
Abstract
Purpose
This phase I study endeavored to estimate the maximum tolerated dose and describe the dose-limiting toxicities (DLTs) of oral irinotecan with gefitinib in children with refractory solid tumors.
Methods
Oral irinotecan was administered on days 1–5 and 8–12 with oral gefitinib (fixed dose, 150 mg/m2/day) on days 1–12 of a 21-day course. The escalation with overdose control method guided irinotecan dose escalation (7 dose levels, range 5–40 mg/m2/day).
Results
Sixteen of 19 patients were evaluable, with serial pharmacokinetic studies in ten patients. Diagnoses included osteosarcoma (N = 5), neuroblastoma (N = 3), sarcoma (N = 3), and others (N = 5). Patients received a median of two courses (range 1–20), with at least two patients treated on dose levels 2–7. Three patients had five DLTs; the most common being metabolic (hypokalemia, N = 2 and hypophosphatemia, N = 1) at dose levels two (10 mg/m2) and four (20 mg/m2). One patient experienced grade 3 diarrhea (40 mg/m2). Irinotecan bioavailability was 2.5-fold higher when co-administered with gefitinib, while the conversion rate of irinotecan to SN-38 lactone was unaffected. The study closed due to poor accrual before evaluation of the next recommended irinotecan dose level (35 mg/m2). Of 11 patients receiving at least two courses of therapy, three had stable disease lasting two to four courses and one patient maintained a complete response through 18 courses.
Conclusions
The combination of oral gefitinib and irinotecan has acceptable toxicity and anti-tumor activity in pediatric patients with refractory solid tumors. Pharmacokinetic analysis confirms that co-administration of gefitinib increases irinotecan bioavailability leading to an increased SN-38 lactone systemic exposure.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Irinotecan is a prodrug of the potent topoisomerase I inhibitor, SN-38 [1], with significant preclinical and clinical activity in pediatric solid tumors [2–14]. Gefitinib, a novel EGFR tyrosine kinase (TK) inhibitor, has led to improved progression-free survival in adults with advanced non-small cell lung cancer with activating mutations of the EGFR TK [15–19] and has synergistic activity with irinotecan in pediatric solid tumor xenograft models independent of ERBB1 expression [20]. Single-agent gefitinib is well tolerated in children, with maximum tolerated dose (MTD) of 400 mg/m2/day limited by rash and elevated transaminases; stable or partial response was documented in five of 25 enrolled patients during a phase I trial [21]. Gefitinib increases the bioavailability of intravenous irinotecan [22] with dose-limiting diarrhea, but further investigation of this interaction with oral medication is warranted given the potential use of this combination in an outpatient regimen.
The primary goal of this study (http://www.cancer.gov, NCT00132158) was to determine the dose-limiting toxicities (DLTs) and the MTD of gefitinib and irinotecan administered orally in children with refractory solid tumors. A secondary aim was to assess the bioavailability and SN-38 systemic exposure of oral irinotecan co-administered with gefitinib.
Materials and methods
Patient eligibility
Eligibility included age ≤21 years at time of study entry, diagnosis of a solid tumor, recurrent and unresponsive to conventional therapy or with no known effective therapy (measurable or unmeasurable disease, including bone marrow involvement), life expectancy of >8 weeks, ECOG performance status ≤2 (or Lansky performance scale ≥50 % for children ≤10 years old), adequate hematologic (hemoglobin >8 g/dL, absolute neutrophil count >1,000/mm3 without growth factor support, and platelet count >50,000/mm3 without transfusion support), liver (bilirubin <1.5× normal for age, AST and ALT <3× normal for age) and renal (serum creatinine <3× normal for age) function, and no evidence of active GVHD or treatment for GVHD. Patients with significant intercurrent illness (including diarrhea or interstitial lung disease) or systemic disease, or who were pregnant or lactating were excluded. Concomitant use of phenytoin, carbamazepine, barbiturates, rifampin, phenobarbital or St. John’s wort was not allowed. Patients were required to be off non-approved or investigational agents for ≥30 days before day 1 of study treatment, though prior exposure to irinotecan or gefitinib was allowed. Females of child-bearing potential were required to utilize birth control during and 30 days following completion of the study. The protocol was approved by the St. Jude Institutional Review Board. Written informed consent (and assent when applicable) was obtained for all patients.
Drug administration and study design
A previous study identified the gefitinib MTD of 112.5 mg/m2/day with intravenous irinotecan (15 mg/m2/day) [22]. Since the starting dose of irinotecan in this study was 5 mg/m2/day, the dose of gefitinib was slightly higher and fixed at 150 mg/m2 (approximately equal to 250 mg/1.73 m2; maximum 250 mg). This dose is less than half the pediatric single-agent MTD of 400 mg/m2/day [21]. Gefitinib began on day one of course 1 and was given daily, 1 hour prior to irinotecan, on days 1–12 of each course. To assess the pharmacokinetic interactions of oral gefitinib and irinotecan, gefitinib was held for the first 2 days of course 2 and then resumed on days 3–14. All gefitinib doses were administered in tablet form, though it is possible to dissolve the tablet in lukewarm water and create a suspension for administration. Irinotecan (20 mg/mL mixed with juice) was administered for five consecutive days followed by a 2-day rest and then another five consecutive days [(daily × 5) × 2]. The irinotecan dose on day 12 of course 1 and day 2 of course 2 was given intravenously to assess bioavailability; all other doses were oral. Each course of therapy was 21 days. Patients continued therapy until they experienced disease progression and/or unacceptable toxicity; no intrapatient dose escalation was allowed.
Patient evaluation
After a thorough initial evaluation for study entry, patients underwent at least weekly laboratory and physical examinations during therapy. Patients had routine physical examinations, laboratory and radiographic testing to evaluate for toxicity and response [using Response Evaluation Criteria in Solid Tumors (RECIST)] after two courses of therapy, then after every three to four courses and at the end of therapy. Toxicities were graded according to the NCI Common Toxicity Criteria (version 3.0). Dose-limiting toxicities (DLTs) at least possibly related to gefitinib and irinotecan were defined in relation to the first course of therapy. Hematologic DLTs (after evaluation of bone marrow to rule out involvement by tumor) included grade 4 neutropenia or grade 4 thrombocytopenia that persisted more than 7 days or grade 4 infection related to drug administration. Non-hematologic DLTs were defined as any grade 3 or 4 toxicity with the specific exclusion of the following: grade 3 nausea/vomiting responsive to antiemetics, grade 3 AST/ALT elevation that returns to ≤grade 1 or baseline within 7 days of interrupting treatment, grade 3 fever or infection and grade 3 diarrhea without administration of loperamide.
Statistical considerations
Dose escalation of irinotecan included two periods: initial dose escalation period and escalation with overdose control (EWOC) [23] dose escalation period. During the initial dose escalation period, one patient was assigned to each dose level beginning at the lowest dose level. This cohort size was maintained until a DLT was observed, when the cohort size was increased to two patients and the EWOC dose escalation period began. The toxicity data of all patients previously enrolled in the trial were used to update the dose–toxicity relationship and to guide the next escalation/de-escalation. During this EWOC dose escalation period, a cohort of two patients was assigned to a dose level, and the next dose level for enrollment was calculated using EWOC software with a target DLT probability of 25 % and overdose controlled to be less than 30 %. The posterior distribution of the MTD and the 85 % confidence interval of the MTD were calculated after each patient’s toxicity report was available. If the magnitude of the change in the estimate of the Bayesian confidence interval of MTD and posterior distribution between successive patients was small, specifically the width of the confidence interval estimated MTD did not change by 5 % between three successive patients, the study would terminate if there were already six patients treated at the estimated MTD or continue until there were six patients treated at that level.
Pharmacokinetic studies
The pharmacokinetics of irinotecan and SN-38 lactone were evaluated during course 1 (with concurrent gefitinib) and at the beginning of course 2 (without gefitinib). On days 1 and 11 of courses 2 and 1, respectively, 2 mL of whole blood was obtained before and 0.25, 1.5, 3 and 6 h after the oral irinotecan dose. On days 2 and 12 of courses 2 and 1, respectively, 2 mL of whole blood was obtained from a site contralateral to the infusion site, before and 0.25, 0.5, 1, 4 and 6 h after the end of the irinotecan infusion. Plasma was immediately separated, and the concentrations of the lactone forms of irinotecan and SN-38 were assessed by high-performance liquid chromatography (HPLC) with fluorescence detection, as previously described [24].
Pharmacokinetic analysis was performed using nonlinear mixed effects modeling (NONMEM). A four-compartment model was used, and estimated model parameters included oral bioavailability of irinotecan (F), absorption rate constant for irinotecan (k a), volume of the central compartment for irinotecan (V CPT11L) and apparent volume of distribution for SN-38 lactone (V SN-38L), the intercompartmental rate constants (k 12 and k 21), conversion of irinotecan to SN-38 (k 13), and the SN-38 elimination rate constant (k 30). Secondary parameters calculated during data fitting included apparent oral clearance of irinotecan, CLCPT11L, and apparent oral clearance of SN-38 lactone, CLSN-38L. Area under the plasma concentration–time curve from zero to infinity (AUC0–∞) for irinotecan and SN-38 lactone was estimated using the trapezoidal rule on the simulated concentration–time curve.
Results
Patient population
Nineteen patients were enrolled, and three were not evaluable for either toxicity or efficacy (two developed early progressive disease and one withdrew before receiving any therapy). Table 1 shows the characteristics of the 16 evaluable patients. The median age at enrollment was 14.7 years (range 5.3–20.7 years). Ten patients (62.5 %) were male, and six (37.5 %) were female. The most frequent diagnoses were osteosarcoma (N = 5), neuroblastoma (N = 3) and sarcoma (N = 3) (not otherwise specified, NOS) (Table 1).
Dose escalation, response and toxicity
Patients received a median of two courses (range 1–20). During the irinotecan dose escalation, only three patients with five DLTs were reported. After identifying a DLT (diarrhea) at 40 mg/m2, the next dosage level of irinotecan was determined to be 35 mg/m2. However, prior to patient enrollment, the study was closed due to poor accrual. All 16 patients received 150 mg/m2/day of gefitinib (Table 1).
Eleven patients received at least two courses of therapy. Three patients developed progressive disease (PD) after one course of therapy and two stopped therapy during course 2 (PD and hypertension with cerebral ischemia). One patient with neuroblastoma (progressive disease in bone marrow and multiple bony sites at study entry) achieved a complete response (CR) after three courses of therapy (Fig. 1). This response was documented every two courses by MIBG scans and bone marrow examinations and lasted for 18 courses; progressive disease was noted prior to course 20. Four patients had stable disease (SD) after two courses; two patients abandoned therapy in favor of other treatments, and two patients had disease progression prior to course 4.

Metaiodobenzylguanidine (MIBG, top panels) and single photon emission computed tomography (SPECT, bottom panels) of a patient with metastatic neuroblastoma (arrows) showing complete response after 10 weeks of therapy with oral irinotecan and gefitinib
Toxicities during course 1 of therapy are shown in Table 2. Only five DLTs (three patients) were reported in course 1, the most common being metabolic (hypokalemia, N = 2 and hypophosphatemia, N = 1), with one patient experiencing grade 3 diarrhea. No DLTs were reported in patients who were evaluated during course 2. Four patients experienced grade 3 non-DLTs in course 2, with one patient experiencing a grade 4 ANC (neutropenia) and grade 4 leukocytes (total WBC) toxicity (Table 3). There were no DLTs reported for patients who were evaluated through course 3 (N = 2), course 4 (N = 1) and course 20 (N = 1) (data not shown). The non-DLTs reported in courses three and 16 were hematologic (grade 3 and 4); no other toxicities were noted.
Pharmacokinetic results
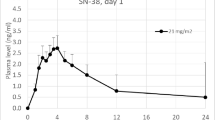
Plasma concentration–time data for irinotecan (oral and i.v.) and SN-38 lactone from four doses in ten patients were included in the population pharmacokinetic model. The median (range) CLCPT11L and V CPT11L were 63.8 L/h/m2 (39.2–92.2) and 57.0 L/m2 (36.2–76.3), respectively, while the median (range) CLSN-38L and V SN-38L were 447.2 L/hr/m2 (22.0–1,498) and 764.3 L/m2 (35.9–2,762), respectively. Figure 2 shows that co-administration of oral gefitinib led to an approximate 2.5-fold increase in irinotecan oral bioavailability, from a median of 0.20 (range 0.03–0.27) without gefitinib to 0.49 (range 0.20–0.75) with gefitinib (p < 0.001; Wilcoxon signed-rank test). By increasing the irinotecan bioavailability, co-administration of oral gefitinib was associated with an increase in the SN-38 lactone metabolite AUC0–∞ from 6.3 ± 3.6 µg/L h to 39.7 ± 39.9 µg/L h (p < 0.001; Wilcoxon signed-rank test). The conversion of irinotecan to SN-38 lactone (k 13) was not significantly altered by gefitinib administration.

Irinotecan oral bioavailability, F, is significantly higher (p < 0.001; Wilcoxon signed-rank test) in pediatric patients dosed daily with gefitinib, than those patients not receiving gefitinib treatment
Discussion
The combination of daily oral gefitinib (150 mg/m2/day) and oral irinotecan up to 30 mg/m2/day [(daily × 5) × 2] is well tolerated in pediatric patients and increases the bioavailability of irinotecan approximately 2.5-fold. Diarrhea was dose limiting in this population.
Due to study closure resulting from poor accrual, a definitive MTD was not established. However, our pharmacokinetic data suggest that a dosage level of 30 mg/m2 is unlikely to differ significantly from 35 mg/m2 owing to the large degree of interpatient variability in systemic exposure (see Fig. 2). Thus, our study supports an MTD of oral irinotecan in the range of 30–35 mg/m2/day when given in combination with 150 mg/m2/day of oral gefitinib. The protracted regimen [(daily × 5) × 2] was chosen to allow for 10 days of continuous gefitinib administration to ensure steady state concentrations were achieved for the purposes of studying the impact of gefitinib on irinotecan bioavailability.
Supportive care with prophylactic antibiotics to ameliorate irinotecan-associated diarrhea has been shown to increase the MTD of single-agent oral irinotecan in children to 60 mg/m2/day given on a protracted schedule [25]. Despite the enhanced bioavailability of irinotecan with co-administration of gefitinib, the first episode of dose-limiting diarrhea was not observed until a dose of 40 mg/m2/day of irinotecan. While the protocol allowed the use of prophylactic cefixime or cefpodoxime following the first course of therapy if the patient developed significant diarrhea, further investigation of the effect of supportive care was not possible due to early study closure.
The pediatric pharmacokinetic model describes the plasma disposition of both irinotecan and SN-38 lactone, with irinotecan apparent oral clearance similar to that observed in prior pharmacokinetic studies conducted in children (34.2–83.1 L/h/m2) [22, 26–28].
The oral irinotecan bioavailability in our population when administered with and without gefitinib is comparable to that previously reported (0.09 without and 0.42 with gefitinib) [22]. Also in agreement with prior studies [29], oral gefitinib treatment significantly increased the oral bioavailability of irinotecan after simultaneous administration (Fig. 1), and, furthermore, systemic exposure of SN-38 lactone was considerably higher than that observed after oral irinotecan dosing without gefitinib. Overall, oral gefitinib co-administration did not influence the rate of clearance of intravenously administered irinotecan or the SN-38 lactone/irinotecan metabolic ratio. The median metabolic ratio of SN-38 lactone to irinotecan (AUCSN-38L/AUCCPT11L) was 0.3, comparable to a published value of 0.2 [26]. Hence, the pharmacokinetic results corroborate earlier preclinical/clinical studies suggesting that the observed increases in SN-38 lactone AUC and irinotecan bioavailability are attributable to enhanced absorption of irinotecan due to gefitinib inhibition of ABCG2 drug transporters expressed in the intestine [29]. Based on simulations using parameters from the pharmacokinetic model, the typical SN-38 lactone exposure at the 30 mg/m2 dosage level with gefitinib co-administration would be 47.2 µg/L h, which would otherwise require an oral irinotecan dose of approximately 74 mg/m2 (assuming linear pharmacokinetics). This exposure is also more than double the typical SN-38 lactone exposure in patients who received irinotecan at the i.v. MTD of 20 mg/m2/day × 5 days for 2 weeks (18.5 µg/L h) [30].
An obvious limitation of this study was early closure due to poor accrual—a challenge faced by many pediatric phase I oncology trials which compete for small numbers of eligible participants with rare diseases [31]. Therefore, it is imperative to safely optimize recruitment and treatment of eligible patients. The Children’s Oncology Group has explored a rolling six design, which reduces the timeline, but not necessarily the number of patients, for pediatric phase I trials in comparison with the traditional 3 + 3 design [32]. Alternatively, we employed EWOC, a trial design that attempts to give a more accurate estimate of MTD while controlling for overdosing. It treats fewer patients at either subtherapeutic or severely toxic dose levels and treats more patients at optimal dose levels [23]. With previous pediatric phase I data to guide the initial dose of each agent, this design provided the potential for more rapid dose escalation [33]. In total, using the EWOC method, we safely and efficiently evaluated seven dose levels with only 16 patients during this study, minimizing time and patient accrual at lower, likely subtherapeutic, dose levels. The large number of dose levels was in part due to the conservative starting dose of irinotecan. While limiting trials to examining four dose levels may be indicated in single-agent pediatric phase I trials [34], this combination drug study with potential overlapping GI toxicity warranted more cautious evaluation. Therefore, our study validates the EWOC scheme as an efficient and safe method that provides dose escalations that more closely approximate the MTD without compromising study duration.
Other protocol-, patient-, and physician-related barriers might have prevented accrual into our study such as availability of other phase I studies with targeted agents [sunitinib (NCT00387920); IGF-1R antagonist (NCT00560144)], as well as a concurrent phase I study of intravenous irinotecan with oral gefitinib (NCT00186979). Additionally, irinotecan became more widely used as initial salvage therapy (especially in combination with temozolomide) and patients who had failed one irinotecan combination might not have been interested in enrolling in another irinotecan trial.
The combination of gefitinib and irinotecan showed promising anti-tumor activity with a CR in one patient (neuroblastoma) and four with SD (neuroblastoma, germ cell tumor, osteosarcoma and fibromyxoid sarcoma). Therefore, this combination of enhanced irinotecan exposure may have appeal in other patient populations where irinotecan has proven beneficial, such as low-grade glioma [35], Ewing sarcoma [36, 37] and hepatoblastoma [38]. Gefitinib co-administration improved irinotecan exposure at lower irinotecan dosages without increased toxicity, which may allow for dose escalation of other active anticancer agents, such as temozolomide or vincristine. In addition, treatment options for patients with high risk, metastatic or recurrent solid tumors are often limited by the cumulative toxicity of active chemotherapy agents, especially cisplatin and doxorubicin (i.e., hepatoblastoma therapy). For these patients, utilizing irinotecan as “maintenance therapy” after surgical resection may provide improved long-term survival without increasing the toxicity of therapy [39, 40]. Furthermore, gefitinib co-administration would enhance the bioavailability of oral irinotecan in a protracted dosing regimen that could be administered in an outpatient setting, potentially improving therapeutic response, cost of therapy and quality of life for these patients.
References
Tsuji T, Kaneda N, Kado K et al (1991) CPT-11 converting enzyme from rat serum: purification and some properties. J Pharmacobiodyn 14:341–349
Bomgaars L, Kerr J, Berg S et al (2006) A phase I study of irinotecan administered on a weekly schedule in pediatric patients. Pediatr Blood Cancer 46:50–55
Bomgaars LR, Bernstein M, Krailo M et al (2007) Phase II trial of irinotecan in children with refractory solid tumors: a children’s oncology group study. J Clin Oncol 25:4622–4627
Dharmarajan KV, Wexler LH, Wolden SL (2013) Concurrent radiation with irinotecan and carboplatin in intermediate- and high-risk rhabdomyosarcoma: a report on toxicity and efficacy from a prospective pilot phase II study. Pediatr Blood Cancer 60:242–247
Mascarenhas L, Lyden ER, Breitfeld PP et al (2010) Randomized phase II window trial of two schedules of irinotecan with vincristine in patients with first relapse or progression of rhabdomyosarcoma: a report from the children’s oncology group. J Clin Oncol 28:4658–4663
Mixon BA, Eckrich MJ, Lowas S, Engel ME (2013) Vincristine, irinotecan, and temozolomide for treatment of relapsed alveolar rhabdomyosarcoma. J Pediatr Hematol Oncol 35:e163–e166
Rodriguez-Galindo C, Crews KR, Stewart CF et al (2006) Phase I study of the combination of topotecan and irinotecan in children with refractory solid tumors. Cancer Chemother Pharmacol 57:15–24
Shitara T, Shimada A, Hanada R et al (2006) Irinotecan for children with relapsed solid tumors. Pediatr Hematol Oncol 23:103–110
Vassal G, Couanet D, Stockdale E et al (2007) Phase II trial of irinotecan in children with relapsed or refractory rhabdomyosarcoma: a joint study of the French Society of Pediatric Oncology and the United Kingdom Children’s Cancer Study Group. J Clin Oncol 25:356–361
Vassal G, Doz F, Frappaz D et al (2003) A phase I study of irinotecan as a 3-week schedule in children with refractory or recurrent solid tumors. J Clin Oncol 21:3844–3852
Blaney S, Berg SL, Pratt C et al (2001) A phase I study of irinotecan in pediatric patients: a pediatric oncology group study. Clin Cancer Res 7:32–37
Cosetti M, Wexler LH, Calleja E et al (2002) Irinotecan for pediatric solid tumors: the Memorial Sloan-Kettering experience. J Pediatr Hematol Oncol 24:101–105
Furman WL, Stewart CF, Poquette CA et al (1999) Direct translation of a protracted irinotecan schedule from a xenograft model to a phase I trial in children. J Clin Oncol 17:1815–1824
Mugishima H, Matsunaga T, Yagi K et al (2002) Phase I study of irinotecan in pediatric patients with malignant solid tumors. J Pediatr Hematol Oncol 24:94–100
Mok TS, Wu YL, Thongprasert S et al (2009) Gefitinib or carboplatin–paclitaxel in pulmonary adenocarcinoma. N Engl J Med 361:947–957
Maemondo M, Inoue A, Kobayashi K et al (2010) Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med 362:2380–2388
Wu YL, Chu DT, Han B et al (2012) Phase III, randomized, open-label, first-line study in Asia of gefitinib versus carboplatin/paclitaxel in clinically selected patients with advanced non-small-cell lung cancer: evaluation of patients recruited from mainland China. Asia Pac J Clin Oncol 8:232–243
Gao Z, Han B, Wang H et al (2012) Clinical observation of gefitinib as a first-line therapy in sixty-eight patients with advanced NSCLC. Oncol Lett 3:1064–1068
Zhang L, Ma S, Song X et al (2012) Gefitinib versus placebo as maintenance therapy in patients with locally advanced or metastatic non-small-cell lung cancer (INFORM; C-TONG 0804): a multicentre, double-blind randomised phase 3 trial. Lancet Oncol 13:466–475
Houghton PJ, Cheshire PJ, Harwood FG (2000) Evaluation of ZD1839 (gefitinib) alone and in combination with irinotecan (CPT-11) against pediatric solid tumor xenografts. In: Proceedings of the 11th NCI-EORTC-AACR symposium, Amsterdam, The Netherlands, November 7–10, 2000. Clin Cancer Res 2000 (Suppl 6):379
Daw NC, Furman WL, Stewart CF et al (2005) Phase I and pharmacokinetic study of gefitinib in children with refractory solid tumors: a children’s oncology group study. J Clin Oncol 23:6172–6180
Furman WL, Navid F, Daw NC et al (2009) Tyrosine kinase inhibitor enhances the bioavailability of oral irinotecan in pediatric patients with refractory solid tumors. J Clin Oncol 27:4599–4604
Babb J, Rogatko A, Zacks S (1998) Cancer phase I clinical trials: efficient dose escalation with overdose control. Stat Med 17:1103–1120
Owens TS, Dodds H, Fricke K et al (2003) High-performance liquid chromatographic assay with fluorescence detection for the simultaneous measurement of carboxylate and lactone forms of irinotecan and three metabolites in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci 788:65–74
Furman WL, Crews KR, Billups C et al (2006) Cefixime allows greater dose escalation of oral irinotecan: a phase I study in pediatric patients with refractory solid tumors. J Clin Oncol 24:563–570
Crews KR, Stewart CF, Jones-Wallace D et al (2002) Altered irinotecan pharmacokinetics in pediatric high-grade glioma patients receiving enzyme-inducing anticonvulsant therapy. Clin Cancer Res 8:2202–2209
Ma MK, Zamboni WC, Radomski KM et al (2000) Pharmacokinetics of irinotecan and its metabolites SN-38 and APC in children with recurrent solid tumors after protracted low-dose irinotecan. Clin Cancer Res 6:813–819
Thompson PA, Gupta M, Rosner GL et al (2008) Pharmacokinetics of irinotecan and its metabolites in pediatric cancer patients: a report from the children’s oncology group. Cancer Chemother Pharmacol 62:1027–1037
Stewart CF, Leggas M, Schuetz JD et al (2004) Gefitinib enhances the antitumor activity and oral bioavailability of irinotecan in mice. Cancer Res 64:7491–7499
Wagner LM, Crews KR, Iacono LC et al (2004) Phase I trial of temozolomide and protracted irinotecan in pediatric patients with refractory solid tumors. Clin Cancer Res 10:840–848
Caldwell PH, Murphy SB, Butow PN, Craig JC (2004) Clinical trials in children. Lancet 364:803–811
Skolnik JM, Barrett JS, Jayaraman B et al (2008) Shortening the timeline of pediatric phase I trials: the rolling six design. J Clin Oncol 26:190–195
Le Tourneau C, Lee JJ, Siu LL (2009) Dose escalation methods in phase I cancer clinical trials. J Natl Cancer Inst 101:708–720
Lee DP, Skolnik JM, Adamson PC (2005) Pediatric phase I trials in oncology: an analysis of study conduct efficiency. J Clin Oncol 23:8431–8441
Gururangan S, Fangusaro J, Poussaint TY et al (2014) Efficacy of bevacizumab plus irinotecan in children with recurrent low-grade gliomas—a pediatric brain tumor consortium study. Neuro Oncol 16:310–317
Wagner LM, McAllister N, Goldsby RE et al (2007) Temozolomide and intravenous irinotecan for treatment of advanced Ewing sarcoma. Pediatr Blood Cancer 48:132–139
Casey DA, Wexler LH, Merchant MS et al (2009) Irinotecan and temozolomide for Ewing sarcoma: the memorial sloan-kettering experience. Pediatr Blood Cancer 53:1029–1034
Zhang YT, Feng LH, Zhong XD et al. Vincristine and irinotecan in children with relapsed hepatoblastoma: A single-institution experience. pediatr hematol oncol 2014
Qayed M, Powell C, Morgan ER et al (2010) Irinotecan as maintenance therapy in high-risk hepatoblastoma. Pediatr Blood Cancer 54:761–763
Trobaugh-Lotrario AD, Katzenstein HM (2012) Chemotherapeutic approaches for newly diagnosed hepatoblastoma: past, present, and future strategies. Pediatr Blood Cancer 59:809–812
Acknowledgments
This work was supported in part by a grant from AstraZeneca (Protocol 1839US/0300), which provided partial funding as well as study drug for participants. The manuscript was reviewed and approved prior to publication. We thank the American Lebanese Syrian Associated Charities for their support, Amy Sanders and Dana Hawkins for facilitating data collection, Dr. Barry Shulkin for providing diagnostic images and Dr. Alberto Pappo for editorial assistance.
Conflict of interest
The authors have declared no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Brennan, R.C., Furman, W., Mao, S. et al. Phase I dose escalation and pharmacokinetic study of oral gefitinib and irinotecan in children with refractory solid tumors. Cancer Chemother Pharmacol 74, 1191–1198 (2014). https://doi.org/10.1007/s00280-014-2593-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-014-2593-7