
Abstract
Purpose
This 2-arm crossover study compared the relative bioavailability of two dovitinib (TKI258) formulations [anhydrate clinical service form (CSF) capsule and monohydrate final market image (FMI) tablet; Arm 1] and determined the effect of food on dovitinib exposure (Arm 2).
Methods
Patients with advanced solid tumors, excluding breast cancer, were enrolled in 1 of the 2 arms of the study. Patients in Arm 1 were randomized to a single 500-mg dose of CSF capsule or FMI tablet followed by 7 days of rest and 500 mg of the other formulation. Patients in Arm 2 received 300 mg of FMI tablet daily and were randomized to follow 1 of 6 meal sequences, each with 3 prandial conditions: low fat (LF), high fat (HF), or no meal (NM).
Results
In Arm 1 (n = 21), 17 patients were evaluable. FMI tablet compared with CSF capsule showed only slight reductions in the adjusted geometric means for area under the plasma concentration–time curve (AUClast; 3 %) and maximum plasma concentration (C max; 1 %). In Arm 2 (n = 42), 19 patients were evaluable. HF meal versus NM showed a 9 % decrease in the adjusted geometric mean for AUClast and an 18 % decrease for C max. Comparison of LF meal versus NM showed a 1 % decrease for AUClast and a 10 % decrease for C max. Common adverse events suspected to be study drug related included vomiting, diarrhea, nausea, and fatigue.
Conclusions
Dovitinib FMI tablet had similar systemic exposure to the CSF capsule and was not affected by food.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Dovitinib (TKI258) is a potent oral inhibitor of receptor tyrosine kinases. Half-maximal inhibitory concentration values of dovitinib for these kinases are in the low nanomolar range: 8–9 nmol/L for fibroblast growth factor receptor (FGFR), 8–13 nmol/L for vascular endothelial growth factor (VEGF) receptor, 27 nmol/L for platelet-derived growth factor (PDGF) receptor, and 2 nmol/L for C-Kit [1]. The signaling pathways activated by these receptors play a role in angiogenesis and the development and progression of cancer [2, 3].
VEGF plays a critical role in tumor angiogenesis and has been implicated in tumor metastasis [4, 5]. Thus, a number of agents that inhibit the VEGF pathway have been studied and approved to treat several different types of cancer [6]. However, cancers eventually develop resistance to these drugs, in part through a process called angiogenic escape, in which neovascularization is reactivated via signaling through alternative angiogenesis pathways. [7]. In addition to the roles they play in angiogenesis, PDGF regulates cell proliferation [8] and FGF plays a key role in proliferation, differentiation, and tumorigenesis and can be tumor protective [9]. Additionally, data have shown that resistance to drugs that inhibit the VEGF pathway can be overcome by inhibition of the FGF pathway, emphasizing the need for multitargeted inhibitors like dovitinib [7]. Dovitinib has shown antitumor activity in patients with advanced metastatic renal cell carcinoma (RCC) [10] and FGFR-positive breast cancer [11]. Dovitinib is currently being investigated in several solid tumor indications, including breast and endometrial cancer and gastrointestinal stromal tumors.
Early clinical studies of dovitinib determined the maximum tolerated dose (MTD) to be 400 mg/day when administered continuously, [12] with steady state achieved by Day 7 [13]. Dose-dependent accumulation was observed at higher doses, but pharmacokinetic (PK) modeling predicted that a 5-days-on/2-days-off schedule would allow the use of higher doses of dovitinib in order to achieve higher therapeutic plasma concentrations [13]. Results from a phase 1 study, in which this intermittent dosing schedule in advanced RCC was used (NCT00715182), confirmed that higher doses were feasible; therefore, the MTD of 500 mg/day using the 5-days-on/2-days-off schedule, which achieved steady state by the second week, was recommended as the phase 2 dose and schedule [14].
Initial phase 1 and 2 trials of dovitinib used an anhydrate capsule formulation, referred to as the clinical service form (CSF), but scaling up production for use in larger clinical trials proved to be technically challenging. Therefore, a monohydrate capsule formulation, referred to as the final market image (FMI), was developed in capsule form for use in subsequent phase 2 and 3 trials and tablet form for use in further clinical trials and commercially. A previous phase 1 study in patients with advanced solid tumors (NCT01030055) showed that the bioavailability of the FMI capsule and CSF capsule formulations is comparable, but the FMI tablet was not assessed [15]. Here, we used a similar study design to compare the bioavailability of the FMI tablet to that of the CSF capsule following a single administration of the recommended phase 2 dose (RP2D) of 500 mg (Arm 1).
The prior study also showed that food did not affect the systemic exposure of dovitinib FMI capsules [15]. Following the same design, we compare here the effect of food on systemic exposure to the FMI tablet (Arm 2). To reduce the duration of the food effect tests, a continuous once-daily dovitinib schedule was used, as the time required to reach steady state is decreased compared with the 5-days-on/2-days-off schedule. Additionally, a reduced dose of 300 mg/day (1 dose level below the MTD) was used in Arm 2, in case food were to increase the bioavailability of dovitinib.
Materials and methods
Study design
This was a 2-arm, phase 1 crossover study of dovitinib in patients with advanced solid tumors. Patients with breast cancer were excluded as a previous trial of dovitinib monotherapy did not proceed to the second stage [11], suggesting the risk/benefit ratio may not be favorable in this advanced patient population. The primary objective of Arm 1 was to determine the relative bioavailability of the planned commercial formulation of dovitinib, the FMI monohydrate tablet, compared with that of the CSF anhydrate capsule formulation. The primary objective of Arm 2 was to determine the effect of food on the bioavailability of dovitinib. Secondary objectives were to characterize the safety and tolerability of dovitinib and evaluate the preliminary evidence of its antitumor activity. The study was registered at www.clinicaltrials.gov, identifier NCT01155713.
The design of this study was similar to the one that compared the bioavailability of the CSF and FMI capsule formulations of dovitinib and determined the effect of food on the bioavailability of the FMI capsule [15]. Briefly, patients enrolled in Arm 1 were randomly assigned to receive a single 500-mg dose of the anhydrate CSF capsule or the monohydrate FMI tablet in Cycle 1, followed by a single 500-mg dose of the other formulation 7 days later. In subsequent cycles, patients could receive the CSF capsule formulation of dovitinib at the RP2D of 500 mg/day on a 5-days-on/2-days-off schedule in 28-day cycles until unacceptable toxicity, disease progression, withdrawal of consent, and/or discretion of the investigator (Fig. 1a).

Overview of Arm 1 (bioavailability) and Arm 2 (food effect). a In Arm 1, patients were randomized to receive a single 500-mg dose of either the anhydrate clinical service form (CSF) dovitinib capsule or the monohydrate final market image (FMI) dovitinib tablet on Day 1. On Day 9 (after 7 days of rest), patients received a single 500-mg dose of the other formulation. After completing Cycle 1, patients could receive the CSF capsule at 500 mg once a day on a 5-days-on/2-days-off schedule in 28-day cycles until unacceptable toxicity, disease progression, withdrawal of consent, and/or discretion of the investigator. b In Arm 2, patients were randomized to 1 of 6 sequences with 3 prandial conditions (high-fat meal, low-fat meal, and no meal). Patients received 300 mg of dovitinib FMI tablet on a continuous once-daily dosing schedule. On Days 8, 15, and 22, patients took their dovitinib under the defined prandial state and blood was drawn for pharmacokinetic analysis. Following Cycle 1, patients in Arm 2 could receive the FMI tablet at 500 mg once a day on a 5-days-on/2-days-off schedule in 28-day cycles until unacceptable toxicity, disease progression, withdrawal of consent, and/or discretion of the investigator
The formulation used in Arm 2 was determined following an interim PK evaluation of the relative bioavailability in Arm 1. In Cycle 1 of Arm 2, patients received 300 mg of dovitinib FMI tablet once daily following 1 of 6 meal sequences, each with 3 prandial conditions: low fat (LF), high fat (HF), or no meal (NM). On days of PK evaluation, dovitinib was administered ≤0.5 h after the end of a LF or NF, or 1 h before or 2 h after a light breakfast (NM). Meal formulations and PK testing dates were as previously described [15]. In subsequent cycles, patients could receive the FMI tablet formulation of dovitinib at the RP2D of 500 mg/day on a 5-days-on/2-days-off schedule in 28-day cycles until unacceptable toxicity, disease progression, withdrawal of consent, and/or discretion of the investigator (Fig. 1b).
In Arm 1, the PK set included all randomized patients who received both of the Cycle 1 CSF capsule and FMI tablet doses (without vomiting within 4 h) and provided evaluable PK data. In Arm 2, the PK set included all patients who received 300 mg of dovitinib on ≥4 consecutive days prior to the food effect tests on Days 8, 15, and 22 of Cycle 1, consumed ≥75 % of each assigned test meal (e.g., LF or HF), and received the planned dose of 300 mg within 30 min after meal consumption ended and did not vomit within 4 h after dosing on Days 8, 15, and 22 of Cycle 1; the PK set provided evaluable PK data.
The study protocol and all amendments were reviewed and approved by the institutional review board for each center. Each patient provided informed written consent before any screening assessments were performed. The study was conducted in accordance with Good Clinical Practice and the ethical principles of the Declaration of Helsinki.
Patients
Patients ≥18 years of age with World Health Organization performance status ≤2 were eligible if they had cytopathologically or histopathologically confirmed diagnosis of an advanced solid tumor (excluding breast cancer) that had progressed despite standard therapy or for which no standard therapy existed. Additional eligibility criteria were as described previously [15].
Pharmacokinetic assessments
Assessment criteria and details were also described previously [15]. Blood samples for PK analyses were obtained on Days 1 and 9 of Cycle 1 in Arm 1 and Days 8, 15, and 22 of Cycle 1 and Day 26 of Cycle 3 in Arm 2. A linear mixed-effect model was fitted to the log-transformed PK parameters: area under the plasma concentration–time curve from time 0 to the last measurable sample time (AUClast), area under the curve from time zero to infinity (AUCinf), and maximum plasma concentration (C max). Included in the model were treatment (Arm 1) or food state (Arm 2), period, and sequence as a fixed factor and patients nested within sequence as a random factor. The FMI tablet was the test treatment in Arm 1, and the LF and HF food states were the test treatments in Arm 2. The 2-sided 90 % confidence interval (CI) for the least-square means of the difference (test – reference) on the log scale was calculated. The data were antilogged to obtain the point estimates and 90 % CI for the ratio of the geometric means on the untransformed scale.
Efficacy/safety assessments
Tumors were assessed at baseline and every 8 weeks by local investigators using the response evaluation criteria in solid tumors version 1.0 [16]. Adverse events (AEs) occurring on treatment or within 28 days after the last dose were assessed according to the Common Terminology Criteria for Adverse Events version 4.0.
Results
Arm 1 (bioavailability)
A single-dose crossover of the anhydrate CSF capsule formulation and the monohydrate FMI tablet formulation of dovitinib was evaluated in Arm 1 of the study (Fig. 1a). Between July 12, 2010, and October 27, 2010, twenty-one patients in Arm 1 were randomized to either CSF capsule followed by FMI tablet (CSF → FMI; n = 10) or FMI tablet followed by CSF capsule (FMI → CSF; n = 11). Patient demographics were similar between the two sequences: about half of all patients were male (52.4 %), patients were predominantly white (81.0 %) and had World Health Organization performance status of 1 (71.4 %), and the median body mass index was 29.9 kg/m2 (Table 1). The median ages for the CSF → FMI and FMI → CSF sequences were 67 years (range 45–79 years) and 59 years (range 36–82 years), respectively.
Two patients discontinued treatment in Cycle 1, one due to an AE and the other withdrew consent (Table 2). Nineteen patients continued to receive dovitinib in subsequent cycles, with one patient ongoing at the time of data cutoff. The remaining 18 patients discontinued due to disease progression (n = 10), investigator decision (n = 4), AEs (n = 3), or withdrawal of consent (n = 1).
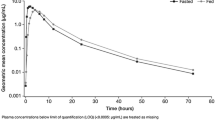
The PK set of Arm 1, Cycle 1 consisted of 17 patients. Based on the linear mixed-effect model, the adjusted geometric means (CSF capsule and FMI tablet, respectively) were 4,819 and 4,670 h ng/mL for AUClast and 192 and 191 ng/mL for C max (Table 3; Fig. 2a). The adjusted geometric mean for AUClast decreased by 3 %, while C max decreased by 1 % for the FMI tablet formulation compared with the CSF capsule formulation. C max was achieved at a mean time to maximum plasma concentration (T max) of 7 h for both the CSF capsule and FMI tablet formulations. The half-life of dovitinib was similar in the CSF and FMI formulations (geometric mean, 18.4 and 16.1 h, respectively). PK parameters such as AUCinf, volume of distribution, and clearance were not reported due to limited sampling and extrapolated AUC values >20 % of the AUClast in a number of patients. In summary, the FMI tablet and CSF capsule formulations of dovitinib had similar systemic exposure.

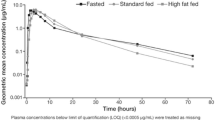
Plasma pharmacokinetic profiles of dovitinib. The arithmetic mean (±SD) of dovitinib plasma concentration is plotted versus time for the a clinical service form (CSF) capsule and final market image (FMI) tablet treatments and b food states [high-fat (HF) meal, low-fat (LF) meal, and no meal (NM)]. SD standard deviation
Arm 2 (food effect)
Based on the similar bioavailability of the two formulations in Arm 1, the FMI tablet was used in Arm 2 in a continuous once-daily schedule to test the effect of food on dovitinib bioavailability (Fig. 1b). Between April 5, 2011, and January 23, 2012, forty-two patients were randomized to 1 of 6 Cycle 1 meal sequences: NM → LF → HF (n = 7), LF → HF → NM (n = 6), HF → NM → LF (n = 8), LF → NM → HF (n = 4), NM → HF → LF (n = 8), and HF → LF → NM (n = 9). Demographic characteristics, including age (median 62.5 years), were similar across the treatment sequences, with the exception of weight (median 75.7 kg) and body mass index (median 26.6 kg/m2), which were imbalanced among the treatment sequences. During Cycle 1, 17 patients discontinued treatment due to AEs (n = 8), investigator decision (n = 3), withdrawal of consent (n = 2), disease progression (n = 2), or death (n = 2; Table 2). Twenty-five patients continued to receive dovitinib in subsequent cycles, with two patients receiving treatment at the time of data cutoff. The remaining 23 patients discontinued treatment due to disease progression (n = 11), AEs (n = 6), investigator decision (n = 5), or withdrawal of consent (n = 1).
In Cycle 1 of Arm 2, the PK set consisted of 19 patients. The adjusted geometric means (NM, LF meal, and HF meal, respectively) were 2,701, 2,662, and 2,464 h ng/mL for AUClast and 185, 166, and 151 ng/mL for C max (Table 3; Fig. 2b). The HF meal versus NM comparison showed a 9 % decrease in the adjusted geometric mean for AUClast and an 18 % decrease in the adjusted geometric mean for C max based on the linear mixed-effect model (Table 3; Fig. 2b). For the LF meal versus NM comparison, 1 and 10 % decreases in the adjusted geometric means for AUClast and C max, respectively, were observed. The median T max was 4.0 h with NM, 6.0 h with an LF meal, and 6.1 h with an HF meal. In summary, these results indicate that food did not affect the systemic exposure of dovitinib.
Safety
Across both arms of the study, the majority of AEs suspected to be study drug related were generally grade 1 or 2 (Table 4), with only one suspected grade 4 AE (increased lipase in Arm 1, Cycle 1). The most common AEs suspected to be study drug related during subsequent cycles in which dovitinib was dosed on a 5-days-on/2-days-off schedule were vomiting (54.5 %), diarrhea (47.7 %), nausea (45.5 %), and fatigue (36.4 %). Two patients (9.5 %) in Arm 1, Cycle 1, 13 patients (31.0 %) in Arm 2, Cycle 1, and 23 patients (52.3 %) in subsequent cycles across both arms experienced a grade 3 or 4 AE suspected to be study drug related. The suspected grade 3/4 AEs with an incidence of >5 % were fatigue (9.5 %) in Arm 2, Cycle 1 and fatigue (11.4 %) and hypertriglyceridemia (11.4 %) in subsequent cycles across both arms.
In Arm 1, Cycle 1, no grade 3 laboratory abnormalities with an incidence of >5 % were reported. In Arm 2, Cycle 1, the grade 3 or 4 laboratory abnormalities with an incidence of >5 % were increased γ-glutamyltransferase (11.9 %), increased lipase (9.5 %), decreased sodium (7.1 %), and increased alkaline phosphatase (7.1 %). In subsequent cycles across both arms, the grade 3 laboratory abnormalities with an incidence of >5 % were increased triglycerides (20.5 %), decreased lymphocytes (13.6 %), increased γ-glutamyltransferase (13.6 %), increased alkaline phosphatase (13.6 %), decreased sodium (11.4 %), and increased lipase (9.1 %). One patient developed grade 4 increased alkaline phosphatase (4.8 %) during Cycle 1 of Arm 1 that was grade 2 at baseline and one patient developed grade 4 decreased lymphocytes (4.0 %) during subsequent cycles of Arm 2 that was grade 3 at baseline.
No patients experienced grade 3 or 4 total bilirubin increases. One case of grade 3 increased alanine aminotransferase was reported. No patients met the criteria for Hy’s law. One instance of Fridericia-corrected QT interval >500 ms was observed in Arm 2, Cycle 1. The single episode, which occurred concurrently with a finding of flat T waves, was reported in a patient who had discontinued dovitinib due to nausea and vomiting 6 days earlier. No patients in either arm of the trial had a Fridericia-corrected QT increase from baseline >60 ms.
Across the entire study, 18 patients (28.6 %) discontinued treatment due to AEs, most commonly fatigue (n = 3) and vomiting (n = 2).
No deaths occurred on treatment or within 28 days of the last dose in Arm 1. Seven on-treatment deaths occurred in Arm 2 (2 in Cycle 1, five within 28 days of treatment discontinuation). Six of these events were associated with disease progression, and one patient with lung cancer died of cardiac arrest. None of the deaths were suspected to be related to study drug.
Efficacy
Of the 63 patients in both arms, no patients achieved a complete response. One patient (1.6 %) in Arm 2 with heavily pretreated adenocarcinoma of the endometrium had an unconfirmed partial response at the first evaluation and progressive disease at the second evaluation. This patient died due to disease progression 10 days after the last dose of dovitinib.
In addition, 29 patients (46.0 %) achieved stable disease (SD; 14 patients in Arm 1, 15 patients in Arm 2). Ten patients who achieved SD were on study drug for >120 days, with durations on treatment as follows: four patients with RCC (129, 131, 162, and 213 days), three patients with thyroid cancer (242, 348, and 590+ days), one patient with gastrointestinal stromal tumor (145 days), one patient with thymoma (130 days), and one patient with bladder cancer (123 days).
Discussion
During the course of its development, dovitinib has been formulated as an anhydrate CSF capsule and as a monohydrate FMI capsule and tablet. In a previous study, the CSF capsule and FMI capsule formulations were shown to have similar bioavailability [15]. However, that study did not include the FMI tablet for comparison. Thus, this 2-arm, phase 1 crossover study investigated the relative bioavailability of the CSF capsule and FMI tablet formulations.
Results from Arm 1 of this study showed comparable bioavailability for these dovitinib formulations. Hence, the preferred formulation, the FMI tablet, was used in Arm 2 of the study to investigate the effect of food on dovitinib pharmacokinetics. The results indicate that food (HF and LF meals) does not affect the systemic exposure of the FMI tablet formulation of dovitinib, which is consistent with results from the prior study, and show that dovitinib can be taken with or without food [15].
Dovitinib had a tolerable safety profile in both study arms. The most frequently reported AEs suspected to be related to study drug were grade 1 and 2 vomiting, diarrhea, nausea, and fatigue, which are known side effects of dovitinib [10, 11, 15].
One patient with heavily pretreated adenocarcinoma of the endometrium had an unconfirmed partial response to single-agent dovitinib, and ten patients who achieved SD remained on study drug for a prolonged period (>120 days). These results show that single-agent dovitinib has clinical activity and support its continued investigation.
In conclusion, the results from this study show that the FMI tablet had similar bioavailability to that of the CSF capsule and that its systemic exposure was not affected by food. Therefore, the FMI tablet formulation can be used going forward and can be taken with or without food. The FMI tablet is currently being used in a phase 2 study of dovitinib or placebo plus fulvestrant in postmenopausal women with human epidermal growth factor receptor 2—negative and hormone receptor—positive breast cancer (NCT01528345) [17].
References
Lee SH, Lopes de Menezes D, Vora J, Harris A, Ye H, Nordahl L, Garrett E, Samara E, Aukerman SL, Gelb AB, Heise C (2005) In vivo target modulation and biological activity of CHIR-258, a multitargeted growth factor receptor kinase inhibitor, in colon cancer models. Clin Cancer Res 11:3633–3641
Cook KM, Figg WD (2010) Angiogenesis inhibitors: current strategies and future prospects. CA Cancer J Clin 60:222–243
Heinrich MC, Blanke CD, Druker BJ, Corless CL (2002) Inhibition of KIT tyrosine kinase activity: a novel molecular approach to the treatment of KIT-positive malignancies. J Clin Oncol 20:1692–1703
Xu B, Shen F, Cao J, Jia L (2013) Angiogenesis in liver metastasis of colo-rectal carcinoma. Front Biosci (Landmark Ed) 18:1435–1443
Welti J, Loges S, Dimmeler S, Carmeliet P (2013) Recent molecular discoveries in angiogenesis and antiangiogenic therapies in cancer. J Clin Investig 123:3190–3200
Hojjat-Farsangi M (2014) Small-molecule inhibitors of the receptor tyrosine kinases: promising tools for targeted cancer therapies. Int J Mol Sci 15:13768–13801
Casanovas O, Hicklin DJ, Bergers G, Hanahan D (2005) Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 8:299–309
Gavalas NG, Liontos M, Trachana SP, Bagratuni T, Arapinis C, Liacos C, Dimopoulos MA, Bamias A (2013) Angiogenesis-related pathways in the pathogenesis of ovarian cancer. Int J Mol Sci 14:15885–15909
Tiong KH, Mah LY, Leong CO (2013) Functional roles of fibroblast growth factor receptors (FGFRs) signaling in human cancers. Apoptosis 18:1447–1468
Motzer RJ, Porta C, Vogelzang NJ, Sternberg CN, Szczylik C, Zolnierek J, Kollmannsberger C, Rha SY, Bjarnason GA, Melichar B, De Giorgi U, Grunwald V, Davis ID, Lee JL, Esteban E, Urbanowitz G, Cai C, Squires M, Marker M, Shi MM, Escudier B (2014) Dovitinib versus sorafenib for third-line targeted treatment of patients with metastatic renal cell carcinoma: an open-label, randomised phase 3 trial. Lancet Oncol 15:286–296
Andre F, Bachelot T, Campone M, Dalenc F, Perez-Garcia JM, Hurvitz SA, Turner N, Rugo H, Smith JW, Deudon S, Shi M, Zhang Y, Kay A, Porta DG, Yovine A, Baselga J (2013) Targeting FGFR with dovitinib (TKI258): preclinical and clinical data in breast cancer. Clin Cancer Res 19:3693–3702
Kim KB, Chesney J, Robinson D, Gardner H, Shi MM, Kirkwood JM (2011) Phase I/II and pharmacodynamic study of dovitinib (TKI258), an inhibitor of fibroblast growth factor receptors and VEGF receptors, in patients with advanced melanoma. Clin Cancer Res 17:7451–7461
Wang X, Kay A, Anak O, Angevin E, Escudier B, Zhou W, Feng Y, Dugan M, Schran H (2013) Population pharmacokinetic/pharmacodynamic modeling to assist dosing schedule selection for dovitinib. J Clin Pharmacol 53:14–20
Angevin E, Lopez-Martin J, Lin CC, Gschwend JE, Harzstark A, Castellano D, Soria JC, Sen P, Chang J, Shi MM, Kay A, Escudier B (2013) Phase I study of dovitinib (TKI258), an oral FGFR, VEGFR, and PDGFR inhibitor, in advanced or metastatic renal cell carcinoma. Clin Cancer Res 19:1257–1268
Sharma S, Britten CD, Mortimer J, Kulkarni S, Quinlan M, Liu A, Scott JW, George D (2014) The effect of formulation and food consumption on the bioavailability of dovitinib (TKI258) in patients with advanced solid tumors. Cancer Chemother Pharmacol 74:867–874
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National CANCER Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92:205–216
Andre F, Neven P, Musolino A, Latini L, Campone M, Cortes J, Barrios C, Squires M, Zhang Y, Deudon S, Gogov S, Blackwell K (2013) Dovitinib, a receptor tyrosine kinase inhibitor in combination with fulvestrant in postmenopausal endocrine resistant human epidermal growth factor receptor 2 negative (HER2-)/hormone receptor-positive (HR+) breast cancer: a phase II, randomized, double blind, placebo-controlled study. Cancer Res 73(24 suppl) (abstract OT2-6-04)
Acknowledgments
Financial support for medical editorial assistance was provided by Novartis Pharmaceuticals Corporation. We thank Swarupa Kulkarni, Suraj Anand, Alaeddin Homsi, and Mariama Diallo for their contributions to the study and Julie M. Shilane, PhD, and Peter J. Simon, PhD, for medical editorial assistance with this manuscript. This study was funded by Novartis Pharmaceuticals Corporation.
Conflict of Interest
Dr. Infante discloses a consultant/advisory role with Novartis (no personal compensation; institution is compensated). Dr. Ramanathan discloses research funding from Novartis. Dr. George discloses remuneration from Novartis, Pfizer, Sanofi and Dendreon; consultant/advisory role with Astellas, AVEO, Bayer, Dendreon, Exelixis, Genentech, Medivation, Novartis, Pfizer, Sanofi, Viamet; funding from BMS, Dendreon, Exelixis, Genentech, Glaxo Smith Kline, Janssen, Millennium, Novartis, Pfizer. Eugene Tan discloses stock ownership in Novartis. Michelle Quinlan has nothing to disclose. Angela Liu and Jeffrey Scott disclose remuneration from and stock ownership in Novartis. Dr. Sharma discloses a consultant/advisory role with Novartis, stock ownership in Salarius, Beta Cat and ConverGene, and funding from Novartis.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Infante, J.R., Ramanathan, R.K., George, D. et al. A randomized, crossover phase 1 study to assess the effects of formulation (capsule vs tablet) and meal consumption on the bioavailability of dovitinib (TKI258). Cancer Chemother Pharmacol 75, 729–737 (2015). https://doi.org/10.1007/s00280-015-2681-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-015-2681-3