
Abstract
Introduction
Patients with triple-class refractory (TCR) multiple myeloma (MM) often need cytoreductive chemotherapy for rapid disease control. Bendamustine is an outpatient-administered, bifunctional alkylator and isatuximab is an anti-CD38 monoclonal antibody with unique cytotoxicity characteristics. We hypothesized that isatuximab-bendamustine-prednisone would be well-tolerated regimen in TCR MM, and conducted single-center, phase Ib, investigator-initiated study.
Patients/Methods
Patients had TCR MM and last daratumumab exposure ≥ 6 weeks. This study was conducted as a 3 + 3 design to establish the maximally tolerated dose (MTD) and/or recommended phase 2 dose (RP2D). Isatuximab 10 mg/kg IV was administered weekly (cycle 1), and every 2 weeks thereafter. Bendamustine was administered on days 1 and 2 at 3 dose levels (DL): 50, 75, and 100 mg/m2. Methylprednisolone was administered as 125 mg on day 1 and prednisone 60 mg days 2–4. Common definitions were used for DLTs, adverse events (CTCAE v 5.0), and disease response.
Results
Fifteen patients were treated (3 DL1, 6 DL2, 6 DL3). Median age was 71, 53% had high-risk cytogenetics, and 34% had prior BCMA-targeting therapy. One DLT was observed at DL2 (Grade 3 thrombocytopenia plus bleeding). There were no Grade 5 treatment-related AEs. The MTD was not reached. The overall response rate was 20% (3/15) including one stringent complete response. The median PFS was 2.5 months (95% CI 0.9–4.1 months).
Conclusion
We demonstrated the safety and tolerability of isatuximab-bendamustine-prednisone. Toxicities were mild and manageable with limited intervention. The study was discontinued due to slow accrual. However, we observed responses even among highly refractory patients.
Clinical trial registration
This study was registered on clinicaltrials.gov as NCT04083898 on 9/6/2019.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Over the past 50 years, prognosis for patients with multiple myeloma has enjoyed steady improvement, with the estimated overall survival of patients improving from a dismal 1–2 years in the 1980s to over a decade today [1]. However, patients with disease progression on at least one immunomodulatory drug (IMID), proteasome inhibitor (PI) and anti-CD38 antibody (i.e. triple class refractory disease (TCR)) still have a grim prognosis with an overall response rate (ORR) of 25% and median PFS of 3.9 in the prospective LocoMMotion study [2, 3]. With results from multiple randomized trials becoming available, frontline quadruplet induction is rapidly becoming the standard-of-care in fit patients and, even in transplant ineligible patients, many patients treated today will meet the criteria of TCR after 1–2 lines of therapy (LOT) [4,5,6]. While bispecific T-cell engagers (TCEs) and chimeric antigen receptor (CAR) T cells have shown impressive results in relapsed/refractory MM, global access is lacking [7,8,9,10,11,12]. In addition, despite improved manufacturing capacity, vein-to-vein time for CAR T cell therapy remain approximately 1–2 months [13]. Consequently, effective therapies for patients with TCR myeloma who are not eligible for or lack access to cutting edge immunotherapy remains an unmet medical need.
While they have been largely supplanted as part of frontline therapy, cytotoxic chemotherapy regimens remain useful tools in highly refractory patients in need of disease control for the purposes cytoreduction, palliation, or bridging to subsequent advanced therapies. However, common chemotherapy regimens such as high-dose cyclophosphamide (HD Cy), dexamethasone-cyclophosphamide-etoposide-cisplatin (DCEP) and other infusional or hyperfractionated regimens generally require inpatient admission and significant supportive care for notable hematologic and non-hematologic toxicities. Our single-center retrospective analysis of DCEP in TCR MM identified a 16% incidence of Grade 3–5 febrile neutropenia/sepsis with a confirmed infectious etiology [14]. Within the same analysis, we observed that bendamustine plus a corticosteroid was active (27% overall response rate) in this population with a more favorable toxicity profile and feasible outpatient administration, highlighting its potential as a backbone regimen for novel chemoimmunotherapeutic combinations.
Isatuximab is an anti-CD38 IgG-κ chimeric monoclonal antibody (mAb) approved in combinations with carfilzomib-dexamethasone and pomalidomide-dexamethasone [15]. It binds an epitope that is distinct from the daratumumab binding site [16]. Additionally, while the cytotoxicity of daratumumab hinges on Fc-dependent immune effector mechanisms, isatuximab can additionally induce direct apoptosis in the absence of secondary crosslinking, as well as inhibit CD38 ectoenzyme activity [17]. While these differences suggest possible activity of isatuximab in a daratumumab-refractory population, a phase 1/2 study examining isatuximab monotherapy in such a population did not yield any objective responses, and such patients were excluded from pivotal trials [18].
Given the mechanistic similarities between rituximab and isatuximab, the success and tolerability of bendamustine-rituximab in non-Hodgkin lymphomas, our experience with bendamustine in heavily-pretreated MM, and the unmet need for a tolerable, outpatient chemoimmunotherapy for MM, we undertook a prospective, single center phase Ib trial to investigate the combination of isatuximab, bendamustine and prednisone in patients with triple class refractory MM.
Materials and methods
Trial design
This trial was designed as a single center phase Ib/II seamless trial design. The phase I portion of the trial employed a 3 + 3 design with a maximum targeted enrollment of 18 patients over three dose levels. The objective of the phase I trial was to demonstrate safety and tolerability and define a maximum tolerated dose (MTD) and/or recommended phase 2 dose (RP2D). By design, the phase II cohort analysis includes the patients in the phase I cohort receiving the MTD (n = 6) and up to 19 additional patients. Due to slow accrual and changing landscape of MM therapies, the study was ended after the phase Ib portion. This investigator-initiated study was funded by Sanofi. The trial was registered at clinicaltrials.gov (NCT04083898) and performed in accordance with the Declaration of Helsinki.
Patients
The study included patients with a history of relapsed/refractory multiple myeloma previously treated with a PI, IMID and daratumumab and were refractory or intolerant to these agents per IMWG criteria. All patients were required to be at least 6 weeks from their last dose of daratumumab at time of study screening, and were required to have adequate bone marrow and organ function, including creatinine clearance > 30 ml/min. Patients with prior exposure to isatuximab or bendamustine were excluded. In addition, patients requiring renal replacement therapy and those with a history of plasma cell leukemia, central nervous system involvement or concurrent malignancy requiring treatment were also excluded. Full inclusion and exclusion criteria are included in the Supplementary appendix.
Procedures
All patients received isatuximab (10 mg/kg) intravenously (IV) on days 1, 8, 15 and 22 of cycle 1 and on days 1 and 15 of subsequent cycles. Standard pre-medications and post-infusion monitoring were administered with each dose of isatuximab (Supplementary appendix). Bendamustine was administered on days 1 and 2 of each cycle at the specified dose level (Level 1: 50 mg/m2, Level 2: 75 mg/m2, Level 3: 100 mg/m2). In addition, all patients received methylprednisolone 100 mg IV on day 1 and prednisone 60 mg daily on days 2–4 of each cycle. All patients were required to receive prophylaxis for herpes simplex/varicella zoster reactivation and peptic ulcer disease during the duration of the study. All other supportive care was performed per institutional practice.
Definitions and outcomes
High-risk cytogenetics were defined as t(4;14), t(14;16), del(17p) and/or amplification of 1q. Treatment refractoriness was defined as progression while on therapy or within 60 days of the last dose of treatment [19]. TCR was defined as disease refractoriness to at least one IMID, PI and anti-CD38 antibody, while penta-drug refractory disease (PDR) required refractoriness to two IMIDs, two PIs and anti-CD38 antibody. Response criteria were defined per the 2016 criteria from the International Myeloma Working Group [20]. Overall response rate (ORR) was defined as a partial response or better as best response. The clinical benefit rate (CBR) was defined as stable disease or better as best response. All time-to-event end points, including progression-free survival (PFS) and overall survival (OS) were defined as time from start of therapy.
Adverse events were defined per the Common Terminology Criteria for Adverse Events (CTCAE), version 5.0. A dose limiting toxicity (DLT) was defined as a treatment emergent toxicity occurring during cycle 1 that met any of the following criteria: grade 4 anemia not explained by the underlying disease, grade 4 thrombocytopenia (or grade 3 with significant bleeding), grade 4 neutropenia (or grade 3 with febrile neutropenia), any grade 3 or greater non-hematologic toxicity and any non-hematologic toxicity requiring dose reduction or treatment delay > 14 days at or prior to cycle 2 day 1. The MTD was to be defined as the dose level below that at which two DLTs occur, assuming six patients were treated at the MTD. The RP2D was defined as the MTD, or in the absence of detected an MTD, the highest dose-level assessed.
All patients who received at least one dose of study treatment were evaluable for toxicity and were followed for 30 days after study completion or death, whichever was sooner. Patients were considered evaluable for disease response unless they discontinued treatment prior to completion of cycle 2 and did not have a disease assessment prior to treatment discontinuation.
Statistical considerations
All analyses were descriptive in nature. The characteristics of the cohort and observed toxicities were summarized. ORR, PFS, and OS were calculated with corresponding 95% confidence intervals (CI).
Results
Patient characteristics
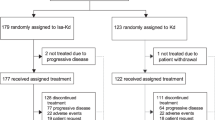
Twenty patients who provided informed consent were screened for study inclusion and 15 were enrolled between June 2020 and February 2023 (Fig. 1). All 15 patients were enrolled in the dose escalation phase.

Consort diagram of patient enrollment
Baseline characteristics are presented in Table 1. In the overall cohort, the median age at registration was 71 years (range: 56–85), 67% were male and 87% were white. The median ECOG was 1 (range: 0–2). Eight patients (53%) had extramedullary disease (EMD) at study entry, including 8 with soft tissue involvement only and one with both soft tissue and paramedullary disease. High-risk cytogenetics were present in 5 patients and 3 additional patients had gain(1q). The median time from diagnosis to trial enrollment was 38 months (range: 9–160).
Nine patients had previously undergone autologous hematopoietic cell transplantation (AHCT), including 6 in first remission and 3 as salvage therapy. None had undergone more than 1 AHCTs, but one patient subsequently received CAR T cells. The median number of prior LOT was 3 (range: 2–9). Thirteen patients (87%) were TCR, two additional patients had IMID intolerance but were refractory to PIs and daratumumab. Eleven patients (73%) were penta-drug exposed and no patients were penta-drug refractory. The median time from prior anti-CD38 antibody therapy to study enrollment was 40 weeks (range: 6–168). Five patients had received prior BCMA-directed therapy, including bispecific antibodies (4) and CAR T cells (1).
Dose escalation and treatment toxicity
During dose escalation, three patients were enrolled on dose level 1 (bendamustine 50 mg/m2) and six patients were enrolled each on dose levels 2 (75 mg/m2) and 3 (100 mg/m2). One DLT was observed at dose level two (grade 3 thrombocytopenia with bleeding complications). No DLTs were observed at dose level three. Consequently, dose level 3 (100 mg/m2) was selected as the RP2D.
All patients had at least one grade 3–4 AE. Two patients discontinued therapy due to AEs, including one patient with the DLT described above and one patient who was diagnosed with hemolymphophagocytic histiocytosis (HLH) thought to be unrelated to study treatment due to presence of HLH symptoms prior to treatment initiation. The other 13 patients discontinued due to disease progression.
The most common grade 3–4 AEs were hematologic, including decreased lymphocytes (93%), leukocytes (53%), neutrophils (27%) and platelets (33%), as well as anemia (33%). Common non-hematologic adverse events grade 3–4 AEs included anorexia (27%), hypertension (27%) and respiratory infections (40%). No other grade 3–4 AEs occurred in more than two study patients. Three patients had isatuximab-related infusion reactions, all during the first dose of isatuximab. Infusion reactions were mild (grade 1 or 2) and responded to supportive measures with resumption of isatuximab the same day. Notably, one patient experienced febrile neutropenia and received growth factor support with G-CSF, but no other patients required growth factors during the study period. Two patients required platelet transfusion, one due to HLH and one due to grade 3 thrombocytopenia with bleeding. Two grade 5 AEs occurred during the study period: one patient presented with a COVID infection three weeks after progression and died of respiratory failure and the patient who came off study for HLH died of sepsis three weeks later. A full summary of the adverse events is available in the Supplementary Material.
Response to therapy
All 15 patients were considered evaluable by study criteria. Enrolled patients received a median of 3 cycles of treatment (range: 1–13). The ORR was 20% (3/15; 95% C.I 4% − 48%) (Figs. 2A and 3A]). No responses were seen on dose level 1 but one (17%) was observed on dose level 2 and two (33%) on dose level 3. All patients achieved their best responses after 1 cycle of therapy. The CBR was 80% (12/15; 95% C.I 52% − 96%) overall, including 100% (3/3) in dose level 1, 66% (4/6) in dose level 2 and 83% (5/6) in dose level 3.

A-C: Response to therapy. A shows response rates by IMWG Criteria. Though no responses were seen at dose level 1, all patients had stable disease after at least one cycle of therapy. Overall response rate (ORR) improved from dose level 1 (0%) to dose level 2 (17%) to dose level 3 (33%). The ORR for the combined cohort was 20%. Analysis of PFS (B) and OS (C) show outcomes comparable with other non-immunotherapy based approaches in relapsed/refractory myeloma, with a median PFS of 2.5 months and OS of 6.9 months

A & B: Treatment response. A shows a swimmer plot for treatment response and duration in the entire cohort. B shows baseline (panel A) and follow up (panel B) imaging for patient IBP-20, a 58-year-old woman with penta-drug exposed disease and prior anti-BCMA CAR T cell therapy who enrolled on the study as sixth line therapy in the setting of extramedullary relapse. The patient had an exceptional response with a stringent CR by IMWG criteria and complete resolution of their extramedullary disease
Of the 8 patients with EMD at study entry, 6 had EMD-specific response assessment by imaging and/or exam. Two patients had stable disease by imaging and 3 patients had a significant (> 50%) reduction in the size of one or more lesion. One patient had a > 50% reduction in the size of the EMD by exam. A second patient had 2 sites of EMD, with marked improvement (> 50%) reduction in one mass and stable disease in the other after 2 cycles of therapy. A third patient had complete resolution of a 7.0 x 3.7 cm trapezius mass on follow up PET scan, but relapsed in her subcutaneous tissue 9.4 months after starting therapy (Fig. 3B).
Relapse, progression-free and overall survival
At time of analysis, 13 patients had relapsed at a median of 2.5 months (range: 0.9–11.7)from initiation of therapy. The median PFS was 2.5 months (95% CI 0.9–4.1 months) (Fig. 2B). At last follow up, 12 patients had died from disease (n = 6), COVID-19 (3) or infection (3). The median OS was 6.9 months (95% C.I 4.7–9.2) within the entire cohort (Fig. 2C).
Subsequent therapy
Following completion of study treatment, 13 patients received subsequent anti-myeloma therapy (Supplemental Table 2) with a median of 1 (range: 1–6) additional lines. In these patients, the median time to next therapy was 3.3 months (range: 1.4–12.5). Six patients received T-cell engager (TCE) therapy as part of a later line of therapy. Response evaluable patients received a median of 8 cycles (range: 1–20) with 3/5 patients (60%) experiencing a response. No patients have gone on to CAR T cell as a later line of therapy.
Discussion
Herein, we report the results of a prospective phase Ib trial of an outpatient chemoimmunoptherapy regimen combining isatuximab with bendamustine and prednisone (IBp). Out of the 15 participants treated during the dose escalation phase, only one experienced a DLT. Therefore, the MTD remained undefined and the RP2D was determined to be bendamustine at 100 mg/m2 on days 1 and 2 of each cycle with isatuximab dosed conventionally. The toxicity profile was predictable, with grade 3–4 AEs predominantly hematologic in nature and manageable, with minimal transfusion or growth factor support. Infectious complications were common (16 infections in 9 patients, 60%) but generally mild (5 grade ≥ 3 infections in 5 patients, 33%) despite patients receiving our institutional standard supportive measures of HSV prophylaxis without anti-bacterial prophylaxis, anti-pneumocystis prophylaxis or pre-emptive immunoglobulin replacement. Respiratory infections, a common AE associated with CD38-targeting antibodies, were the majority of the infectious complications (9 infections (including 3 COVID-19 infections) in 6 patients, 56% of total infections) including severe infections (4 grade ≥ 3 infections in 4 patients, 80% of severe infections).
Three patients (20%) achieved an objective response, which is markedly inferior to outcomes with TCE and CAR T cell therapy. Two of six patients (33%) receiving the RP2D responded, which is similar to other traditional combination approaches in relapsed/refractory multiple myeloma. Notably, most patients (80%) experienced a period of disease stability, with the median PFS of 2.5 months. A high proportion of participants had EMD which did not decrease in size by 50% but remained stable or improved modestly with treatment. We did observe one patient with an exceptional response, who achieved a stringent complete response lasting 9.4 months and complete resolution of their EMD, as well as two other patients with documented improvement in at least one site of EMD. In patients without disease progression, multiple cycles of therapy were tolerable, with 5 patients (33%) remaining on study for > 3 months and only 1 study patient (7%) discontinuing therapy due to treatment toxicity.
Conventional cytotoxic chemotherapy has a limited role in the current management of MM. Predominantly, chemotherapy regimens are deployed for rapid cytoreduction in the setting of high marrow burden or symptomatic EMD, for the purposes of palliation or more recently for bridging to more promising advanced therapies such as CAR T and TCEs. Responses tend to be transient and toxicities significant. Additionally, there is paucity of quality evidence as most data are derived from small, retrospective studies. In a retrospective study of salvage DCEP among patients refractory to PIs and IMIDs, Park et al. (2013) reported a 91.5% incidence of Grade 3–4 neutropenia with treatment-related mortality (TRM) of 14.8%, mainly infectious [21]. A single-institution retrospective study of regimens with hyperfractionated cyclophosphamide backbone demonstrated reasonable responses with PFS of about 3 months, although rehospitalization rates of 52–77% and TRM up to 19% [22]. Griffin et al. retrospectively compared three infusional chemotherapy regimens which had comparable efficacy and safety, all associated with relatively high rates of rehospitalization (34%), febrile neutropenia (37%), and TRM (7%) [23]. These experiences highlight the unfilled niche for an active MM cytoreductive regimen that provides a better balance between efficacy, tolerability, and resource utilization. Our study suggests lower doses of bendamustine (50 and 75 mg/m2) are associated with low response rates (11%, 1/9) in this combination and patients should be treated with ≥ 100 mg/m2 to maximize the chance of response, which was 33% (2/6 patients) in our study. We observed responses regardless of cytogenetic risk, EMD and prior therapy, including one exceptional responder with t(4;14), EMD and prior CAR T cell therapy. Unfortunately, due to slow accrual, our clinical trial of IBp did not advance to the phase II portion to further evaluate the efficacy signal. Our study opened April 2020 and slow accrual was multifactorial, driven initially by limited trial enrollment during the COVID-19 pandemic and the FDA approval of multiple other agents for patients with RRMM.
Significant questions regarding this regimen remain, which could be evaluated in future retrospective and prospective studies. In particular, more data is needed on the impact of bendamustine-based therapy on subsequent TCE and CAR T cell therapy. Bendamustine is generally avoided prior to leukapheresis for CAR T cell manufacturing due to reports of poor outcomes with this combination (cite). However, while only five patients in our trial received subsequent TCE after disease progression, their ORR (60%) to TCE was comparable to that seen in pivotal trials, suggesting that the bendamustine exposure does not impact subsequent TCE efficacy [11, 12, 24]. Future retrospective analyses are needed to better define the risk for poor outcomes with T-cell based immunotherapy after bendamustine exposure and the optimal washout period.
Prospective studies of bendamustine-based regimens (including IBp) may still have a place in the new age of myeloma immunotherapy. We and others have used bendamustine as part of a lymphodepleting chemotherapy prior to CAR T during times of fludarabine shortage, without a clear detrimental impact on subsequent CAR T outcomes [25, 26]. Since CD38 is restricted to mature plasma cells, the addition of isatuximab to bendamustine for pre-CAR T cell lymphodepletion could augment T cell response and is a concept that may warrant prospective study. In addition, patients who do not respond or progress rapidly after these therapies continue to have dismal outcomes and novel approaches are needed, which may include chemoimmunotherapy combinations.
In conclusion, we demonstrated the safety and tolerability of isatuximab-bendamustine-prednisone with a RP2D of bendamustine at 100 mg/m2 on days 1 and 2 of each cycle with conventionally dosed isatuximab and minimal supportive care requirements. The ORR at the R2PD (33%) was similar to other non-T cell immunotherapy approaches in this space, but the study was closed early due to poor accrual. Our experience highlights the challenges of performing clinical trials in this rapidly evolving field, where sequential FDA approvals have led to a marked increase in available standard-of-care agents for patients with RRMM from the initiation (2019) to closure (2023) of this study. This embarrassment of riches is an unabashed victory for patients with myeloma and physicians who care for them. Design of clinical trials in the future should explore novel approaches (such as platform designs) to incorporate advances in treatment during the period of study enrollment and keep pace with the changing standard of care.
Data availability
Requests for de-identified patient level data should be directed to the senior author and will be evaluated on a case-by-case basis.
References
Rajkumar SV (2022) Multiple myeloma: 2022 update on diagnosis, risk stratification, and management. Am J Hematol 97:1086–1107. https://doi.org/10.1002/ajh.26590
Costa LJ, Hungria V, Mohty M, Mateos MV (2022) How I treat triple-class refractory multiple myeloma. Br J Haematol 198:244–256
Mateos MV, Weisel K, De Stefano V et al (2022) LocoMMotion: a prospective, non-interventional, multinational study of real-life current standards of care in patients with relapsed and/or refractory multiple myeloma. Leukemia 36:1371–1376. https://doi.org/10.1038/s41375-022-01531-2
Sonneveld P, Dimopoulos MA, Boccadoro M et al (2023) PERSEUS: Daratumumab, Bortezomib, Lenalidomide, and Dexamethasone for multiple myeloma. N Engl J Med. https://doi.org/10.1056/NEJMoa2312054
Gay F, Roeloffzen W, Dimopoulos MA et al (2023) Results of the Phase III Randomized Iskia Trial: Isatuximab-Carfilzomib-Lenalidomide-Dexamethasone vs Carfilzomib-Lenalidomide-Dexamethasone as Pre-transplant induction and post-transplant consolidation in newly diagnosed multiple myeloma patients. Blood 142:4. https://doi.org/10.1182/blood-2023-177546
Voorhees PM, Kaufman JL, Laubach J et al (2020) GRIFFIN: Daratumumab, lenalidomide, bortezomib, and dexamethasone for transplant-eligible newly diagnosed multiple myeloma: the GRIFFIN trial. Blood 136:936–945. https://doi.org/10.1182/blood.2020005288
Martin T, Usmani SZ, Berdeja JG et al (2022) CARTITUDE-1 2YFU: Ciltacabtagene Autoleucel, an Anti–B-cell maturation Antigen chimeric Antigen receptor T-Cell therapy, for Relapsed/Refractory multiple myeloma: CARTITUDE-1 2-Year Follow-Up. J Clin Oncol. https://doi.org/10.1200/jco.22.00842
San-Miguel J, Dhakal B, Yong K et al (2023) CARTITUDE-4: Cilta-Cel or Standard Care in Lenalidomide-Refractory multiple myeloma. N Engl J Med 1–13. https://doi.org/10.1056/nejmoa2303379
Rodriguez-Otero P, Ailawadhi S, Arnulf B et al (2023) KarMMa-3: Ide-cel or standard regimens in relapsed and refractory multiple myeloma. N Engl J Med 388:1002–1014. https://doi.org/10.1056/nejmoa2213614
Munshi NC, Anderson LD, Shah N et al (2021) KarMMa: Idecabtagene Vicleucel in Relapsed and Refractory multiple myeloma. N Engl J Med 384:705–716. https://doi.org/10.1056/nejmoa2024850
Moreau P, Garfall AL, van de Donk NWCJ et al (2022) MajesTEC-1 P1/P2: Teclistamab in relapsed or refractory multiple myeloma. N Engl J Med 387:495–505. https://doi.org/10.1056/nejmoa2203478
Bahlis NJ, Costello CL, Raje NS et al (2023) MagnetisMM-1: Elranatamab in relapsed or refractory multiple myeloma: the MagnetisMM-1 phase 1 trial. Nat Med 29:2570–2576. https://doi.org/10.1038/s41591-023-02589-w
Firestone RS, Mailankody S Current use of CAR T cells to treat multiple myeloma
Goldsmith SR, Fiala MA, Wang B et al (2020) DCEP and bendamustine/prednisone as salvage therapy for quad- and penta-refractory multiple myeloma. Ann Hematol 99:1041–1048. https://doi.org/10.1007/s00277-020-03970-2
Goldsmith SR, Liu L, Covut F (2021) Isatuximab for the treatment of multiple myeloma. Drugs Today 57:665–675. https://doi.org/10.1358/dot.2021.57.11.3343690
Martin TG, Corzo K, Chiron M et al (2019) Therapeutic opportunities with pharmacological inhibition of CD38 with isatuximab. Cells 8
Van De Donk NWCJ, Usmani SZ (2018) CD38 antibodies in multiple myeloma: mechanisms of action and modes of resistance. Front Immunol 9
Mikhael J, Belhadj-Merzoug K, Hulin C et al (2021) A phase 2 study of isatuximab monotherapy in patients with multiple myeloma who are refractory to daratumumab. Blood Cancer J 11
Rajkumar SV, Harousseau JL, Durie B et al (2011) Consensus recommendations for the uniform reporting of clinical trials: report of the International Myeloma Workshop Consensus Panel 1. Blood 117:4691–4695. https://doi.org/10.1182/blood-2010-10-299487
Kumar S, Paiva B, Anderson KC et al (2016) International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol 17:e328–e346
Park S, Lee SJ, Jung CW et al (2014) DCEP for relapsed or refractory multiple myeloma after therapy with novel agents. Ann Hematol 93:99–105. https://doi.org/10.1007/s00277-013-1952-5
Shank BR, Primeaux B, Yeung EK et al (2023) Hyperfractionated cyclophosphamide and dexamethasone alone or in combination with Daratumumab and/or Carfilzomib for the treatment of relapsed or refractory multiple myeloma: a single-Center Retrospective Analysis. Clin Lymphoma Myeloma Leuk 23:279–290. https://doi.org/10.1016/j.clml.2022.12.004
Griffin PT, Ho VQ, Fulp W et al (2015) A comparison of salvage infusional chemotherapy regimens for recurrent/refractory multiple myeloma. Cancer 121:3622–3630. https://doi.org/10.1002/cncr.29533
Chari A, Minnema MC, Berdeja JG et al (2022) MonumenTAL-1: Talquetamab, a T-Cell-redirecting GPRC5D bispecific antibody for multiple myeloma. N Engl J Med. https://doi.org/10.1056/NEJMoa2204591
Sidana S, Hosoya H, Jensen A et al (2023) Bendamustine vs. fludarabine/cyclophosphamide lymphodepletion prior to BCMA CAR-T cell therapy in multiple myeloma. Blood Cancer J 13
Ong SY, Pak S, Mei M et al (2023) Bendamustine lymphodepletion is a well-tolerated alternative to fludarabine and cyclophosphamide lymphodepletion for axicabtagene ciloleucel therapy for aggressive B-cell lymphoma. Am J Hematol 98:1751–1761. https://doi.org/10.1002/ajh.27069
Acknowledgements
We would like to thank the Alvin J. Siteman Cancer Center at Washington University School of Medicine and Barnes-Jewish Hospital in St. Louis, Missouri, for the use of the Clinical Trials Core which provided protocol development and statistical support services. The Siteman Cancer Center is supported in part by an NCI Cancer Center Support Grant #P30 CA91842.
Funding
This study was performed as an investigator-initiated trial with support from Sanofi. The study was designed and data was collected and analyzed by the research team. The manuscript was written by the listed authors without assistance from a medical writer or external editorial consultant. Sanofi provided high level review of the manuscript and decision to submit for publication was made after agreement between the authors and the study sponsor.
Author information
Authors and Affiliations
Contributions
SRG, MAF, and RV designed the clinical trial and conducted trial oversight. SRG, ZDC, MAS, KG, and RV enrolled and managed patients on the trial. MH contributed to trial oversight, toxicity and response assessments, data management. MJS conducted statistical analysis and contributed to trial oversight. SRG and MJS wrote the main manuscript. All authors reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Ethical approval
Prior to study initiation, this study underwent review by and received approval from the Protocol Review and Monitoring Committee (Siteman Cancer Center) and the Institutional Review Board (Washington University in Saint Louis). All patients provided informed consent at time of study enrollment. The study was performed in accordance with the Declaration of Helsinki.
Competing interests
SRG reports honoraria from Oncovalent, Wugen Inc, Sanofi-Genzyme, Janssen Pharmaceuticals, and Adaptive Biotechnologies and research funding from BMS. MJS reports research support from Secura Bio, Rapid Novor and Pfizer. MF reports no relevant conflicts of interest. MH reports no relevant conflicts of interest. ZDC reports research funding and honoraria from Bioline. MAS reports honoraria from Advarra, Equillium Inc, GSK, Incyte, Inmagene Bio, Novo Nordisk, and StemLine. KSG reports research support from Sanofi. RV reports research support from BMS, Takeda, and Sanofi and honoraria from BMS, Sanofi, Janssen, Pfizer, Regeneron, and Karyopharm.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Goldsmith, S.R., Slade, M.J., Fiala, M. et al. A phase Ib trial of isatuximab, bendamustine, and prednisone in relapsed/refractory multiple myeloma. Ann Hematol (2024). https://doi.org/10.1007/s00277-024-05975-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00277-024-05975-7