
Abstract
Purpose
To evaluate safety of balugrastim, a recombinant human serum albumin and granulocyte colony-stimulating factor (G-CSF), administered over a range of therapeutic doses in women with breast cancer receiving doxorubicin plus docetaxel chemotherapy.
Methods
The phase I, sequential dose-escalation first segment compared subcutaneous balugrastim 50, 150, 300, and 450 µg/kg during chemotherapy cycles 0–2. The randomized (2:2:1), open-label, phase IIa second segment compared balugrastim 300 or 450 µg/kg with pegfilgrastim 6 mg during chemotherapy cycles 1 and 2.
Results
In the phase I segment, balugrastim was escalated to 450 µg/kg in 13 patients without dose-limiting toxicity. Three (9.7 %) of the 31 adverse events (AEs) reported in nine patients were grade 3 (agranulocytosis, vomiting, hypertension); none was grade 4. In the open-label phase IIa segment (N = 51), the majority of the 64 AEs reported in 31 (75.6 %) balugrastim-treated patients were grade 1 (59.4 %), with 39.1 % grade 2, 1.6 % grade 3 (one AE of vomiting), and none grade 4. Of the 16 AEs reported in seven (70.0 %) pegfilgrastim-treated patients, 87.5 % were grade 1, 6.3 % were grade 2, 6.3 % were grade 3 (one AE of thrombocytopenia), and none were grade 4. Overall, there were six bone pain AEs reported, one in the balugrastim 300 µg/kg group and five in the balugrastim 450 µg/kg group. No AEs in either study necessitated treatment interruption/discontinuation. The incidence and duration of grade 3–4 neutropenia were similar between balugrastim- and pegfilgrastim-treated patients.
Conclusions
Balugrastim was well tolerated in this small population of breast cancer patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neutropenia, a decrease in the levels of neutrophils in the peripheral blood, is commonly observed in cancer patients receiving myelosuppressive chemotherapy and is associated with an increased risk of serious or life-threatening infections. Therefore, neutropenia has been identified as a major dose-limiting toxicity for many cytotoxic chemotherapy regimens and may necessitate delaying a subsequent cycle of chemotherapy [1]. Taxanes and anthracyclines are widely used chemotherapeutic agents for treatment of both early and advanced breast cancer [2]. For breast cancer patients who receive doxorubicin and docetaxel, a mean duration of severe neutropenia, generally defined as an absolute neutrophil count (ANC) <0.5 × 109/L of 3.8 days during the first chemotherapy cycle, has been reported [3], with previous studies demonstrating that 33 to 48 % of patients develop febrile neutropenia [4, 5].
Recombinant granulocyte colony-stimulating factors (G-CSFs) induce neutrophil proliferation and differentiation and enhance the effector function of mature neutrophils, reducing the duration and incidence of chemotherapy-induced neutropenia and febrile neutropenia [6, 7]. Standard G-CSFs such as filgrastim require daily subcutaneous injections, while longer-lasting G-CSFs require less frequent dosing, presenting a more convenient alternative for patients. Pegfilgrastim (Amgen, Thousand Oaks, CA, USA) is a covalent conjugate of recombinant methionyl human G-CSF (filgrastim) and monomethoxypolyethylene glycol used to decrease the incidence of infection, as manifested by febrile neutropenia, in patients with non-myeloid malignancies receiving myelosuppressive anti-cancer drugs [8]. The addition of the PEG moiety extends the elimination half-life, allowing for once-per-cycle dosing [9].
Balugrastim (Teva Pharmaceutical Industries, Ltd., Netanya, Israel) was developed as a long-acting recombinant human G-CSF human serum albumin using a proprietary recombinant DNA expression process and is expressed in a highly engineered yeast strain, Saccharomyces cerevisiae. Albumin was chosen as a carrier because it is the most naturally occurring blood protein and due to its long half-life (approximately 19 days in humans) [10]. This technology has allowed for the design of a long-lasting agent suitable for once-per-cycle administration. This paper reports safety data from a phase I/IIa study of balugrastim administered to breast cancer patients scheduled to receive doxorubicin plus docetaxel. The primary objectives were to evaluate the safety profile of balugrastim administered over a range of potential therapeutic doses (phase I); and to compare the safety profile of balugrastim at doses confirmed to be safe in the phase I portion of the study with that of pegfilgrastim (active moiety filgrastim; phase IIa). Efficacy, pharmacokinetics (PK), and immunogenicity measures also were collected throughout the study.
Materials and methods
Study design and treatment
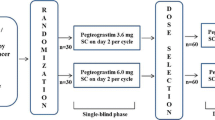
This 2-phase study comprised phase I and phase IIa portions (Fig. 1) conducted at three centers in Hungary and Poland between May 2007 (first patient visit) and September 2008 (last patient visit).

Study design
Phase I, an open-label, sequential dose-escalation study, comprised four dose cohorts (balugrastim 50, 150, 300, or 450 µg/kg administered subcutaneously), with three patients planned for each group. Patients received balugrastim at least 2 weeks before the start of chemotherapy (cycle 0), for an initial assessment of safety and effects on ANC. Chemotherapy consisted of doxorubicin 50 mg/m2 and docetaxel 75 mg/m2 administered sequentially by intravenous infusion on day 1 of a 21-day cycle for up to two cycles. After a minimum follow-up of 2 weeks, patients within a given dose cohort could receive the same dose of balugrastim the day following chemotherapy in cycles 1 and 2 if there were no dose-limiting adverse events (AEs) considered by study physicians to be related to balugrastim in cycle 0, and the patient continued to meet all eligibility criteria.
Within each cohort, initial study drug administration to each patient was separated by a minimum of 24 h, to allow monitoring for acute AEs. The decision to proceed to the next dose level was based on a review of the safety data for at least 7 days after the initial dose within the preceding dose cohort. If none of the three patients at a given dose experienced a dose-limiting toxicity (DLT), dose escalation would continue, with the enrollment of three new patients at the next dose level. A DLT was defined as any grade 2 or higher clinically significant AE considered by the investigator to be possibly, probably, or definitely related to the study agent, with the exception of grade 2 medullary bone pain. If one of three patients in a cohort exhibited evidence of a DLT, another three patients were to be recruited at the same dose level, for a total of six patients at that dose level. If only one of six patients experienced a DLT, dose escalation continued. If two of six patients developed a DLT, dose escalation was stopped and no further balugrastim treatments were administered. The remaining patients completed their scheduled safety, PK, and pharmacodynamic (PD) evaluations. The duration of phase I was 65 days, including follow-up at a minimum of 30 days after cycle 2.
In the open-label, randomized, parallel-group phase IIa portion, patients with breast cancer who were scheduled to receive doxorubicin plus docetaxel were randomized 2:2:1 to receive balugrastim 300 µg/kg, balugrastim 450 µg/kg, or pegfilgrastim 6 mg once per cycle for two chemotherapy cycles. At randomization, patients were assigned a unique subject number. Randomization was performed by two clinical research organizations (Chiltern International, Slough, UK, and Nexus, Roslin, UK) using an Internet-based interaction randomization system (WebEZ; Clinical Trial Services, Craigavon, UK). Patients who withdrew from the study after randomization retained their subject number, and replacement patients were given a new subject number and received the same treatment assignment as the patient being replaced. The duration of phase II was 21 days plus a minimum follow-up period of 30 days.
Patients were considered treatment failures if they experienced febrile neutropenia or persistent severe neutropenia (ANC <0.5 × 109/L for >5 days); such patients were removed from the study. Patients also were removed from the study if they experienced severe hypersensitivity reactions or nonhematologic toxicities that precluded further cycles of chemotherapy. Such patients were to complete follow-up, and those with febrile neutropenia or persistent severe neutropenia were to receive standard supportive care, including growth factor support, at the discretion of the investigator.
Inclusion and exclusion criteria
Patients at least 18 years of age with histologically or cytologically confirmed breast cancer, an ANC >1.5 × 109/L and platelet count >100 × 109/L, and scheduled to receive chemotherapy with doxorubicin and docetaxel were included in this study. Additional inclusion criteria were adequate hepatic and renal function (serum creatinine <2.0 mg/dL); total bilirubin within normal limits; alanine transaminase (ALT) and aspartate transaminase (AST) <1.5 upper limit of normal (ULN); alkaline phosphatase <2.5 ULN; eligible to receive doxorubicin based on left ventricular ejection fraction within normal limits; and an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1 [11].
Patients were excluded from the study if they received ≥1 prior chemotherapy regimen, any chemotherapy or immunotherapy ≤4 weeks prior to study entry, or a cumulative anthracycline dose that would preclude two full-dose cycles of doxorubicin in this study. In addition, patients who had used any nitrosourea (l,3-bis[2-chloroethyl]-l-nitrosourea, l-[2-chloroethyl]-3-cyclohexyl-nitrosourea, or mitomycin-C) within 6 weeks of study chemotherapy or any investigational agent during the 30 days before randomization were excluded, as were patients who had undergone surgery or radiation therapy within the previous 2 weeks or who had used myeloid (G-CSF or granulocyte macrophage colony-stimulating factor) growth factors within 4 weeks of study chemotherapy. Known brain metastases also precluded participation in this study.
Concomitant medications
Permitted concomitant medications included oral corticosteroids (for 3 days, starting one day before docetaxel administration), prophylactic oral antibiotics (e.g., ciprofloxacin) following each course of chemotherapy, and antiemetic agents or other premedication (at the discretion of the physician). Cytokines, other hematopoietic growth factors, and prophylactic antibiotics were not permitted unless prolonged neutropenia or febrile neutropenia occurred.
End points
Primary end point: safety
In both phases, safety was assessed by measuring the frequency and National Cancer Institute Common Terminology Criteria (CTC) severity grade. Serious AEs (SAEs) were defined as life-threatening AEs; AEs that resulted in death, hospitalization, or prolongation of hospitalization, persistent or significant disability or incapacity, congenital anomaly, or birth defect; or AEs that were otherwise medically important.
Complete blood count (CBC) with differential was measured at screening; on days 1, 2, 4, 8, 10, and 14 of cycle 0 (phase I, prior to chemotherapy); at baseline screening and on days −1, 4, 6, 8, 10, 12, and 15 of cycle 1 of phase II; and on day −1, once between days 4–6, day 8, once between days 10–12, and on day 15 of cycles 1 and 2 of phase I and cycle 2 of phase II. In the event of any grade 4 neutropenia, laboratory tests were obtained daily until the ANC was >0.5 × 109/L.
Serum chemistry was assessed at screening; on days 1 and 8 of cycle 0; on days −1, 8, and 15 of cycles 1 and 2 in phase I; at baseline screening and on days −1, 8, and 15 of cycle 1; and on days −1, 8, and 15 of cycle 2 in phase II. Urinalysis was performed at screening, on day 1 of cycle 0 and day −1 of cycles 1 and 2 in phase I, as well as at baseline screening and on day −1 of cycles 1 and 2 of phase II. Vital signs were measured at screening, prior to and 1 h after docetaxel administration, and prior to and 1 h after study drug administration; additional assessments were made if medically indicated. Follow-up assessments, performed 30 days after last study dose, included hematology and chemistry evaluation and urinalysis, as well as recording any AEs.
Secondary end points: pharmacokinetics, immunogenicity, and efficacy
Serum balugrastim concentrations were determined using a validated sandwich enzyme-linked immunosorbent assay with a lower limit of quantification of 6.3 ng/mL.
Pharmacokinetic parameters were evaluated during cycle 0 of phase I and cycle 1 of phases I and II. During cycle 0 of phase I, samples were collected before balugrastim administration and 0.5, 1, 3, 6, 24, 72, 168, 216, and 312 h after administration. During cycle 1 of both phases I and II, samples were collected before balugrastim administration and 0.5, 1, 3, 6, 24, 48, 144, and 192 h after administration. Pharmacokinetic parameters included maximal serum concentration (C max), time to C max (T max), elimination half-life (t ½, elim), absorption half-life (t ½, abs), area under the concentration curve (AUC0−∞), and apparent clearance (CL/F). Parameters were calculated for cycle 0 and cycle 1 for balugrastim dose groups 150, 300, and 450 µg/kg using non-compartmental modeling techniques, with the exception of t ½, abs, which was determined using a first-order absorption, first-order elimination one-compartment model. Analysis was performed with WinNonlin Professional version 5.0.1 (Pharsight; Sunnyvale, CA).
Immunogenicity was assessed in serum samples collected from patients treated with balugrastim before dosing during every cycle of phase I and phase II, and at the end-of-treatment visit (≥14 days after the last dose), to detect antidrug antibodies against balugrastim or anti-albumin antibodies. If samples were confirmed positive for anti-balugrastim antibodies, samples were further characterized to determine antibody titer and neutralization activity in a cell-based assay. Patients testing/confirmed positive for anti-drug antibodies were to have a follow-up sample obtained 6 months after the last dose of balugrastim.
Efficacy outcomes including incidence and duration of grade 4 neutropenia, incidence and duration of grade 3–4 neutropenia, nadir ANC, time to nadir ANC, time to ANC recovery, and incidence of febrile neutropenia were evaluated in phase II.
Statistical methodology
No strict statistical power requirement was used to select the sample size for this study. A study with a power of 80 % to demonstrate non-inferiority of balugrastim to pegfilgrastim at a significance level of 5 % was calculated to require approximately 37 patients per treatment group. The calculated patient size of 37 per treatment group was considered larger than appropriate for a phase I/IIa safety study; therefore, smaller sample sizes (n = 20 per balugrastim treatment group and n = 10 per pegfilgrastim group) were used in phase II, and efficacy trends were evaluated.
Safety was assessed for all patients who received at least one dose of study drug using descriptive statistics; no formal statistical analyses were performed. Efficacy analyses were performed on a modified intent-to-treat (mITT) population consisting of all patients who were treated and received the correct treatment.
Chi-square tests were used to test for differences in the incidence of febrile neutropenia, severe neutropenia, and persistent severe neutropenia across treatments. If the Chi-square test was deemed significant, 95 % confidence intervals (CIs) for relative risk between treatment pairs were calculated.
Analysis of variance (ANOVA) techniques were used where appropriate. Alternatively, the nonparametric equivalent Kruskal–Wallis test was used to test for differences across treatments. If the overall ANOVA/Kruskal–Wallis test was deemed significant, individual pairwise differences were inspected using either t tests or the Wilcoxon rank sum test, as appropriate. Statistical tests were two-sided, with a significance level set at 0.05. All statistical analyses were performed using the SAS system (SAS Institute, Inc., Cary, NC).
Results
Patients
Thirteen patients were enrolled and completed phase I of the study (n = 3 for balugrastim 50, 150, and 450 µg/kg, and n = 4 for balugrastim 300 µg/kg; Online Resource, Supplemental Figure 1A) and 51 were enrolled in phase II (n = 20 for balugrastim 300 µg/kg, n = 21 for balugrastim 450 µg/kg, and n = 10 for pegfilgrastim; Online Resource, Supplemental Figure 1B). One patient in the phase II balugrastim 300-µg/kg group was discontinued because of a protocol violation when the patient was given epirubicin instead of doxorubicin. Nine additional patients across both phases had protocol violations that called for discontinuation but were allowed to remain on study. These included three patients with severe neutropenia (ANC <0.5 × 109/L) for at least 5 days and one patient with hypertension in phase I, and three patients with severe neutropenia for at least 5 days, one patient who received epirubicin instead of doxorubicin, and one patient with febrile neutropenia in phase II. All enrolled patients were included in the ITT and safety analyses.
Patient demographics are summarized in Table 1. All participants in both phases of the study were white women. Cancer stages ranged from IIA to IV in both phases, but the phase I study had a larger proportion of patients with stage II disease. In phase II, more patients treated with balugrastim than pegfilgrastim had received prior chemotherapy.
Safety
Overall, balugrastim was well tolerated in this patient population. The most commonly reported AEs by body system for patients treated with balugrastim 300 µg/kg, balugrastim 450 µg/kg, and pegfilgrastim 6 mg (phases I and II combined) are summarized in Table 2.
Phase I: adverse events
No DLTs were observed during phase I, and the balugrastim dose was successfully escalated to 450 µg/kg. Of the 31 reported AEs, 16 (51.6 %) were CTC grade 1, 12 (38.7 %) were grade 2, and three (9.7 %) were grade 3; no grade 4 AEs occurred. The three grade 3 events included agranulocytosis and vomiting in one patient treated with balugrastim 150 µg/kg, and hypertension in one patient treated with balugrastim 450 µg/kg. One patient experienced the only two SAEs, consisting of severe vomiting and moderate vomiting in two consecutive chemotherapy cycles, both resulting in hospitalization. None of the events necessitated treatment interruption or discontinuation.
The safety of balugrastim in the absence of chemotherapy was assessed by considering AEs occurring during cycle 0. Four patients experienced six AEs during this pre-chemotherapy cycle, two patients in the balugrastim 300 µg/kg group (urinary tract infection, grade 1 and pyrexia, grade 1), two patients in the balugrastim 450 µg/kg group (one patient with two events of grade 2 bone pain and one of grade 2 headache, and one patient with grade 3 hypertension). All events were resolved with concomitant medication.
Phase II: adverse events
Of the 64 events reported in the balugrastim and pegfilgrastim groups, only two events were grade 3: vomiting in the balugrastim 450 µg/kg group and thrombocytopenia in the pegfilgrastim group (Table 2). Two SAEs requiring hospitalization were reported in the balugrastim 450 µg/kg group (severe vomiting and febrile neutropenia). Pain was the most frequently occurring AE (five events; one grade 1 and four grade 2) in four (9.8 %) balugrastim-treated patients. No AE required treatment interruption or discontinuation. No deaths were related to AEs.
Phase I and II: effects on laboratory parameters, chemistry, and vital signs
White blood cell (WBC) counts and ANC in balugrastim-treated patients increased during study phase I, peaking between days 2 and 6 and returning to baseline between days 14 and 15. During cycles 0 and 1 of study phase I, all patients (n = 13) recorded grade 0 results for WBC at baseline. During chemotherapy cycle 1, five patients experienced grade 3 and three patients experienced grade 4 WBC results. During cycle 2, of the 11 patients with grade 0 WBC at baseline, two experienced grade 2, eight experienced grade 3, and one experienced grade 4 WBC results. Of the two patients with grade 1 WBC at baseline of cycle 2, one experienced grade 2 and one experienced grade 4 WBC results. Absolute neutrophil counts were graded at 0 for 12 patients and graded at 1 for one patient at baseline of chemotherapy cycle 0, study phase I. No deterioration of ANC grades was noted during cycle 0. All 13 patients began cycles 1 and 2 with grade 0 ANC. During cycle 1, two patients experienced grade 2, five experienced grade 3, and six experienced grade 4 ANC results. During cycle 2, one patient experienced grade 2, two experienced grade 3, and ten experienced grade 4 ANC results.
Patients treated with balugrastim and pegfilgrastim during phase II experienced peak levels of WBC and ANC between days 4 and 6, which decreased and fell below baseline levels between days 6 and 8 and returned to approximate baseline levels between days 10 and 12 (Fig. 2). Of the 41 patients treated with balugrastim with grade 0 WBC at baseline of cycle 1, study phase II, three remained at grade 0, six experienced grade 1, 13 experienced grade 2, 14 experienced grade 3, and five experienced grade 4 WBC results during cycle 1. At baseline of cycle 2, 38 patients treated with balugrastim recorded grade 0 and two recorded grade 1 WBC. Of the 38 patients with grade 0 at baseline of cycle 2, eight remained at grade 0, four experienced grade 1, eight experienced grade 2, 14 experienced grade 3, and four experienced grade 4 WBC results during cycle 2. Of the two patients at grade 1 at baseline of cycle 2, one experienced grade 2 and one experienced grade 4 WBC results during cycle 2. Forty patients treated with balugrastim started cycle 1 with baseline ANC rated at grade 0 and one patient at grade 1. During cycle 1, of the 40 patients at grade 0 at baseline, five remained at grade 0, four experienced grade 1, four experienced grade 2, 13 experienced grade 3, and 14 experienced grade 4 ANC results. The one patient with ANC grade 1 at baseline experienced grade 4 ANC during cycle 1. At the start of chemotherapy cycle 2, phase II, 39 patients treated with balugrastim had grade 0 ANC. During cycle 2, of these 39 patients, eight remained at grade 0, two experienced grade 1, five experienced grade 2, ten experienced grade 3, and 14 experienced grade 4 ANC results. No noteworthy changes were observed in clinical chemistry parameters or vital signs for either balugrastim- or pegfilgrastim-treated patients throughout both phases of this study.

Balugrastim concentrations and median absolute neutrophil count in phase II, cycle 1 after 450 µg/kg
ANC absolute neutrophil count, LLOQ lower limit of quantitation
Pharmacokinetics
Data from patients in the phase I and II portions of the study were combined for PK analysis. Balugrastim serum concentrations for all patients in the 50 µg/kg group were below the lower limit of quantitation (LLOQ). Thus, PK parameters were not calculated for this dose group. Calculated PK parameters for all patients treated with balugrastim 150, 300, and 450 µg/kg in cycles 0 and 1 are summarized in Table 3.
Drug exposure was higher in cycle 1 compared with cycle 0 (pre-chemotherapy) within each dose group. Virtually no cycle-to-cycle drug accumulation was observed. Of the 51 patients sampled, 1 had serum balugrastim concentrations above the LLOQ of 6.3 mg/mL at 192 h (day 8) in cycle 1.
Efficacy
Phase II study
The incidence, severity, or duration of neutropenia or ANC nadir was similar among treatment groups (Table 4). One patient treated with balugrastim 450 µg/kg experienced mild febrile neutropenia during cycle 1; however, by the end of the study, this event was resolving. One patient in the balugrastim 300 µg/kg group, two patients in the balugrastim 450 µg/kg group, and one patient in the pegfilgrastim group were considered treatment failures.
The PK/PD profile of balugrastim was demonstrated in phase II study patients receiving balugrastim 450 µg/kg after doxorubicin plus docetaxel treatment in cycle 1 (Fig. 2). Balugrastim C max was achieved within the first day of treatment, then gradually fell to undetectable levels by day 10. Following balugrastim administration, ANC rose to a peak on day 4, fell to a nadir on day 8, and returned to normal by day 10.
Immunogenicity
None of the patients had detectable anti-balugrastim antibodies or antibodies against the albumin domain of balugrastim as a result of receiving balugrastim.
Discussion
Treatment with myelosuppressive chemotherapy may result in neutropenia and, potentially, febrile neutropenia, which may delay chemotherapy and require treatment with antibiotics or hospitalization. Therefore, preventing this adverse event ultimately benefits the patient and has the potential to reduce the cost of treatment by decreasing healthcare costs. Current treatment guidelines recommend prophylactic use of G-CSFs for patients at high risk (>20 %) of febrile neutropenia, taking into account disease characteristics, myelotoxicity of the chemotherapy regimen, patient-related risk factors, and treatment intent (curative vs palliative) [12–14]. Treatment with G-CSFs may be considered for those with patient-related risk factors between 10 and 20 %, as well as for patients with clinical factors that could predispose them to complications in the setting of prolonged neutropenia [12–14]. Furthermore, G-CSFs are recommended for the treatment of febrile neutropenia if the patient is at high risk of infection-associated complications [12, 13].
Filgrastim and pegfilgrastim are currently approved in Europe and in the USA for the reduction of infection associated with febrile neutropenia in patients receiving myelosuppressive anti-cancer drugs. Filgrastim requires daily injections or infusions dosed by weight until ANC recovery [12, 15], while pegfilgrastim is administered just once per chemotherapy cycle at a fixed 6-mg dose [8, 12].
In this first-in-human study, balugrastim was well tolerated. In the present study, bone pain and hypertension occurred in five and one patients, respectively. None of the ten patients treated with pegfilgrastim experienced bone pain in the present study [8]. Usually, bone pain associated with these therapies is mild to moderate and can be controlled with non-narcotic analgesics [12, 15]. While the pegfilgrastim and filgrastim prescribing information contains warnings regarding the potential for splenic rupture, acute respiratory distress syndrome, and serious allergic reactions [8, 15], no such events occurred among the small number of balugrastim-treated patients in this study, and there were no signs of immunogenicity.
Pharmacokinetic analysis demonstrated that balugrastim serum concentrations were detectable across dose groups in most patients (45 out of 50 patients) for at least 144 h post-dose. Drug exposure was higher in cycle 1 compared with cycle 0 (i.e., with versus without chemotherapy), most likely because chemotherapy reduces the number of neutrophils, which play an important role in the clearance of balugrastim. In cycle 1, balugrastim was detected out to 144 h in most patients (45/50 sampled) in the 150, 300, and 450 µg/kg dose groups, supporting once-per-cycle dosing. The median t ½, elim of balugrastim in cycle 1 was approximately 36 h for the 300 µg/kg dose group and 30 h for the 450 µg/kg dose group. This is longer than the reported t ½, elim for filgrastim (3–4 h), indicating that the addition of human serum albumin supports a longer-lasting G-CSF [15]. However, while the half-life of balugrastim is longer than that of filgrastim, it is not as long as albumin (~19 days in humans) [10]. G-CSFs are primarily cleared through receptor-mediated endocytosis by neutrophils and renally [16], while albumin is distributed between vascular and extravascular compartments and is degraded in a number of tissues including liver, muscle, and skin [17]. With a molecular weight of approximately 85 kDa, renal clearance of balugrastim should be reduced, while the presence of G-CSF allows for clearance via the receptor-mediated route, thereby resulting in a half-life between that of filgrastim and albumin [18].
At the highest dose levels tested (300 and 450 µg/kg), treatment with balugrastim showed preliminary efficacy similar to that of pegfilgrastim 6 mg in the prevention of chemotherapy-induced neutropenia in this small population of breast cancer patients. The incidence of grade 3 or 4 neutropenia or duration of severe neutropenia was similar among the treatment groups. In phase I, cycle 0, balugrastim induced a dose-dependent increase in WBCs and ANC before the administration of chemotherapy. The ANC increases in cycle 0 were comparable to historical data for pegfilgrastim at equimolar doses [19]. As expected, WBCs and ANC declined after chemotherapy, but recovery from nadir in cycle 1 was mediated by balugrastim treatment such that ANC recovered by day 10, 2 days after nadir. In patients who do not receive prophylactic G-CSF treatment, time to reach recovery of ANC post-nadir is historically 5–7 days [6].
Results of this study support further clinical development of balugrastim. Phase II/III and phase III studies of balugrastim in breast cancer patients receiving doxorubicin plus docetaxel have been completed (NCT00837265; NCT01126190). The phase III trial evaluated a fixed 40-mg dose, which, if supported, will further simplify dosing and administration.
Results of this study should be interpreted in the context of its limitations. In particular, this study had limited statistical power to determine between-treatment differences in the phase II portion, given the small sample size. This limitation will be addressed in the larger phase II/III and phase III trials. All of the studies to date were conducted in patients with breast cancer who were receiving doxorubicin plus docetaxel; further studies are needed to determine whether the results are generalizable to patients with other tumor types and to those receiving other myelosuppressive chemotherapy regimens.
Conclusions
Balugrastim was well tolerated in this first-in-human study. In addition, the balugrastim dose-dependent increase in ANC was similar to that observed for pegfilgrastim based on historical data. In addition, the ANC recovery and safety profiles for the balugrastim 450 µg/kg group were similar to those for the pegfilgrastim 6 mg group, supporting the choice of a starting fixed dose of 30 mg (equivalent to 450 µg/kg) for further clinical trial assessment. Results from further studies in which 30, 40, and 50 mg fixed doses of balugrastim were conducted to select the appropriate dose will be available soon.
References
Pettengell R, Schwenkglenks M, Leonard R et al (2008) Neutropenia occurrence and predictors of reduced chemotherapy delivery: results from the INC-EU prospective observational European neutropenia study. Support Care Cancer 16:1299–1309
Joerger M, Thurlimann B (2013) Chemotherapy regimens in early breast cancer: major controversies and future outlook. Exp Rev Anticancer Ther 13:165–178
del Giglio A, Eniu A, Ganea-Motan D, Topuzov E, Lubenau H (2008) XM02 is superior to placebo and equivalent to Neupogen in reducing the duration of severe neutropenia and the incidence of febrile neutropenia in cycle 1 in breast cancer patients receiving docetaxel/doxorubicin chemotherapy. BMC Cancer 8:332–338
Alba E, Martin M, Ramos M et al (2004) Multicenter randomized trial comparing sequential with concomitant administration of doxorubicin and docetaxel as first-line treatment of metastatic breast cancer: a Spanish Breast Cancer Research Group (GEICAM-9903) phase III study. J Clin Oncol 22:2587–2593
Nabholtz JM, Falkson C, Campos D et al (2003) Docetaxel and doxorubicin compared with doxorubicin and cyclophosphamide as first-line chemotherapy for metastatic breast cancer: results of a randomized, multicenter, phase III trial. J Clin Oncol 21:968–975
Crawford J, Ozer H, Stoller R et al (1991) Reduction by granulocyte colony-stimulating factor of fever and neutropenia induced by chemotherapy in patients with small-cell lung cancer. N Engl J Med 325:164–170
Roberts AW (2005) G-CSF: a key regulator of neutrophil production, but that’s not all! Growth Factors 23:33–41
Neulasta [package insert] (2014) Amgen Inc., Thousand Oaks, CA
Yang BB, Kido A (2011) Pharmacokinetics and pharmacodynamics of pegfilgrastim. Clin Pharmacokinet 50:295–306
Elsadek B, Kratz F (2012) Impact of albumin on drug delivery–new applications on the horizon. J Control Release 157:4–28
Oken MM, Creech RH, Tormey DC et al (1982) Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am J Clin Oncol 5:649–655
NCCN Clinical Practice Guidelines in Oncology (2014) Myeloid Growth Factors v.2.2014. National Comprehensive Cancer Network. www.nccn.org/professionals/physician_gls/f_guidelines.asp. Accessed 23 Dec 2014
Smith TJ, Khatcheressian J, Lyman GH et al (2006) 2006 update of recommendations for the use of white blood cell growth factors: an evidence-based clinical practice guideline. J Clin Oncol 24:3187–3205
Aapro MS, Bohlius J, Cameron DA et al (2011) 2010 update of EORTC guidelines for the use of granulocyte-colony stimulating factor to reduce the incidence of chemotherapy-induced febrile neutropenia in adult patients with lymphoproliferative disorders and solid tumours. Eur J Cancer 47:8–32
Neupogen [package insert] (2013) Amgen Inc., Thousand Oaks, CA
Molineux G, Kinstler O, Briddell B et al (1999) A new form of Filgrastim with sustained duration in vivo and enhanced ability to mobilize PBPC in both mice and humans. Exp Hematol 27:1724–1734
Sleep D, Cameron J, Evans LR (2013) Albumin as a versatile platform for drug half-life extension. Biochim Biophys Acta 1830:5526–5534
Halpern W, Riccobene TA, Agostini H et al (2002) Albugranin, a recombinant human granulocyte colony stimulating factor (G-CSF) genetically fused to recombinant human albumin induces prolonged myelopoietic effects in mice and monkeys. Pharm Res 19:1720–1729
Johnston E, Crawford J, Blackwell S et al (2000) Randomized, dose-escalation study of SD/01 compared with daily filgrastim in patients receiving chemotherapy. J Clin Oncol 18:2522–2528
Acknowledgments
Medical writing and editorial assistance was provided by Ruth Sussman, Chameleon Communications, New York, NY, and Lisa Feder, PhD, Peloton Advantage, Parsippany, NJ, and was funded by Teva Branded Pharmaceutical Products R&D, Inc. The authors also acknowledge Scott Newcomer, MS, Teva Pharmaceuticals, Frazer, PA, and Pippa Loupe, PhD, Teva Pharmaceuticals, Kansas City, MO, for their medical writing and editorial assistance in the development of this manuscript. Teva provided a full review of the article. This study was sponsored by Teva Biopharmaceuticals USA (formerly CoGenesys, Inc).
Conflict of interest
Noa Avisar and Liat Adar are employees of Teva Pharmaceuticals, Netanya, Israel. Laurie Pukac, Jason Bock, and Steve Barash are employees of Teva Biopharmaceuticals, Rockville, MD, and Udo Müller is an employee of Teva GmbH, Ulm, Germany, both subsidiaries of Teva Pharmaceuticals. David Shen was an employee of Teva Pharmaceuticals at the time of this study and during manuscript preparation.
Ethical standard
This study was approved by the appropriate ethics committee at each study site and was conducted in accordance with the International Conference on Harmonisation guidelines for Good Clinical Practice and the current version of the Declaration of Helsinki (Tokyo, 2004). All study participants provided written informed consent prior to their participation.
Author information
Authors and Affiliations
Corresponding author
Additional information
EudraCT Number: 2006-005997-28
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Avisar, N., Adar, L., Bock, J. et al. First-in-human, phase I/IIa dose-escalation and safety study of balugrastim in breast cancer patients receiving myelosuppressive chemotherapy. Cancer Chemother Pharmacol 75, 929–939 (2015). https://doi.org/10.1007/s00280-015-2703-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-015-2703-1