
Abstract
Molecular detection of JAK2 mutation (V617F or exon 12) is included as a major diagnostic criterion for polycythemia vera (PV) by the WHO 2016 guidelines. JAK2 exon 12 mutations are seen in about 2–5% of JAK2V617F-negative cases of PV. Mutations in JAK2 cause constitutive activation of JAK-STAT pathway which results in variable phenotypes. PV patients with exon 12 mutations in JAK2 present characteristically with erythrocytosis. There are limited reports describing the spectrum of JAK2 exon12 mutations in myeloproliferative neoplasms (MPNs). Here, we describe the characteristics of a series of MPN patients with mutations in exon 12 of JAK2 of which two were novel variants associated with polycythemia. Interestingly, we noted two patients presenting as myelofibrosis having JAK2 exon 12 mutations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Philadelphia-negative myeloproliferative neoplasms (MPNs) are a group of clonal hematopoietic disorders characterized by an over production of differentiated cells in the peripheral blood. The three main diseases included in this group are polycythemia vera (PV), essential thrombocytosis (ET), and primary myelofibrosis (PMF). They are clinically characterized by nonspecific symptoms such as fatigability, pruritus, early satiety due to splenomegaly, increased risk of infections, and thrombotic events. Disease morbidity is associated with thromboembolic and hemorrhagic events. Complication of the disease by transformation to myelofibrosis is seen to a greater extent with PV than in ET and in secondary AML with PMF than with PV [1, 2].
In the vast majority of MPN cases, mutation in codon 617 of JAK2, resulting in the replacement of the amino acid valine with phenylalanine [V617F, JAK2 NM_004972.3:c.1849G > T (p.Val617Phe)] is found. This gain of function mutation is present in approximately 96% of PV cases, 55% of ET cases, and 65% of PMF cases [3]. Mutations in CALR and MPL gene account for the remaining driver mutations contributing to the pathogenesis of MPN [4].
JAK2 associates with the cytoplasmic portions of various receptors for key hematopoietic cytokines, such as erythropoietin (EPO), thrombopoietin (THPO), and granulocyte colony-stimulating factor (G-CSF). Constitutive activation of JAK2 by either point mutation or fusion protein causes activation of the JAK-STAT pathway. JAK2V617F mutation in the JH2 domain disrupts the inhibitory function of the pseudo-kinase [JH2] leading to constitutive tyrosine phosphorylation activity [JH1] that promotes cytokine hypersensitivity [5].
Exon 12 is present in the region between SH2 and JH2 domains of JAK2. Mutations in this region contribute to approximately 3% of PV cases [6, 7]. This exon codes for amino acids 505 to 547. Among these, mutations have been identified in codons spanning from 533 to 547. Rare JAK2 mutations have also been reported in exons 12, 13, 14, and 15 [6].
Patients with exon 12 mutations typically present with isolated erythrocytosis and suppressed erythropoietin. In contrast to the trilineage hyperplasia characteristic of patients with V617F mutation, the bone marrow from patients with exon 12 mutations often exhibits nonspecific morphology, with isolated erythroid proliferation and absence of prominent megakaryocyte atypia and clustering. Demonstration of exon 12 mutations in these patients is particularly helpful for ruling out reactive erythrocytosis [7].
JAK2 exon 12 mutations are included as a major criterion for diagnosis of PV in the most recent World Health Organization guidelines [8]. There are limited reports describing the mutation spectrum of JAK2 exon12 mutations [9,10,11]. Here, we describe a series of MPN cases with JAK2 exon 12 mutations identified in our patient cohort.
Patients and methods
A total of 1272 cases were tested by the MPN panel (JAK2V617F by allele-specific PCR (AS-PCR), JAK2 exon 12 by sequencing, CALR by fragment analysis, and MPL by AS-PCR) during January 2016 to July 2019. All patients had their diagnoses made locally based on an integrated review of clinical, laboratory, and histopathological information as a multidisciplinary team. Review of patient’s electronic records was performed to collect the clinical and laboratory characteristics at the time of diagnosis or referral. These were subsequently classified as MPN based on the WHO 2016 criteria. Cases which did not meet these criteria and those with inadequate data were excluded from further analysis.
Methodology
DNA was extracted from whole blood samples (from patients with the clinical suspicion of MPN) using Gentra Puregene blood DNA kit (QIAGEN, Maryland, USA) as per the manufacturer’s protocol. A 453 base pairs PCR product containing the 128 bp of the exon 12 was amplified as described previously [7] followed by direct sequencing using the Big Dye Terminator v1.1 Cycle Sequencing kit (Applied Biosystems, Foster City, CA, USA) and an ABI Prism 3130 Genetic Analyzer (Applied Biosystems). Sequences were aligned to the reference sequence, and mutations, if any, were tested for pathogenicity by using bioinformatic tools and public databases. Mutation detection for JAK2V617F, CALR, and MPL was also done in the two cases of myelofibrosis by previously described methods [3, 12, 13].
Results
Among the samples analyzed, a clinical phenotype of polycythemia was seen in ~ 44%, ET in ~ 14%, PMF in ~ 25%, and the rest were classified as MPN-MDS and MPN-NOS categories. Of the 307 cases of JAK2V617F-negative MPN cases, exon 12 mutations were identified in 8 patients. The clinical features and demographics of these patients are represented in Table 1.
JAK2 exon 12 mutations in PV
All the six cases categorized as PV at presentation, presented as idiopathic erythrocytosis at diagnosis. Bone marrow morphology was panmyelosis with abnormal clustering of variable-sized megakaryocytes which is characteristic of PV. Isolated erythroid hyperplasia that is described in cases with exon 12 mutations, however, was not seen in these cases. None of the patients in this cohort had low or suppressed erythropoietin (sEPO) levels (median of 9.9; range, 5.2–10.9 mIU/mL; normal range, 3.7 to 31.5 mIU/mL). Two patients had thrombotic events. One developed deep vein thrombosis and another had splenic vein thrombosis. There was no organomegaly at diagnosis.
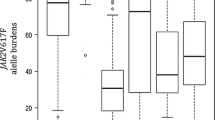
The spectrum of JAK2 exon 12 mutations identified in JAK2V617F-negative PV included complex insertion deletions [n = 2; c.1613_1616delinsT (p.H538_K539delinsL) and c.1619_1627delinsGAA (p.I540_E543delinsRK)], in frame deletions [n = 3; c.1624_1629del (p.N542_E543del), c.1622_1627del (p.R541_E543delinsK), and c.1612_1616del (p.H538Nfs*4)] and substitution [n = 1; c.1586C > A (p.P529H)]. (Fig. 1).

Spectrum of mutations identified in JAK2 exon 12 in our study
Two of these were novel mutations: p.P529H and p.I540_E543delinsRK (Fig. 1). Most of these mutations affected the codons between 538 and 543 present between SH2 and the pseudo-kinase JH2 region (Fig. 2). Pathogenicity of the novel missense substitution determined using bioinformatic tools suggested these mutations to be disease causing.

a Missense substitution c.1586C > A (p.P529H), b complex insertion deletion mutation c.1619_1627delinsGAA (p.I540_E543delinsRK) in JAK2 exon 12. Both the novel mutations were identified in patients presenting as polycythemia
Myeloproliferative Neoplasm Symptom Assessment Form Total Symptom Score (MPN-SAF TSS) [14] available for only 1 patient (MPN 218) was 5 (fatigue, 0; early satiety, 0; abdominal discomfort, 0; inactivity, 0; lack of concentration, 0; night sweats, 0; pruritus, 5; bone pain, 0; fever, 0; unintentional weight loss, 0).
JAK2 exon 12 mutations in myelofibrosis
JAK2 exon 12 mutations were seen in 2 cases with myelofibrosis at presentation. Clinical and demographic details are represented in Table 2.
Case 1 was a 37-year-old male who was symptomatic since 1 year, presented with pancytopenia and massive splenomegaly. He had young onset hypertension, chronic liver disease, and a history of receiving multiple transfusions following blood loss. On repeat evaluation at our center, a leucoerythroblastic blood picture, bone marrow morphology consistent with myelofibrosis, and lymph node biopsy that showed extra medullary hematopoiesis triggered molecular testing.
Case 2 was a 57-year-old male who was symptomatic for a month and was evaluated for anemia with massive splenomegaly. He had a history of blood transfusions before being evaluated at our center. Peripheral smear showed leucoerythroblastic picture, and bone marrow morphology was consistent with myelofibrosis. MPN-SAF TSS was 24 (fatigue, 10; early satiety, 4; abdominal discomfort, 0; inactivity, 10; lack of concentration, 0; night sweats, 0; pruritus, 0; bone pain, 0; fever, 0; unintentional weight loss, 0). No thrombotic or cardiac events were present in both the patients.
Molecular testing with a panel of genes for MPN which included hotspot mutations in JAK2 (both V617F and exon 12), CALR, and MPL genes was done in both cases of myelofibrosis as a part of MPN workup. Complex deletion insertion mutations in exon 12 were identified [c.1614_1616delinsATT (p.H538_K539delinsQL) and c.1613_1617delinsTC (p.H538_K539delinsL)]. Variants identified in both cases are reported in public database (COSM24438 and COSM29084).
Discussion and conclusion
In addition to the single hotspot JAK2V617F mutation that is seen in nearly 95% of PV cases, mutations in exon 12 account for the remaining 2–5% cases of JAK2V617F-negative PV cases. While mutations in JAK2 are reported in numerous MPN phenotypes [5], exon 12 mutations specifically result in erythrocytosis due to increased EPO signaling [5]. This is explained by differential coupling of mutant JAK2 with cytokine receptors. Exon 12 mutations result in stronger ligand-independent signaling through JAK2 and generate higher levels of JAK2, ERK1, and ERK2 phosphorylation than does the V617F mutation [7]. Although erythrocytosis was the most common presentation in this study, we also identified JAK2 exon 12 mutation in 2 patients who presented with myelofibrosis (Tables 1 and 2).
There are reports of higher frequency of JAK2 exon 12 mutations in patients with PV from Asia [11, 15]. Previous reports suggest that PV patients with JAK2 exon 12 mutations have lower platelet and WBC counts at presentation and may also present at a younger age with higher hemoglobin levels as compared with JAK2 V617F mutated patients. However, the clinical outcome in terms of survival and disease related complications like fibrotic or leukemic transformation and thrombosis seems to be similar between the two groups [16, 17].
Marrow morphology in exon 12 mutated cases is described to have isolated erythroid hyperplasia in contrast to the panmyelosis picture seen in V617F mutation [7]. However, the bone marrow morphology in our cohort was predominantly consistent with panmyelosis and typical megakaryocyte morphology described in V617F mutation.
A low serum EPO level is a minor diagnostic criterion in the WHO classification of PV. Study by Scott et al. showed that only 50% of PV cases with exon 12 mutations had low serum EPO [7]. However, we observed that none of the cases with PV had low serum EPO level.
Around 90 exon 12 mutations have been identified to date, including amino acid substitution, deletions, and duplications (http://cancer.sanger.ac.uk/ cancergenome/projects/cosmic). Most frequent exon 12 mutations involve an in-frame deletion of six nucleotides at codons 542 and 543 (N542_E543del) [6]. In-frame deletions were the most common type of mutation identified in our cohort as well, and this mutation was one among the three in-frame deletions identified. Other mutations in our study include complex insertion deletions and a novel missense substitution.
JAK2 exon 12 mutations are generally restricted to PV cases at presentation [7, 18]. However, similar to a previous report [19], we identified JAK2 exon 12 mutations in two cases of myelofibrosis at presentation. A documented MPN-SAF TSS was available only for one of these cases which was high. Exon 12 mutations have been reported in post-PV myelofibrosis [11]. However, in view of the short clinical history before diagnosis, the cases of myelofibrosis in our case series seem to be primary myelofibrosis as against post-PV myelofibrosis.
Limitations of this study are mutation detection by DNA sequencing which has a sensitivity limit of 10–20%. Patients with low mutant allele burden can be missed by this method. As there is no hotspot mutation, more sensitive techniques such as AS-PCR cannot be applied. Future direction is application of targeted NGS that allows broad multigene coverage in a single assay which also allows the detection of mutant burden based on allele frequency.
References
Szuber N, Mudireddy M, Nicolosi M, Penna D, Vallapureddy RR, Lasho TL, Finke C, Begna KH, Elliott MA, Hook CC, Wolanskyj AP, Patnaik MM, Hanson CA, Ketterling RP, Sirhan S, Pardanani A, Gangat N, Busque L, Tefferi A (2019 Apr 1) 3023 Mayo Clinic patients with myeloproliferative neoplasms: risk-stratified comparison of survival and outcomes data among disease subgroups. Mayo Clin Proc 94(4):599–610
Iurlo A, Cattaneo D, Gianelli U (2019) Blast transformation in myeloproliferative neoplasms: risk factors, biological findings, and targeted therapeutic options. Int J Mol Sci 13:20(8)
Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, Vassiliou GS, Bench AJ, Boyd EM, Curtin N, Scott MA, Erber WN, Green AR, Cancer Genome Project (2005 Mar 19) Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 365(9464):1054–1061
Vainchenker W, Kralovics R (2017) Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood. 129(6):667–679
Nangalia J, Green AR (2017) Myeloproliferative neoplasms: from origins to outcomes. Blood 130(23):2475–2483
Gong JZ, Cook JR, Greiner TC, Hedvat C, Hill CE, Lim MS, Longtine JA, Sabath D, Wang YL, Association for Molecular Pathology (2013) Laboratory practice guidelines for detecting and reporting JAK2 and MPL mutations in myeloproliferative neoplasms: a report of the Association for Molecular Pathology. J Mol Diagn JMD 15(6):733–744
Scott LM, Tong W, Levine RL, Scott MA, Beer PA, Stratton MR, Futreal PA, Erber WN, McMullin M, Harrison CN, Warren AJ, Gilliland DG, Lodish HF, Green AR (2007) JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med 356(5):459–468
Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J (2017) WHO classification of tumours of haematopoietic and lymphoid tissues, revised fourth edition. IARC, Lyon
Butcher CM, Hahn U, To LB, Gecz J, Wilkins EJ, Scott HS et al (2008) Two novel JAK2 exon 12 mutations in JAK2V617F-negative polycythaemia vera patients. Leukemia. 22(4):870–873
Pietra D, Li S, Brisci A, Passamonti F, Rumi E, Theocharides A, Ferrari M, Gisslinger H, Kralovics R, Cremonesi L, Skoda R, Cazzola M (2007) Somatic mutations of JAK2 exon 12 in patients with JAK2 (V617F)-negative myeloproliferative disorders. Blood. 111(3):1686–1689
Yeh Y-M, Chen Y-L, Cheng H-Y, Su W-C, Chow N-H, Chen T-Y, Ho CL (2010) High percentage of JAK2 exon 12 mutation in Asian patients with polycythemia Vera. Am J Clin Pathol 134(2):266–270
Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD, Them NC, Berg T, Gisslinger B, Pietra D, Chen D, Vladimer GI, Bagienski K, Milanesi C, Casetti IC, Sant'Antonio E, Ferretti V, Elena C, Schischlik F, Cleary C, Six M, Schalling M, Schönegger A, Bock C, Malcovati L, Pascutto C, Superti-Furga G, Cazzola M, Kralovics R (2013) Somatic mutations of Calreticulin in Myeloproliferative neoplasms. N Engl J Med 369(25):2379–2390
Furtado LV, Weigelin HC, Elenitoba-Johnson KSJ, Betz BL (2013) Detection of MPL mutations by a novel allele-specific PCR-based strategy. J Mol Diagn JMD. 15(6):810–818
Emanuel RM, Dueck AC, Geyer HL, Kiladjian J-J, Slot S, Zweegman S, te Boekhorst PA, Commandeur S, Schouten HC, Sackmann F, Kerguelen Fuentes A, Hernández-Maraver D, Pahl HL, Griesshammer M, Stegelmann F, Doehner K, Lehmann T, Bonatz K, Reiter A, Boyer F, Etienne G, Ianotto JC, Ranta D, Roy L, Cahn JY, Harrison CN, Radia D, Muxi P, Maldonado N, Besses C, Cervantes F, Johansson PL, Barbui T, Barosi G, Vannucchi AM, Passamonti F, Andreasson B, Ferrari ML, Rambaldi A, Samuelsson J, Birgegard G, Tefferi A, Mesa RA (2012) Myeloproliferative neoplasm (MPN) symptom assessment form total symptom score: prospective international assessment of an abbreviated symptom burden scoring system among patients with MPNs. J Clin Oncol 30(33):4098–4103
Park C-H, Lee K-O, Jang J-H, Jung CW, Kim J-W, Kim S-H, Kim HJ (2016 Aug) High frequency of JAK2 exon 12 mutations in Korean patients with polycythaemia vera: novel mutations and clinical significance. J Clin Pathol 69(8):737–741
Tefferi A, Lavu S, Mudireddy M, Lasho TL, Finke CM, Gangat N, Pardanani A, Hanson CA, Mannarelli C, Guglielmelli P, Vannucchi AM (2018) JAK2 exon 12 mutated polycythemia vera: Mayo-Careggi MPN Alliance study of 33 consecutive cases and comparison with JAK2V617F mutated disease. Am J Hematol 93(4):E93–E96
Passamonti F, Elena C, Schnittger S, Skoda RC, Green AR, Girodon F, Kiladjian JJ, McMullin M, Ruggeri M, Besses C, Vannucchi AM, Lippert E, Gisslinger H, Rumi E, Lehmann T, Ortmann CA, Pietra D, Pascutto C, Haferlach T, Cazzola M (2011) Molecular and clinical features of the myeloproliferative neoplasm associated with JAK2 exon 12 mutations. Blood. 117(10):2813–2816
Schnittger S, Bacher U, Haferlach C, Geer T, Müller P, Mittermüller J, Petrides P, Schlag R, Sandner R, Selbach J, Slawik HR, Tessen HW, Wehmeyer J, Kern W, Haferlach T (2009) Detection of JAK2 exon 12 mutations in 15 patients with JAK2V617F negative polycythemia vera. Haematologica. 94(3):414–418
Abstract 37: an unusual case of myelofibrosis with a JAK2 H538QK539L mutation associated with nephrotic syndrome | Clinical Cancer Research [Internet]. [cited 2019 Sep 20]. Available from: https://clincancerres.aacrjournals.org/content/23/24_Supplement/37#
Funding
This study is partly funded by a grant from Department of Biotechnology, India BT/COE/34/SP13432/2015 to P.B.
P.B. is supported by a senior fellowship program of Wellcome DBT India Alliance (IA/S/15/1/501842) New Delhi, India. VM is supported by senior fellowship program of Wellcome DBT India Alliance (IA/CPHS/18/1/503930), New Delhi, India. UK is supported by an early career fellowship program of Wellcome DBT India Alliance (IA/CPHE/17/1/503351), New Delhi, India. AV is supported by DBT JRF (DBT/2018/CMC/1138).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interest.
Ethical approval
This is a retrospective analysis using DNA extracted from EDTA blood sample that has been collected for routine molecular diagnostic procedures (JAK2, CALR, MPL) in patients with myeloproliferative neoplasms in our institution. No additional sample was collected from any patient for this study. All procedures performed in this study involving human participants were in accordance with the ethical standards of the institutional research committee and with the 1964 Helsinki declaration and its later amendments.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Maddali, M., Kulkarni, U.P., Ravindra, N. et al. JAK2 exon 12 mutations in cases with JAK2V617F-negative polycythemia vera and primary myelofibrosis. Ann Hematol 99, 983–989 (2020). https://doi.org/10.1007/s00277-020-04004-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-020-04004-7