
Abstract
Purpose
Ibrutinib is an oral Bruton’s tyrosine kinase inhibitor, recently approved for the treatment of mantle cell lymphoma (MCL) and chronic lymphocytic leukemia (CLL) patients with at least one prior therapy. We developed a population pharmacokinetic (PK) model for ibrutinib in patients.
Methods
Ibrutinib PK data (3,477 observations/245 patients) were available from the following clinical studies: (1) A phase I dose-escalation study in recurrent B cell malignancies (dose levels of 1.25–12.5 mg/kg/day and fixed dose of 560 mg/day); (2) a phase II study in MCL (fixed dose level of 560 mg/day); (3) a phase Ib/II dose-finding study in CLL (fixed dose levels of 420 and 840 mg/day). Different compartmental PK models were explored using nonlinear mixed effects modeling.
Results
A two-compartment PK model with sequential zero–first-order absorption and first-order elimination was able to characterize the PK of ibrutinib. The compound was rapidly absorbed, had a high oral plasma clearance (approximately 1,000 L/h) and a high apparent volume of distribution at steady state (approximately 10,000 L). PK parameters were not dependent on dose, study, or clinical indication. The fasting state was characterized by a 67 % relative bioavailability compared with the meal conditions used in the trials and administration after a high-fat meal. Body weight and coadministration of antacids marginally increased volume of distribution and duration of absorption, respectively.
Conclusions
The proposed population PK model was able to describe the plasma concentration–time profiles of ibrutinib across various trials. The linear model indicated that the compound’s PK was dose independent and time independent.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Ibrutinib (Imbruvica®) is an orally administered, covalent inhibitor of Bruton’s tyrosine kinase (BTK), recently approved for the treatment of mantle cell lymphoma (MCL) and chronic lymphocytic leukemia (CLL) patients [1, 2]. BTK is involved in B cell antigen receptor (BCR) signaling, responsible for the regulation of cell proliferation, differentiation, survival, migration, and adhesion [3]. The BCR pathway was found to be involved in the pathogenesis of several B cell malignancies, including MCL and B cell CLL or small lymphocytic lymphoma (SLL) [3, 4]. Given the central role of BTK in BCR signaling, inhibition of BTK by ibrutinib was hypothesized to represent a promising therapeutic target for treatment of these hematological malignancies [5]. Ibrutinib has been recently reported to induce clinically significant objective responses in patients with B cell malignancies in a phase I study [6] and a high frequency of durable remission in patients with relapsed CLL in a phase Ib/II study [1]. The toxicity profile as observed in these studies demonstrated a favorable risk–benefit profile: Most adverse events were of grade 1–2 [1, 6] and dose-limiting events were not observed [6]. Based on the clinical findings [2], the US Food and Drug Administration (FDA) approved ibrutinib as a single agent for the treatment of patients with MCL and CLL who have received at least one prior therapy.
Following oral dosing to patients with B cell malignancies, ibrutinib plasma pharmacokinetics (PK) showed a peak at 1–2 h after dosing and decreased thereafter, approximately following a biexponential decline. The terminal half-life was estimated between 4 and 8 h [1, 6]; however, this estimate was typically obtained between 6 and 24 h post-dose and may represent an underestimation. The accumulation on repeated daily dosing was reported to be less than 2. Plasma exposure, calculated as the area under the plasma concentration–time curve (AUC), increased in direct proportion with the dose [6]. A dihydrodiol metabolite was observed in the systemic circulation with exposures and a plasma concentration–time profile similar to that of the parent compound.
The pharmacodynamics of ibrutinib was determined by monitoring the BTK active site occupancy in patients’ peripheral blood mononuclear cells before and after ibrutinib treatment. BTK occupancy was measured by performing probe assays where cell lysates were incubated with labeled probes. The probes are chemical derivatives of ibrutinib with similar covalent binding characteristics. Under the conditions of the assays, the probes bind to BTK if the Cys-481 residue in the active site is not already covalently modified by ibrutinib [7]. The BTK occupancy assay determined what doses and systemic drug levels were required to achieve maximal occupancy of BTK and, by inference, maximal inhibition of BCR signaling. Pharmacodynamic (PD) studies indicated that BTK occupancy >95 % was achieved in patients receiving daily doses of ≥2.5 mg/kg, with daily AUC values exceeding 160 ng.h/mL [6]. A PK–PD modeling and simulation approach based on these data showed that 65–74 % of subjects are predicted to maintain ≥95 % BTK engagement at the proposed therapeutic dose levels of 420 and 560 mg daily, respectively [8].
The aim of this analysis was to develop a nonlinear mixed effects PK model to describe ibrutinib’s plasma concentration–time data obtained in patients with various B cell malignancies given oral ibrutinib and explore clinically relevant relationships of PK parameters with demographic and pathophysiological covariates.
Methods
Patient population
For the population PK modeling exercise, the PK data obtained in three clinical studies in patients suffering from different B cell malignancies were analyzed. Study 1 was a phase I, dose-escalation study in different types of B cell malignancies. Dose levels used were 1.25, 2.5, 5, 8.3, and 12.5 mg/kg (given once daily for 28 days followed by 7 days of drug holidays), 8.3 mg/kg (given once daily, on a continuous basis), and 560 mg (on a continuous basis). Study 2 was a phase II trial in MCL patients, given continuous daily doses of 560 mg. Study 3 was a phase Ib/II in CLL/SLL patients given continuous daily doses of 420 or 840 mg. In studies 2 and 3, multiple plasma samples for PK assessments were collected on day 1 and 8 (only on day 1 for study 1). Additional limited PK sampling was obtained on subsequent days (8, 15, 28 and subsequent cycles for study 1; days 15 and 22 for study 2; days 15, 22, and 28 for study 3). In all cases, ibrutinib had to be taken at least 30 min before or at least 2 h after a meal (which is the condition adopted in all clinical trials in cancer patients, abbreviated hereafter as “30 min before/2 h after meal”). The only exceptions were two occasions (day 8 and 15) in a 16-patient cohort of study 3, in which patients were randomized to receive ibrutinib either in fasting condition or after a high-fat meal in a crossover design. The assessment was performed to evaluate the potential effects of these conditions on the PK of ibrutinib.
An independent ethics committee or Institutional Review Board at each study site approved the protocols of the studies included in this analysis. The studies were conducted in accordance with the ethical principles originating in the Declaration of Helsinki and ICH Good Clinical Practice guidelines, applicable regulatory requirements, and in compliance with the protocols. All patients (and their legally acceptable representatives, if required by local regulations) provided written informed consent.
Bioanalysis
The bioanalytical methods used for ibrutinib and its metabolite in plasma consisted of liquid chromatography coupled to tandem mass spectrometry (LC–MS/MS). Plasma samples were extracted using protein precipitation with acetonitrile and subsequently analyzed. For detection, a mass spectrometer in the multiple reaction monitoring (MRM) mode was used. The lower limit of quantification (LLOQ) for ibrutinib concentrations ranged from 0.05 to 0.5 ng/mL. The validated concentration ranges varied for the different laboratories to support the different types of studies.
PK analysis
Compartmental PK models were fitted to the ibrutinib plasma concentration–time data using nonlinear mixed effects modeling (NONMEM) software [9].
Candidate structural PK models were assessed through the analysis of model fit and selected for further study if significant improvements in NONMEM objective function values (OFV) were obtained. A drop of 7.9 points (corresponding to p < 0.005) in OFV was considered significant for the inclusion of an additional parameter in nested models [10]. An exponential model was assumed for the estimation of between-subject variability (BSV) of model parameters. The log-transform-both-sides approach was applied to simplify the minimization of the NONMEM runs [11]. The residual unexplained variability (RUV) was modeled using an additive model on natural log-transformed data (which corresponds to an exponential model on untransformed data).
The effect of body weight, dose level, and food was assessed as part of the base model. The search for additional covariates to be included in the PK model was limited to those expected to have physiological and clinical relevance. The effect of a categorical covariate was not assessed if less than 10 % of the overall population was represented in that category [12]. In addition, if two or more continuous covariates were highly correlated, the practically relevant covariate was tested, and the other covariate(s) were discarded [13]. Eventually, study, sex, race, gender, creatinine clearance, total bilirubin, aspartate aminotransferase, alanine aminotransferase, albumin, total serum proteins, lactic acid dehydrogenase (LDH), comedications, and cohort (bortezomib-naïve or bortezomib-experienced) were assessed as potential meaningful covariates. For the covariate analysis, a model-based approach was applied. A univariate search was first performed (criterion: drop in OFV >7.9, corresponding to p < 0.005). All significant covariates were included in the complete model at once. A backward elimination process was then applied, in which the covariates were removed from the model, one at the time, starting from the least significant: The covariate was removed from the model if the relative OFV increase was <10.8 [14]. Categorical covariates were analyzed considering different parameter values for each category. A power model was used to describe the relationship between parameters and continuous variables.
A schematic describing the modeling approach is presented in Fig. 1. Diagnostic graphics, exploratory analyses, and post-processing of NONMEM output were performed using the R software, version 2.14.2 or higher [15].

Schematics of the model analysis overview. Key: Exp BSV exponential model for between-subject variability; Additive RUV additive model for residual unexplained variability; KOKA sequential zero–first-order absorption; F1 relative bioavailability; gof goodness of fit (plots); VPC visual predictive checks; NONMEM nonlinear mixed effects modeling
Results
Overall, PK data were available for a total of 3,477 observations in 245 patients from Studies 1, 2, and 3. Patient demographics showed that the vast majority of them were men (72.2 %) and White (89.4 %), with no other individual race achieving the threshold of 10 %; the covariate race was therefore tested as White versus all other races. Summary statistics of covariates are presented in Table 1. The covariate “coadministration with CYP3A inhibitors” was populated by scanning the whole list of coadministered medications and by extracting all compounds that, in the Drug Interaction Database of the Washington University, were reported to have CYP3A inhibitory properties (both in vitro and in vivo, irrespective of their potency) [16]. High correlation was expected for all body-size-related covariates, so that only the most practically relevant, body weight, was accounted for in the covariate analysis. High correlation was found between transaminase values and between total serum proteins and albumin; the covariate assessment was thus limited to aspartate aminotransferase and total proteins, respectively.
Model development
Only 20 post-dose plasma samples had ibrutinib concentration below LLOQ: Therefore, the exclusion of these data (method M1) was adopted [17]. A two-compartment disposition model with sequential zero–first-order absorption and a lag time (Fig. 2) described the data appropriately. Exponential BSV term was included on all parameters, with the exception of the absorption rate constant (which was negligible and thus fixed to zero). Also, correlation terms were explored, but their inclusion in the model did not improve the model significantly (for instance, including the correlation between CL/F and V/F caused a drop in the objective function of only 1.727). An additional run was performed to examine the effect of the bioanalytical assay for measuring ibrutinib in plasma as a covariate on residual error. This run, including two values of residual variability, did not improve significantly the model fit compared with the model with a single residual variability term, which was therefore preferred.

Schematics of the model structure. Key: ALAG1 = temporal delay (lag time) before absorption process is started; CL/F = apparent (oral) drug clearance; D1 = duration of zero-order input; F1 = relative bioavailability; ka = first-order absorption rate constant (KA in NONMEM code); V2/F = apparent central volume of distribution, V3/F = apparent peripheral volume of distribution, Q/F = apparent inter-compartmental flow; F = absolute oral bioavailability; k20, k23, k32 kinetic microconstants
The effect of food intake, body weight, clinical trial, and ibrutinib dose level was further assessed on this model.
The effect of food conditions [i.e., the condition used in clinical trials (i.e., ibrutinib dosed at least 30 min before or at least 2 h after a meal), overnight fasted, or fed (high-fat breakfast)] on the absorption of ibrutinib was assessed, based on the overall PK dataset, using categorical covariates on the duration of zero-order input (D1) and the relative bioavailability (F1) parameters. After using different values of these parameters for each of the tested conditions, the model was reduced. The reduced model included therefore two values for both D1 and F1. A lower value for the typical relative bioavailability (approximately 67 %) was estimated when the compound was given in the fasted condition versus an arbitrary 100 % bioavailability assumed in the condition used in the clinical trials and following the dosing after a high-fat meal. In addition, following the administration after the high-fat meal, the duration of the zero-order absorption was longer than in the overnight fasting state and in the condition used in the clinical trials. Compared with the initial version of the model, the inclusion of these effects of food on PK parameters led to a significant better fit (OFV drop of 77.295 points).
The dependency of disposition parameters on body weight was estimated using allometric relationships. The effect on CL/F and Q/F was negligible (the estimated allometric coefficient was 0.0495, and the OFV drop was not significant). The coefficient on the apparent volumes was retained despite a nonsignificant drop in OFV of 6.004 was achieved, as the estimated allometric coefficient (0.673) was relatively consistent with the theoretical one [18].
The effect of clinical trial and dose levels on the PK parameters of ibrutinib was assessed, but none of the model alternatives improved the model fit significantly. The base model, on which the covariate analysis was implemented, was therefore a 2-compartment model with sequential zero–first-order absorption, with an effect of food on D1 and F1, and an allometric relationship implemented on the volume terms. This model achieved successful minimization and passed the covariance step.
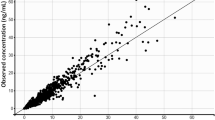
The subsequent covariate analysis [including age, gender, race, comedications, patient pretreatment (cohort), clinical chemistry data (e.g., transaminases, creatinine clearance, LDH, and total proteins), and drug product manufacturing process of ibrutinib capsules] suggested no significant effects on ibrutinib PK parameters. The only exception was the effect of the coadministration with antacid drugs on D1, which caused a 13.921 points drop in the OFV and an increase of the zero-order process duration of 61 % compared with the administration without antacid comedications. The model with the identified significant covariate minimized successfully, and the covariance step was achieved. The parameter estimates obtained with the final population PK model are shown in Table 2. They were consistent with the parameter estimates provided by the base model previously described; only the apparent volume of the central compartment showed a 25 % decrease compared with the base model estimate. The model was able to describe the data well, as can be seen both from the goodness of fit plots (Fig. 3a) and the conditional weighted residuals (CWRES) vs time and population predictions (Fig. 3b). In particular, no trend was found between CWRES and time as well as between CWRES and concentration. Furthermore, the visual predictive checks (Fig. 3c) show that the model was able to explain the inter-individual variability well. More in detail, day 1 was characterized by approximately 15, 13, and 19 % of the observed values outside the predicted intervals for the dose of 420, 560, and 840 mg, respectively, whereas approximately 8, 16, and 6 % of observations were outside the predicted intervals obtained in day 8 for 420, 560, and 840 mg, respectively. In both cases, the observations were evenly distributed below the 2.5th and above the 97.5th prediction intervals. Relatively high shrinkage values were observed for all the parameter estimates (from 35.3 to 67.8 % for Q and D1, respectively), with the exception of the relative bioavailability (shrinkage less than 10 %).


a Goodness of fit plots. Population predictions (PRED, left panel) and Individual predictions (IPRED, right panel) against observed data (DV). Solid line smoother; dashed line unity slope. b Plots of conditional weighted residuals versus time (left panel) and predictions (right panel). c Visual predictive checks (VPCs) for day 1 (left column) and day 8 (right column). From top to bottom: dose levels of approximately 420, 560 and 840 mg are reported
Discussion
The mixed effects PK model of ibrutinib, a 2-compartment linear model, was able to accommodate the observations obtained from patients treated under different conditions and at different dose levels. The absorption of the compound was relatively complex and was described using a sequential zero–first-order model with a lag time. Absorption was also dependent on the meal conditions [ibrutinib given at least 30 min before or 2 h after a meal (conditions used in the clinical trials), overnight fasted, administration after a high-fat meal]. The administration of ibrutinib after a high-fat breakfast resulted in an increased duration of the zero-order absorption process (D1, from 1.1 to 3.29 h), with a corresponding change in the peak time. Simulations indicated that this change slightly decreased the maximal plasma concentrations in the fed compared with the 30 min before/2 h after meal condition (Fig. 4). Relative bioavailability F1 was approximately 67 % when the compound was given after an overnight fast, compared with that of both the condition used in the clinical trials and after a high-fat meal.

Population plasma concentration–time profiles for the different conditions (meal ± antacids). Key: Mod Fast 30 min before/2 h after meal
Although the estimate of the apparent volume of the central compartment V2/F did not appear particularly stable (it showed some dependency on the adopted structural model and on the initial values), the apparent volume of distribution at steady state (sum of the volumes of the central and peripheral compartment) was stable and indicated that ibrutinib is widely distributed within the body (with apparent volumes of distribution at steady state being in the order of 10,000 L). Apparent clearance was also high, in the order of 1,000 L/h. In line with this, the absolute bioavailability (F), in fasted condition, was recently determined to be 2.9 % in 8 healthy participants and it increased more than twofold when ibrutinib was dosed 30 min prior to a standard breakfast [19]. The intravenous plasma clearance was thus confirmed to be efficient with 62 and 76 L/h in fasted and non-fasted condition, respectively, close to liver blood flow. Volume of distribution at steady state was 683 L.
Covariate analysis indicated that the coadministration of ibrutinib with antacid drugs had a significant effect on the prolongation of the duration of the zero-order process (+61 %), without affecting other PK parameters of ibrutinib. This might be due to a potential delayed dissolution rate caused by the higher pH of the GI tract. This effect has, however, to be considered minor and of no clinical relevance as can be observed from the minimal change of the typical plasma concentration–time curve shown in Fig. 4. The model also included a minimal effect of body weight on volumes; this effect was retained, despite a nonsignificant drop in OFV of 6.004 was achieved, as the estimated allometric coefficient (0.673) was relatively consistent with the theoretical unit one [18]. However, this effect of body weight on ibrutinib PKs was not considered relevant from a clinical standpoint and, therefore, no dose adjustment for the bodyweight of patients was warranted.
The analysis suggested no significant effect of other covariates [including dose, study, age, gender, patient pretreatment, clinical chemistry data (e.g., transaminases, creatinine clearance, LDH, and total proteins), and drug product manufacturing process of ibrutinib capsules] on ibrutinib PK parameters. As the majority of patients were White (with none of the other individual races represented by at least 10 % of the study population), the observed lack of effect of race on ibrutinib PK should be interpreted with caution. Considering that ibrutinib is almost exclusively metabolized via CYP3A [20], the lack of a significant effect of the coadministration of medications known to inhibit CYP3A activity also appeared inconclusive, as combining all categories of CYP3A inhibitors in one comedication category likely prevented the chance to observe a relevant effect of CYP3A inhibitors on ibrutinib PK. Moreover, only mild and moderate inhibitors were administered concomitantly during the PK sampling intervals (see Table 1). While detailed dosing data were not available for all inhibitors, chances are that in most cases the dose and/or the administration timing did not constitute conditions of maximal interaction. This was also the outcome of an analysis on individual patient data taking CYP3A inhibitors [21].
Simulations indicated that the terminal half-life (t 1/2) was approximately 14 h, longer than that calculated through noncompartmental analysis. In the literature, the concept of functional half-life was proposed to describe the rate of decline of plasma concentrations with time for compounds with polyexponential PKs [22]. This half-life, determined by model-based simulations of the peak and trough plasma concentrations at steady state, is calculated as
for a compound given once daily. The functional half-life of ibrutinib, which was more influenced by the half-life of the initial than the terminal phase, was found to be approximately 4–6 h, supporting the observation of a relatively limited accumulation of the compound upon repeated daily dosing.
All PK parameters of the final model were affected by a substantial BSV (CV % of V2/F was 153 % and those of V3/F, Q/F and F1 were ≥47 %; the exception was CL/F, characterized by a CV% of approximately 22 %). Also the RUV was high (CV% approximately 73 %). The uncertainty on the disposition parameter estimates was relatively small, with the exception of the standard error of the mean BSV on CL/F, which was approximately 51 %. Also, the high estimated shrinkage was likely due to the combination of the sparse nature of the data and the fact that the biexponential decline was not characterized for more than 24 h after a single dose.
Some difficulties were encountered in the identification of the parameters. This finding was likely due to the fact that the sparse plasma concentration data might be insufficient to accurately characterize these PK parameters. This could also be due to some over-parameterization of the model; however, the condition number, calculated as the ratio of the largest to the smallest eigenvalues of the NONMEM correlation matrix was 494, which is suggestive of the absence of severe ill-conditioning [23]. Another possible cause of these findings may be related to the specifics of the structural model; for instance, the relative proximity of the rates describing the absorption and the rapid disposition processes may confound the different PK phases in different individuals. These complexities, combined with the relatively limited sampling schedule, might prevent the precise estimation of these PK parameters. The adoption of a more mechanistically based model, for instance including the process of irreversible interaction with BTK, may be helpful to address these issues in the future.
To summarize, the proposed population PK model described the plasma concentration–time profiles of ibrutinib from trials in different indications well. The linear model indicated that the PKs were dose independent and time independent and there were no significant PK differences in patients suffering from different B cell malignancies. Also, the average ibrutinib plasma concentrations following dosing at least 30 min before or at least 2 h after a meal (the dosing condition used in the clinical trials) were similar to those obtained after a high-fat meal, while intake of ibrutinib in the fasting state was associated with lower plasma concentrations.
References
Byrd JC, Fuman RR, Coutre SE, Flinn IW, Burger JA, Blum KA et al (2013) Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med 369:32–42
Wang ML, Rule S, Martin P, Goy A, Auer R, Kahl BS et al (2013) Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med 369:507–516
Woyach JA, Johnson AJ, Byrd JC (2012) The B-cell receptor signaling pathway as a therapeutic target in CLL. Blood 120:1175–1184
Wiestner A (2013) Targeting B-cell receptor signaling for anticancer therapy: the Bruton’s tyrosine kinase inhibitor ibrutinib induces impressive responses in B-cell malignancies. J Clin Oncol 31:128–130
Herman SEM, Gordon AL, Hertlein E, Ramanunni A, Zhang X, Jaglowski S et al (2011) Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-3276. Blood 117:6287–6296
Advani RH, Buggy JJ, Sharman JP, Smith SM, Boyd TE, Grant B et al (2013) Bruton tyrosine kinase inhibitor Ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol 31:88–94
Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B et al (2010) The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci USA 107:13075–13080
Poggesi I, Sardu ML, Marostica E, Sukbuntherng J, Chang BY, de Jong J, Woot de Trixhe X, Vermeulen A, De Nicolao G, O’Brien SM, Byrd JC, Advani RH, James DF, Deraedt W, Beaupre D, Wang M (2014). Population pharmacokinetic-pharmacodynamic (PKPD) modeling of ibrutinib in patients with B-Cell malignancies. Accepted for AACR meeting “Hematological malignancies: Translating discoveries in novel therapies” September 20–23, 2014 Sheraton Philadelphia Downtown Philadelphia, PA, USA
Beal S, Sheiner LB, Boeckmann A, Bauer RJ, NONMEM (2009) User’s guides. Icon Development Solutions, Ellicott City
Ette Ene I, Williams Paul J (2007) Pharmacometrics: the science of quantitative pharmacology. Wiley-Interscience, New York
Oberg A, Davidian M (2000) Estimating data transformations in nonlinear mixed effects models. Biometrics 56:65–72
Bonate PL (2011) Pharmacokinetic-pharmacodynamic modeling and simulation, 2nd edn. Springer, New York
Bonate PL (1999) The effect of collinearity on parameter estimates in nonlinear mixed effect modeling. Pharm Res 16:709–717
Wahlby U, Jonsson EN, Karlsson MO (2002) Comparison of stepwise covariate model building strategies in population pharmacokinetic-pharmacodynamic analysis. AAPS PharmSci 4(4):27
R Development Core Team (2012) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. ISBN 3-900051-07-0, (URL http://www.R-project.org)
DIDB: drug interaction DataBase: University of Washington (www.druginteractioninfo.org), Accessed 20 March 2013
Xu SX, Dunne A, Kimko H, Nandy P, Vermeulen A (2011) Impact of low percentage of data below the quantification limit on parameter estimates of pharmacokinetic models. J Pharmacokinet Pharmacodyn 38:423–432
Anderson BJ, Holford NHG (2008) Mechanism-based concepts of size and maturity. Annu Rev Pharmacol Toxicol 48:303–332
Jong de J, Sukbuntherng J, Skee D, Murphy J, O’Brien S, Byrd JC, et al (2014) Evaluation of the pharmacokinetics and food effect of oral ibrutinib in healthy subjects and chronic lymphocytic leukemia patients. AACR annual meeting. Abstract #4637: April 5–9
Imbruvica prescribing information (2013) http://www.imbruvica.com/downloads/Prescribing_Information.pdf. Accessed 24 February 2013
Sukbuntherng J, Jong de J, Skee D, Pak Y, Fardis M, O’Brien S et al (2014) Pharmacokinetics and safety of ibrutinib with concomitant use of CYP3A inhibitors in patients with B-cell malignancies. ACCP annual meeting
Hsu A, Granneman GR, Witt G, Locke C, Denissen J, Molla A et al (1997) Multiple-dose pharmacokinetics of ritonavir in human immunodeficiency virus-infected patients. Antimicrob Agents Chemother 41:898–905
Montgomery DC, Peck EA (1982) Introduction to linear regression analysis. Wiley, London, p 277
Acknowledgments
The authors thank Purvi Jejurkar (Pharmacyclics) for bioanalytical support and Rishabh Pandey (SIRO Clinpharm Pvt. Ltd.) for additional editorial assistance. The authors also thank the study participants, without whom this study would not have been accomplished. The authors received financial support from Janssen Research and Development and Pharmacyclics.
Conflict of interest
Juthamas Sukbuntherng, David Loury, and Jesse McGreivy are employees of Pharmacyclics; Jan de Jong, Xavier Woot de Trixhe, An Vermeulen, and Italo Poggesi are employees of Janssen R&D.
Author information
Authors and Affiliations
Corresponding author
Additional information
Trial registration: Studies included in this analysis are registered on clinicaltrials.gov (NCT01105247; NCT01236391; NCT00849654).
Rights and permissions
About this article

Cite this article
Marostica, E., Sukbuntherng, J., Loury, D. et al. Population pharmacokinetic model of ibrutinib, a Bruton tyrosine kinase inhibitor, in patients with B cell malignancies. Cancer Chemother Pharmacol 75, 111–121 (2015). https://doi.org/10.1007/s00280-014-2617-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-014-2617-3