobimetinib is metabolized via CYP3A and UGT2B7 [3], no difference in CL/F (Fig. 2) and exposure metrics was observed between patient subgroups by co-administration of weak-to-moderate CYP3A inducers or inhibitors in our analysis. Given the small percentage of patients receiving moderate CYP3A inhibitors (N = 16 [3 %]) and moderate inducers (N = 1 [0.2 %]), these results should be interpreted with caution. Vemurafenib was tested separately instead of being pooled with other CYP3A inducers because 77 % patients received vemurafenib. The lack of drug–drug interaction (DDI) effect with weak inducers was consistent with the lack of DDI with vemurafenib (a weak CYP3A inducer) co-administration. However, the current dataset provided limited information on the impact of moderate and strong CYP3A inducers or inhibitors on cobimetinib exposure, which has been assessed in a dedicated DDI study in healthy volunteers.
Creatinine clearance was not a significant covariate on cobimetinib clearance. In the sensitivity analysis, renal function had minimal impact on PK parameters and exposure of cobimetinib although effect of severe renal impairment could not be adequately tested due to the limited number of patients (N = 2). This finding is consistent with the fact that cobimetinib is predominantly excreted in feces with only 18 % of the dose recovered in urine [15]. It was expected that race (Caucasian vs. non-Caucasian) had minimal impact on cobimetinib PK parameters and exposure because there has been little evidence that the variation in CYP3A expression and activity between Caucasians and non-Caucasians led to a significant difference in PK and drug exposure. However, the result of race effect was only exploratory due to the small number of non-Caucasian patients. In addition, ECOG performance score, hepatic function, region, cancer type (melanoma vs. non-melanoma), BRAF V600 mutation subtype (V600E vs. V600K, only available in GO28141) and other covariates evaluated all exhibited minimal impact on PK parameters and exposure of cobimetinib (Supplementary Table 1 and Supplementary Figures 2 and 3). Based on these results, no dose adjustment appears necessary based on these variables.
In conclusion, the population PK model adequately described cobimetinib PK. Age and body weight were the only statistically significant variables influencing cobimetinib PK parameters, but showed minimal impact on steady-state exposure, suggesting that no dose adjustment is needed for the recommended cobimetinib dosing regimen (60 mg once daily with 21 days on followed by 7 days off) based on these covariates.
References
Hoeflich KP, Merchant M, Orr C et al (2012) Intermittent administration of MEK inhibitor GDC-0973 plus PI3K inhibitor GDC-0941 triggers robust apoptosis and tumor growth inhibition. Cancer Res 72:210–219
Larkin J, Ascierto PA, Dréno B, Atkinson V, Liszkay G, Maio M, Mandalà M, Demidov L, Stroyakovskiy D, Thomas L, de la Cruz-Merino L, Dutriaux C, Garbe C, Sovak MA, Chang I, Choong N, Hack SP, McArthur GA, Ribas A (2014) Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med 371(20):1867–1876
Musib L, Choo E, Deng Y, Eppler S, Rooney I, Chan IT, Dresser MJ (2013) Absolute bioavailability and effect of formulation change, food, or elevated pH with rabeprazole on cobimetinib absorption in healthy subjects. Mol Pharm 10(11):4046–4054
Deng Y, Musib L, Choo E, Chapple M, Burke S, Johnson J, Eppler S, Dean B (2014) Determination of cobimetinib in human plasma using protein precipitation extraction and high-performance liquid chromatography coupled to mass spectrometry. J Chromatogr B Anal Technol Biomed Life Sci 1(972):117–123
Musib L, Eppler S, Choo E, Deng A, Miles D, Hsu B, Rosen L, Sikic B, LoRusso P, Ma W, Goldman J, Fisher G, Weise A, Dy G, Chan I, Ware J (2011) Clinical pharmacokinetics of GDC-0973, an oral MEK inhibitor, in cancer patients: data from a phase 1 study. Presented at American Association for Cancer Research, 102nd annual meeting; April 2–6, 2011; Orlando, FL
Ribas A, Gonzalez R, Pavlick A, Hamid O, Gajewski TF, Daud A, Flaherty L, Logan T, Chmielowski B, Lewis K, Kee D, Boasberg P, Yin M, Chan I, Musib L, Choong N, Puzanov I, McArthur GA (2014) Combination of vemurafenib and cobimetinib in patients with advanced BRAF(V600)-mutated melanoma: a phase 1b study. Lancet Oncol 15(9):954–965
R Development Core Team (2008) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. ISBN 3-900051-07-0. http://www.R-project.org
Karlsson MO, Savic RM (2007) Diagnosing model diagnostics. Clin Pharmacol Ther 82:17–20
Guidance for Industry Population Pharmacokinetics, U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER), Feb 1999. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM072137.pdf
Committee for Medicinal Products for Human Use (CHMP), Guideline on reporting the results of population pharmacokinetic analyses, Doc. Ref. CHMP/EWP/185990/06, London, 21 June 2007. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003067.pdf
Bergstrand M, Hooker AC, Wallin JE, Karlsson MO (2011) Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J 13(2):143–151
Ette EI (1997) Stability and performance of a population pharmacokinetic model. J Clin Pharmacol 37:486–495
Savic RM, Karlsson MO (2009) Importance of shrinkage in empirical Bayes estimates for diagnostics: problems and solutions. AAPS J 11:558–569
US Food and Drug Administration (2010) guidance for industry: pharmacokinetics in patients with impaired renal function—study design, data analysis, and impact on dosing and labeling. US Food and Drug administration, Rockville. http://www.fda.gov/downloads/Drugs/guidanceComplianceregulatoryInformation/guidances/UCM204959.pdf
Choo E, Takahashi R, Rooney I, Gates M, Deng Y, Musib L (2013) Assessing human absorption, metabolism, routes of excretion and the contribution of intestinal metabolism to the oral clearance of cobimetinib, a MEK inhibitor. Mol Cancer Ther 12:B160
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
Kelong Han, Jin Jin, Stephen Eppler, Nicholas Choong, Stephen P Hack, Nalin Tikoo, Mark Dresser, Luna Musib and Nageshwar Budha receive salary from Genentech and hold stocks of Roche Pharmaceuticals; in addition, Jin Jin and Luna Musib also hold stock in Eli Lilly; Mathilde Marchand and Rene Bruno receive salary from Pharsight Consulting Services.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Fig. 1
Correlation between cobimetinib pharmacokinetic parameters from the base model and age and body weight. Blue lines represent the locally weighted scatterplot smoothing (LOESS). CL/F = apparent clearance (L/day); V2/F = apparent central volume of distribution (L). (PDF 116 kb)
Supplementary Fig. 2
Correlation between cobimetinib apparent clearance CL/F (L/day) from the final model and clinically relevant covariates that were not statistically significant. (PDF 179 kb)
Supplementary Fig. 3
Correlation between cobimetinib apparent central volume of distribution V2/F (L) from the final model and clinically relevant covariates that were not statistically significant. (PDF 173 kb)
Supplementary Fig. 4
Goodness-of-fit plots for the final PK model of cobimetinib. Gray lines: LOESS (locally weighted scatterplot smoothing; some of the smooth lines were constrained to rich data). (PDF 1357 kb)
Rights and permissions
About this article

Cite this article
Han, K., Jin, J.Y., Marchand, M. et al. Population pharmacokinetics and dosing implications for cobimetinib in patients with solid tumors. Cancer Chemother Pharmacol 76, 917–924 (2015). https://doi.org/10.1007/s00280-015-2862-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-015-2862-0