
Abstract
Acute myeloid leukemia (AML) is the most common acute leukemia in adults. Chemotherapy with cytotoxic agents is the standard of care, but is associated with a high rate of adverse events. Elderly patients are frequently intolerant to such treatment, presenting a very poor prognosis. The hypomethylating agents (HMA) azacitidine or decitabine represent one of the main therapeutic alternatives for these patients. Isocitrate dehydrogenase inhibitors (IDH) constitute another therapeutic class with DNA methylation effects in AML. In this article, we review the use of first- and second-generation HMA and IDH inhibitors in AML. The data collected demonstrated that HMA are generally considered effective and safe for AML patients who are not eligible for standard chemotherapy. The combination of azacitidine or decitabine with venetoclax was recently approved by the US Food and Drug Administration (FDA) for older AML patients and those unfit for intense chemotherapy. IDH inhibitors also showed encouraging results for relapsed/refractory AML patients harboring an IDH mutation and received FDA approval. Therefore, recent studies have led to the emergence of new therapeutic options using HMA and IDH inhibitors for specific groups of AML patients, representing an important step in the treatment of this aggressive malignancy. New options should emerge from the ongoing studies in the coming years.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acute myeloid leukemia (AML) is caused by molecular alterations in hematopoietic stem and progenitor cells that favor greater cellular proliferation and reduced differentiation [1]. The median age at diagnosis is 68–72 years, and the highest mortality rates occur in older adults [2]. In the US, more than 10,000 deaths were estimated to have occurred due to AML in 2015 [3].
The genetic profile of leukemia cells influences the classification, prognosis, and treatment choice [4]. Patients with chromosomal translocations involving transcription factors that compose the core binding factor (CBF), i.e.: t(8;21)(q22;q22) RUNX1/RUNX1T1 and inv (16)(p13q22) CBFB/MYH11, are usually associated with a favorable prognosis, as patients with a mutated nucleophosmin (NPM1). These patients usually experience favorable outcomes without the need of allogeneic hematopoietic stem-cell transplantation (HSCT). On the other hand, patients with complex cytogenetics, monosomal karyotype; t(9;22)(q34.1;q11.2) BCR-ABL1, inv.(3)(q21q26) RPN1/MECOM; t(6;9)(p23;q34.1) DEK-NUP214; t(v;11q23.3); − 5 or del (5q); − 7; − 17/abn(17p); mutated RUNX1; ASXL1 or TP53, and FLT3-ITD with a high allele burden and wild-type NPM1 have dismal outcomes, with median overall survival (OS) of 6–12 months, even with HSCT. Finally, patients without any of the aforementioned cytogenetics findings have an intermediate outcome, usually benefiting from HSCT [5].
Epigenetic changes also contribute to AML pathogenesis [6]. In fact, some of the AML recurring mutations occur in genes involved in the regulation of DNA methylation (DNMT3A, DNMT3B, TET, IDH1, IDH2) or histone acetylation (EZH2, MLL, ASXL1) [7]. These findings, together with the fact that epigenetic modifications are reversible, supported the rationale for the development of key clinical researches of novel epigenetic therapies in AML [6].
AML treatment initially consists of attempting to obtain complete remission (CR) with induction chemotherapy, which usually involves daunorubicin at the dose of 60 or 90 mg/m2 or idarubicin at the dose of 10–12 mg/m2 for the first 3 days and infusion of 100–200 mg/m2 of cytarabine daily from days 1 to 7, a regimen known as “7 + 3” [8, 9]. However, induction failures can occur in up to 40% of patients, depending on the patient’s age [10], with a CR rate of 65–73% in young newly diagnosed AML patients and a CR rate of only 38–62% in patients over 60 years [11]. In order to eliminate possible remaining leukemia cells and prevent relapse, a consolidation (or post-remission) therapy is recommended with the following options: (a) a new course of chemotherapy or (b) HSCT. These strategies may be used separately or in combination, depending on the condition of the patient and the availability of a compatible donor [11]. For patients with a favorable prognosis, consolidation chemotherapy may be the preferred choice [8]. For the intermediate or adverse prognostic groups, HSCT remains the most effective long-term strategy [8]. Whether HSCT should be preceded by a consolidation course is still open for debate, particularly with the incorporation of measurable residual disease in clinical practice, with evidence suggesting either no benefit in patients who achieve CR after induction [12, 13] or an improvement in OS with a consolidation course prior to HSCT [14].
Patients unfit to receive standard chemotherapy may be treated with low dosages of cytarabine (LDAC) or hypomethylating agents (HMA) [15, 16]. LDAC have been the standard of care for patients ineligible to intensive therapy for more than 30 years. Although with CR rates around 20% and median overall survival (OS) around 90 days, complete responders seem to derive a benefit, with overall survival exceeding 18 months [16], which has led to the use of LDAC in several clinical trials assessing the efficacy of new drugs [17,18,19].
HMA are used as a standard treatment for patients with high-risk myelodysplastic syndromes (MDS) [20], and some of them have already been approved by the international regulatory agencies or are in an advanced phase of clinical trials to treat AML.
Despite the available treatments, a 5-year survival is only achieved by 40–45% of young patients and by less than 10% of elderly patients [21]. The development of new therapies is therefore of utmost importance for the reduction of refractory/resistant AML cases and to increase the quality of life and survival of patients [22, 23].
The aim of this review was to summarize the scientific literature on the use of HMA for AML, either approved or in clinical trials. We have reviewed HMA mechanisms of action and main results from clinical trials with AML patients, firstly focusing on decitabine and azacitidine and then newer HMA.
DNA methylation
DNA methylation consists in the enzymatic addition of a methyl (-CH3) group at the C5 position in cytosine [24]. The addition is catalyzed by the action of three enzymes: DNA methyltransferase 1 (DNMT1) – for the maintenance of methylation; and DNMT3A and DNMT3B – for de novo methylation [25].
Demethylation can occur through oxidation reactions catalyzed by the enzyme ten-eleven translocation (TET), more specifically the subtypes TET1, TET2, and TET3 [26, 27].
Cancer cells exhibit an abnormal methylation pattern which involves global hypomethylation and hypermethylation of promoter regions [28, 29]. In addition, mutations in the DNMT3A gene are present in approximately 10–25% of AML patients [7]. The most frequent mutation occurs at arginine 882 (R882), which is associated with increased proliferation of hematopoietic stem cells and resistance to conventional anthracycline chemotherapy [30].
Hypomethylating agents
DNMT inhibitors
The first DNMT inhibitors were studied in animal models for hematological diseases in the 1960s [31,32,33], and since then, research on this pharmacological class has been intensified, leading to the discovery of their multiple antineoplastic effects. These inhibitors were initially used as cytotoxic agents at high doses [34, 35] and later as low-dose epigenetic modifying agents [36,37,38].
Decitabine
Decitabine is a Food and Drug Administration (FDA)-approved drug for the treatment of adult patients with newly diagnosed or secondary AML.
Decitabine is a pro-drug analogous to nucleoside cytosine that generates the active metabolite decitabine triphosphate [36]. The metabolite is incorporated into the DNA during the S phase of the cell cycle for the formation of a covalent adduct with the DNMT enzymes and consequent inhibition of their activity. In addition, decitabine interferes in the synthesis of new DNA, damaging the cell proliferation and causing apoptosis [39]. Reduction of angiogenesis and accumulation of reactive oxygen species have also been reported [39, 40].
Clinical trials
Kantarjian et al. conducted a Phase 3 clinical trial of decitabine in AML elderly patients. Between January 2006 and April 2009, four hundred and eighty-five patients aged 65 or older were randomized into the decitabine group (n = 242) or the physician’s treatment of choice (TC) group (n = 243: 215 for cytarabine and 28 for supportive care) [41].
Two analyses were performed at different time points, both with the participants being evaluated by intention to treat (ITT). The first analysis occurred in October 2009 with a median OS in the decitabine group of 7.7 months (95% CI, 6.2 to 9.2 months) compared to median OS in the TC group of 5 months (95% CI, 4.3 to 6.3 months, p = 0.108) and Hazard Ratio (HR) for death of 0.85 (95% CI, 0.69 to 1.04). In a second analysis with mature survival and updated safety data (October 2010), a statistically and clinically significant reduction in risk of death of 18% was observed in the decitabine group (HR = 0.82, 95% CI, 0.68–0.99, p-nominal = 0.0373). In addition, complete remission (CR) rate plus the CR rate without complete platelet recovery (CRp) was 17.8% in the decitabine group in comparison to 7.8% in the TC group. The incidence and severity of adverse events were similar between both groups, most of them related to myelosuppression [41].
Welch et al. evaluated the relationship between clinical response and mutation status in 84 AML or MDS patients. An extension cohort was included with 32 adult individuals with AML (24 and 8 patients received decitabine 20 mg/m2 respectively for 10 days and 5 days in 28-day cycles) [42].
The CR rate and overall response rate (ORR) were reached by 15 (13%) and 53 (46%) patients, respectively. Exome or targeted gene panel sequencing was performed in 99 of the 116 patients. All 21 patients with TP53 mutation presented clearance of bone marrow blasts (complete remission, complete remission with incomplete count recovery, or morphological leukemia-free state) compared to 32 of 78 patients (41%) with wild-type TP53 (p < 0.001). Patients with unfavorable cytogenetic risk also showed a higher response rate compared to patients with intermediate or favorable cytogenetic risk (67% vs. 34%, p < 0.001). However, responses were not durable, and relapse in patients with TP53 mutation occurred with the outgrowth of a preexisting subclone. The most common adverse events were related to neutropenia and thrombocytopenia [42]. Overall, decitabine treatment seemed effective and well tolerated in patients who do not tolerate intensive chemotherapy or who harbored TP53 mutation. Observational studies will be important to confirm these findings in a real-life setting.
Regarding maintenance therapy, Blum et al. [43] evaluated decitabine in young adults in complete remission who had previously been treated with induction or consolidation chemotherapy. One hundred and thirty-four patients received decitabine, and most of them presented favorable or intermediate cytogenetic risk. Despite severe and frequent adverse events, there was no death-related toxicity. In addition, even with 79% of the patients presenting grade 4 neutropenia, only 4% had grade ≥ 3 infections. The authors then concluded that, overall, patients tolerated the therapy well. Disease-free survival (DFS) in 1 year was 79% (95% CI, 71–85%) and in 3 years was 54% (45–62%); OS was 96% (90–98%) in 1 year and 68% (59–75%) in 3 years. When compared with a historical reference group, decitabine as maintenance therapy showed no apparent benefit. According to the authors, the chosen dosage may have diminished the efficacy of the drug [43]. It is worth pointing out that the treatment schedule according to the package label is 20 mg /m2 in 5 days, repeated every 4 weeks, and the schedule used in the study was 4–5 days of 20 mg/m2decitabine every 6 weeks.
He et al. conducted a meta-analysis about the efficacy and safety of decitabine as monotherapy in previously untreated AML patients over 60 years old. Nine clinical studies, published between 2010 and 2016, with different therapeutic regimens were included (a total of 718 patients). Myelosuppression was the most common adverse event. Infection rate was 36%, and the rate of early death (ED) was 7% at 30 days and 17% at 60 days [44].
The combined estimate of the studies resulted in values of 27% of CR (95% CI, 19–36%), ORR of 37% (95% CI, 28%–47%), and OS of 8.09 months (95% CI, 5.77–10.41). The outcomes varied according to the dosage adopted, and the administration of decitabine for 10 days every 4 weeks presented the best responses. The CR rate in this group was 45% (95% CI, 37–54%), ORR of 53% (95% CI, 37–70%), and OS of 11.30 months (CI 95%, 8.26–14.34). Subgroup analysis by age, cytogenetic risk (intermediate versus adverse), type of AML (de novo versus secondary), and percentage of bone marrow blasts (< 30% versus > 30%) revealed no statistical differences, indicating a homogeneous action of decitabine. A limitation of the study was lack of comparative data with conventional therapy (low-dose chemotherapy) [44].
Azacitidine
Azacitidine is a cytidine nucleotide analog, a substrate of kinases for the generation of active metabolites: 5-azacitidine-5′-triphosphate (5-aza-CTP) and decitabine triphosphate (5-azaC – dCTP) [45]. Unlike decitabine, azacitidine also has effects on RNA. Its action is related to the inhibition of the methylation of the tRNA by the decrease in the levels of tRNA methyltransferase (DNMT2), impacting the protein synthesis and causing apoptosis [46, 47].
Clinical trials
We have selected two multicenter Phase 3 studies employing azacitidine subcutaneously in AML.
Fenaux et al. evaluated azacitidine effects in a trial including 358 patients with high-risk MDS (maximum 30% bone marrow blasts) or chronic myelomonocytic leukemia (CMMoL). However, according to the WHO classification for AML (starting from 20% of blasts in the bone marrow), approximately one-third of the selected patients had in fact AML with low blast counts. One hundred and seventy-nine subjects were randomized to receive azacitidine, and the same number of patients received conventional treatment (supportive care, low-dose cytarabine, or intensive chemotherapy). The median age of the patients was 69 years, with a predominance of elderly individuals over 65 years and intermediate or adverse cytogenetic risk [48].
Considering the entire group (MDS, CMMoL, and AML patients), the median OS was 24.5 months for the azacitidine group and 15 months for the conventional treatment group, resulting in a difference of 9.4 months with a hazard ratio for OS of 0.58 (95% CI, 0.43–0.77, p = 0.0001). HMA benefits were observed in the three cytogenetic groups: favorable, intermediate, and poor risk. Time to disease progression, relapse after complete or partial remission, and death were statistically longer in the test group (median of 14.1 months, IQR 4.2–27.6) than in the control group (median 8.8 months, 3.8 – not reached; log-rank p = 0.047). Neutropenia, thrombocytopenia and anemia were the most common grade 3 or 4 adverse events. The authors concluded that the HMA therapy was effective in patients with MDS and AML with less than 30% of blasts. In the subgroup analysis of treatment (azacitidine vs. supportive care, azacitidine vs. low-dose cytarabine, and azacitidine vs. intensive chemotherapy), azacitidine showed a statistically significant difference in OS when compared to the low-dose cytarabine and supportive care. Azacitidine showed no statistical difference in OS and CR compared to intensive chemotherapy; however, the number of patients in this group was reduced (n = 25), limiting the power of the comparison [48].
Dombret et al. conducted a study to evaluate the effects of azacitidine in 488 elderly AML patients with blast count above 30% and intermediate or adverse cytogenetic risk. Two hundred and forty-one patients were randomized to receive the test drug, and 247 to receive the conventional treatment (supportive care, standard induction, or low-dose cytarabine chemotherapy) [49].
The median OS for azacitidine group was 10.4 months (95% CI, 8.0–12.7 months) compared to 6.5 months in the control group (95% CI, 5.0–8.6 months) (HR = 0.85, 95% CI, 0.69–1.03, p = 0.1009). In a second analysis, in which patients who received subsequent therapies were censored, the median OS in the first group was 12.1 months (95% CI, 9.2–14.2 months) vs. 6.9 months (95% CI, 5.1–9.6 months) in the second group (stratified HR, 0.76; 95% CI, 0.60–0.96; P = 0.0190). The 1-year survival rate was 50.7% with azacitidine vs. 37.7% for conventional treatment, with a difference of 13% (95% CI, 3.3%–22.7%). The CR + CR rate with incomplete recovery of blood cells was 27.8% vs. 25.1% with no statistical difference. The drug was considered well tolerated. Importantly, in the univariate and multivariate regression analysis, the median OS of patients with adverse risk was twice the observed in the conventional treatment group (6.4 vs. 3.2 months, p = 0.0185). Overall, the authors concluded that azacitidine induced a clinically significant improvement and may be an additional treatment option for elderly patients [49].
The data presented above emphasizes how azacitidine and decitabine use in clinical practice represented a progress in the treatment of AML, particularly in the elderly population. They were the first drugs in AML to target epigenetic mechanisms, with excellent responses in subsets of high-risk patients, such as those with TP53 mutations. However, these responses have not been sustained, and most patients have not achieved a complete hematological response. Furthermore, azacitidine and decitabine have not been directly compared against each other in a randomized controlled trial in the setting of AML. Combination therapy of HMAs with recently approved agents, including epigenetically targeted therapies, is a promising approach which has yet to be tested in clinical trials.
Venetoclax (ABT-199) plus decitabine or azacitidine
Venetoclax is an oral inhibitor of the proapoptotic protein B cell lymphoma 2 (BCL-2) that has showed a synergistic effect when combined to azacitidine in myeloid cells in vitro [50].
Clinical trials
A multicenter, phase 1b dose-escalation and expansion study included 145 elderly treatment-naive AML patients and ineligible for intensive chemotherapy to evaluate the combination of venetoclax with decitabine or azacitidine. Decitabine (20 mg/m2 for 5 days) or azacitidine (75 mg/m2 for 7 days) was administered with venetoclax (at doses of 400, 800, or 1200 mg per day). The rate of CR + CR with incomplete count recovery was 67% in all venetoclax doses and 73% in the venetoclax 400 mg plus HMA cohort. The median OS was 17.5 months (12.3 – not reached) for all patients, and it was not reached (NR) (11.0 – NR) for the venetoclax 400 mg + HMA cohort. The most common adverse events were gastrointestinal and hematological. The association was considered well tolerated and effective [51].
Due to the promising results and important unmet medical need, FDA granted accelerated approval for venetoclax + azacitidine or decitabine or low-dose cytarabine for elderly patients with AML (≥ 75 years) or who have comorbidities that preclude the use of intensive induction chemotherapy. It is of utmost importance to validate these findings in a randomized, controlled clinical trial, and two Phase 3 trials are ongoing (NCT02993523 and NCT03069352).
Guadecitabine (SG1-110)
Guadecitabine is a second generation HMA structurally developed to be more resistant to cytidine deaminase (responsible for HMA degradation) and consequently increase the rate of responders and treatment efficacy [52].
Clinical trials
An open-label, randomized, Phase 1 trial evaluated the safety and the most appropriate posology for the treatment of patients with MDS (n = 19) or refractory or relapsed AML (n = 74) after standard treatment by dose escalation. The study showed that guadecitabine was generally well tolerated, and the most appropriate regimens chosen for the Phase 2 study were 60 or 90 mg/m2/day for 5 consecutive days in 28-day cycles [53].
A multicenter, open-label, randomized, Phase 2 trial evaluated different dosages of guadecitabine in treatment-naïve elderly patients (≥ 65 years) with relapsed or refractory MDS or AML who did not tolerate standard chemotherapy. The subjects were randomized to receive guadecitabine 60 mg/m2 during 5 days (n = 26); guadecitabine 90 mg/m2 (n = 28) during 5 days; or guadecitabine 60 mg/m2 during 10 days (n = 53) [54].
Both groups of 5 and 10 days presented similar OS (10.5 vs. 9.5 months, respectively); however, the 5-day regimen group appeared to have benefited more due to the lower incidence of serious adverse events [54].
A Phase 3 randomized study (NCT02348489) comparing guadecitabine to treatment choice (azacitidine, decitabine, or low-dose ARA-C) has recently been completed. The trial included 815 treatment-naïve AML patients not eligible for intense chemotherapy. It failed to meet the primary endpoints of statistically significant advantage of guadecitabine over treatment choice for CR rate and OS, suggesting that guadecibine has an overall similar efficacy and safety compared to standard therapy [55].
In the two arms, the patients received a median of five treatment cycles. However, many patients (approximately 40% in both arms) received ≤ 3 treatment cycles mainly due to early death or progression. Survival analysis of the patients who received more than 3 treatment cycles showed a potential benefit of guadecitabine vs. treatment choice. Median, 1-year OS, and 2-year OS were 15.6 months, 60 and 29% on the guadecitabine group versus 13 months, 52 and 20% on the treatment choice group; log-rank p value = 0.02, HR 0.78, 95%CI, 0.64–0.96. A potential benefit was also observed in patients who achieved any CR (CR, CRp, or CRi) [55].
Other Phase 3 trials with guadecitabine (SGI-110) in patients with relapsed and refractory acute myeloid leukemia are in progress.
Although the aformentioned Phase 3 trial failed, it may be possible that some specific group could benefit from the drug. Therefore, further studies will be important to elucidate this issue.
IDH inhibitors
IDH1 and IDH2 are considered important therapeutic targets since mutations in these genes are found in approximately 20% of AML patients [56]. IDH inhibitors aim to reduce the amount of mutant IDH proteins and, consequently, reducing the levels of the oncometabolite 2-hydroxyglutarate (2-HG). The conversion of a-ketoglutarate (α-KG) to 2-HG oncometabolite by mutant IDH proteins inhibits α-ketoglutarate-dependent enzymes such as TET proteins that mediate DNA demethylation, indirectly leading to a dysregulation of methylation [57].
Enasidenib (IDH2 inhibitor) and ivosidenib (IDH1 inhibitor) have already been approved by FDA. Other IDH inhibitors are in initial clinical development.
Enasidenib
Enasidenib (AG-221) is an oral, first-in-class IDH2 inhibitor which is currently undergoing Phase 3 clinical trials (clinicaltrials.gov). Due to promising results, the FDA has granted the approval of Idhifa® (enasidenib) registry in August 2017 for patients with relapsed or refractory AML harboring the mutant gene [58].
The first clinical study was an open-label phase 1/2 trial to assess safety and dosage. The dose-escalation phase included 113 adult patients with AML or MDS with refractory anemia and excess blasts harboring an IDH2 mutation. The maximum tolerated dose (MTD) was not reached. However, 5 out of 7 patients in the 650 mg dose group presented significant adverse events, which led to dose reduction. Based on the pharmacokinetics, clinical activity, and inhibition of 2-HG levels, the dose of 100 mg once a day was chosen to be administered during the second part of the study (expansion phase) [59]
The expansion phase included 126 patients divided into four cohorts: (1) elderly (> 60 years) with recurrent or refractory AML or at any age when relapsing after HSCT; (2) patients < 60 years with refractory disease or relapse without history of HSCT; (3) elderly (> 60 years) with untreated AML and ineligible for induction chemotherapy; and (4) patients ineligible for the previous groups. Considering all the participants in the study (n = 239), the majority presented intermediate cytogenetic risk with ECOG performance between 0 and 2 and median age of 70 years [59].
All 239 patients were evaluated for safety outcomes. Treatment-related grades 3 to 4 adverse events were hyperbilirubinemia, hematological events, differentiation syndrome associated with the IDH inhibitor (IDH-SD), and infections. Clinical outcomes were described for the relapsed or refractory AML subgroup. In this subgroup, ORR was achieved by 40.3% (95% CI, 33.0–48.0%) of the patients (considering Phases 1 and 2) and 38.5% (95% CI, 29.4–48.3%) of the patients who received a dose of 100 mg per day. Median OS was 9.3 months (95% CI, 8.2–10.9 months). Importantly, the results suggested that enasidenib exerts activity on both IDH2 mutations (IDH2-R140 and IDH2-R172) [59].
Ten percent of the patients proceeded to bone marrow transplantation. In a subsequent analysis, clinical The authors believe that the drug could be a bridge for a potential cure. It is noteworthy that the majority of patients who exhibited reduced levels of 2-HG did not reach clinical response, suggesting that 2-HG decrease is not a predictive of response. Hematologic toxicity was low and may be an advantage over intensive chemotherapy and other treatments [59].
In a subsequent analysis, clinical response to enasidenib reflected the number of mature neutrophils and a low index of infection. In addition, patients who achieved CR or PR harbored fewer mutations than the nonresponders (p < 0.001) and individuals with mutations in NRAS achieved less CR (p = 0.0114), whereas patients with mutations in the MAPK signaling pathway (mPTPN11) did not respond to treatment [60]. Other clinical trials, including Phase 3 are ongoing (clinicaltrials.gov).
Enasidenib has some advantages compared to the standard chemotherapy, presenting clinical response with differentiation of malignant cells and oral form. It is the first targeted therapy for AML with IDH2 mutation, representing an important step in the precision medicine and a possible adjuvant to chemotherapy in some patients. Importantly, part of the patients proceeded to bone marrow transplantation. Despite the promising results obtained in this Phase 2 trial, a randomized and controlled Phase 3 trial remains to be completed. There are some ongoing clinical trials evaluating the association of enasidenib with other drugs, which will be useful to determine potential synergistic effects.
Ivosidenib
The first-in class small molecule IDH1 inhibitor ivosidenib (AG-120) received FDA approval for the treatment of relapsed or refractory AML with susceptible IDH1 mutation after the results achieved in a Phase 1 trial [61, 62]. The study was multicenter, non-randomized, open label, single arm, dose escalation, and dose expansion. Two hundred and sixty-eight patients were enrolled, of which 179 had relapsed or refractory AML and received the starting dose of 500 mg of ivosidenib daily [61].
Ivosidenib was quickly absorbed and the mean plasma 2-HG levels reached similar levels of healthy individuals after multiple doses of 500 mg daily. MTD was not defined. The majority (98.9%) of the patients with relapse or refractory AML presented an adverse event, including IDH differentiation syndrome, grade 3 or higher leukocytosis and a serious adverse event of prolongation of the QT interval. No treatment-related adverse event leading to death was observed in these patients [61].
CR or CR with partial hematologic recovery (CRh) was reached by 30.2% (95% CI, 23.5–37.5) of the 179 patients with relapse or refractory AML. Median length of response was 6.5 months (95% CI, 5.5–11.1) and median time to reach CR or CRh was 2 months (range: 0.9–5.6 months). CR or CRh responders showed decreased levels of IDH1 mutations in bone marrow mononuclear cells and neutrophils during the course. All the 7 patients with clearance of IDH1 mutations achieved complete remission [61]. Transfusion independence was achieved by approximately 37% of the patients who previously presented red blood cell and/or platelet transfusion-dependence [61].
In conclusion, ivosidenib monotherapy was considered well tolerated and effective for relapsed or refractory AML patients harboring IDH1 mutation. Like enasidenib, this is an oral first-in class drug that represents a major advance in the targeted AML treatment. QT prolongation and IDH differentiation syndrome must be monitored during treatment. Randomized, comparative Phase 3 trials are necessary to confirm the efficacy and safety. Double-blind, randomized and placebo-controlled Phase 3 trials are ongoing to evaluate the combination of ivosidenib with other drugs (clinicaltrials.gov).
Conclusion
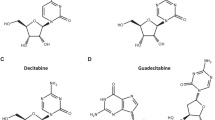
Dysregulation of DNA methylation is a hallmark of AML and numerous preclinical and clinical studies testing DNA methylation modifiers in this malignancy have been conducted, including hypomethylating agents and IDH inhibitors. Here we have briefly reviewed the mechanism of action (Fig. 1) and the results from clinical trials of different HMA and IDH inibitors in AML patients.

Mechanism of action of the reviewed HMA and IDH inhibitors. a Decitabine, azacitidine and guadecitabine metabolites are incorporated into the DNA to form an adduct with DNMT, inhibiting the DNA methylation. b Azacitidine metabolite incorporates into tRNA and also inhibits tRNA methylation. c Ivosidenib and d enasidenib respectively inhibit the mutated forms of IDH1 and IDH2 (IDH1m and IDH2m), which induce the abnormal production of the oncometabolite 2-hydroxyglutarate (2HG)
HMA have demonstrated efficacy in clinical studies, prolonging survival when compared to low-dose chemotherapy or best supportive care. The combination of HMA plus venetoclax yielded a high response rate in a Phase 1b trial with older AML patients ineligible for intense chemotherapy. IDH inhibitors were also effective for relapse or refractory AML patients in a Phase 1/2 trial. The drugs herein being evaluated presented several adverse events; nevertheless, they were generally considered tolerable.
Further clinical trials are needed for a clear evaluation of HMA and IDH inhibitors in AML. Considering the unique mutation profile of each patient, the future approach to AML and other cancers is more likely to be through the combination of different therapeutic classes, in order to target diverse molecular pathways and prevent the emergence of pharmacological resistance.
References
van den Boom V, Horton SJ, Schuringa JJ (2012) Genetic and epigenetic alterations that drive leukemic stem cell self-renewal. J Stem Cells 7(3):155–179
Howlader N NA, Krapcho M, Miller D, Brest A, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, Chen HS, Feuer EJ, Cronin KA (eds) SEER Cancer Statistics Review, 1975-2016, National Cancer Institute.Bethesda, MD, https://seer.cancer.gov/csr/1975_2016/, based on November 2018 SEER data submission, posted to the SEER web site, April 2019
Siegel RL, Miller KD, Jemal A (2015) Cancer statistics, 2015. CA Cancer J Clin 65(1):5–29. https://doi.org/10.3322/caac.21254
Mrozek K, Marcucci G, Nicolet D, Maharry KS, Becker H, Whitman SP, Metzeler KH, Schwind S, Wu YZ, Kohlschmidt J, Pettenati MJ, Heerema NA, Block AW, Patil SR, Baer MR, Kolitz JE, Moore JO, Carroll AJ, Stone RM, Larson RA, Bloomfield CD (2012) Prognostic significance of the European LeukemiaNet standardized system for reporting cytogenetic and molecular alterations in adults with acute myeloid leukemia. J Clin Oncol 30(36):4515–4523. https://doi.org/10.1200/JCO.2012.43.4738
Dohner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Buchner T, Dombret H, Ebert BL, Fenaux P, Larson RA, Levine RL, Lo-Coco F, Naoe T, Niederwieser D, Ossenkoppele GJ, Sanz M, Sierra J, Tallman MS, Tien HF, Wei AH, Lowenberg B, Bloomfield CD (2017) Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 129(4):424–447. https://doi.org/10.1182/blood-2016-08-733196
Fennell KA, Bell CC, Dawson MA (2019) Epigenetic therapies in acute myeloid leukemia: where to from here? Blood 134(22):1891–1901. https://doi.org/10.1182/blood.2019003262
Ohgami RS, Arber DA (2015) The diagnostic and clinical impact of genetics and epigenetics in acute myeloid leukemia. Int J Lab Hematol 37(Suppl 1):122–132. https://doi.org/10.1111/ijlh.12367
Saultz JN, Garzon R (2016) Acute myeloid leukemia: a concise review. J Clin Med 5(3). https://doi.org/10.3390/jcm5030033
Dombret H, Gardin C (2016) An update of current treatments for adult acute myeloid leukemia. Blood 127(1):53–61. https://doi.org/10.1182/blood-2015-08-604520
Thol F (2018) Can we forecast induction failure in acute myeloid leukemia? Haematologica 103(3):375–377. https://doi.org/10.3324/haematol.2018.187575
Medinger M, Lengerke C, Passweg J (2016) Novel prognostic and therapeutic mutations in acute myeloid leukemia. Cancer Genomics Proteomics 13(5):317–329
Tallman MS, Rowlings PA, Milone G, Zhang MJ, Perez WS, Weisdorf D, Keating A, Gale RP, Geller RB, Laughlin MJ, Lazarus HM, Luger SM, McCarthy PL, Rowe JM, Saez RA, Vowels MR, Horowitz MM (2000) Effect of postremission chemotherapy before human leukocyte antigen-identical sibling transplantation for acute myelogenous leukemia in first complete remission. Blood 96(4):1254–1258
Warlick ED, Paulson K, Brazauskas R, Zhong X, Miller AM, Camitta BM, George B, Savani BN, Ustun C, Marks DI, Waller EK, Baron F, Freytes CO, Socie G, Akpek G, Schouten HC, Lazarus HM, Horwitz EM, Koreth J, Cahn JY, Bornhauser M, Seftel M, Cairo MS, Laughlin MJ, Sabloff M, Ringden O, Gale RP, Kamble RT, Vij R, Gergis U, Mathews V, Saber W, Chen YB, Liesveld JL, Cutler CS, Ghobadi A, Uy GL, Eapen M, Weisdorf DJ, Litzow MR (2014) Effect of postremission therapy before reduced-intensity conditioning allogeneic transplantation for acute myeloid leukemia in first complete remission. Biol Blood Marrow Transplant 20(2):202–208. https://doi.org/10.1016/j.bbmt.2013.10.023
Rashidi A, Linden MA, DeFor TE, Warlick E, Bejanyan N, Yohe S, Weisdorf DJ, Ustun C (2017) History of consolidation is prognostic in acute myeloid leukemia patients undergoing allogeneic hematopoietic cell transplantation in minimal residual disease-negative first complete remission. Am J Hematol 92(10):1032–1036. https://doi.org/10.1002/ajh.24834
Gardin C, Dombret H (2017) Hypomethylating agents as a therapy for AML. Curr Hematol Malig Rep 12(1):1–10. https://doi.org/10.1007/s11899-017-0363-4
Burnett AK, Milligan D, Prentice AG, Goldstone AH, McMullin MF, Hills RK, Wheatley K (2007) A comparison of low-dose cytarabine and hydroxyurea with or without all-trans retinoic acid for acute myeloid leukemia and high-risk myelodysplastic syndrome in patients not considered fit for intensive treatment. Cancer 109(6):1114–1124. https://doi.org/10.1002/cncr.22496
Burnett AK, Hills RK, Hunter AE, Milligan D, Kell WJ, Wheatley K, Yin J, McMullin MF, Dignum H, Bowen D, Russell NH (2013) The addition of gemtuzumab ozogamicin to low-dose Ara-C improves remission rate but does not significantly prolong survival in older patients with acute myeloid leukaemia: results from the LRF AML14 and NCRI AML16 pick-a-winner comparison. Leukemia 27(1):75–81. https://doi.org/10.1038/leu.2012.229
Cortes JE, Heidel FH, Hellmann A, Fiedler W, Smith BD, Robak T, Montesinos P, Pollyea DA, DesJardins P, Ottmann O, Ma WW, Shaik MN, Laird AD, Zeremski M, O'Connell A, Chan G, Heuser M (2019) Randomized comparison of low dose cytarabine with or without glasdegib in patients with newly diagnosed acute myeloid leukemia or high-risk myelodysplastic syndrome. Leukemia 33(2):379–389. https://doi.org/10.1038/s41375-018-0312-9
Dennis M, Russell N, Hills RK, Hemmaway C, Panoskaltsis N, McMullin MF, Kjeldsen L, Dignum H, Thomas IF, Clark RE, Milligan D, Burnett AK (2015) Vosaroxin and vosaroxin plus low-dose Ara-C (LDAC) vs low-dose Ara-C alone in older patients with acute myeloid leukemia. Blood 125(19):2923–2932. https://doi.org/10.1182/blood-2014-10-608117
Gil-Perez A, Montalban-Bravo G (2019) Management of myelodysplastic syndromes after failure of response to hypomethylating agents. Ther Adv Hematol 10:2040620719847059. https://doi.org/10.1177/2040620719847059
Siveen KS, Uddin S, Mohammad RM (2017) Targeting acute myeloid leukemia stem cell signaling by natural products. Mol Cancer 16(1):13. https://doi.org/10.1186/s12943-016-0571-x
Przespolewski A, Szeles A, Wang ES (2018) Advances in immunotherapy for acute myeloid leukemia. Future Oncol 14(10):963–978. https://doi.org/10.2217/fon-2017-0459
Wu M, Li C, Zhu X (2018) FLT3 inhibitors in acute myeloid leukemia. J Hematol Oncol 11(1):133. https://doi.org/10.1186/s13045-018-0675-4
Chowdhury B, Cho IH, Irudayaraj J (2017) Technical advances in global DNA methylation analysis in human cancers. J Biol Eng 11:10. https://doi.org/10.1186/s13036-017-0052-952
Schulze I, Rohde C, Scheller-Wendorff M, Baumer N, Krause A, Herbst F, Riemke P, Hebestreit K, Tschanter P, Lin Q, Linhart H, Godley LA, Glimm H, Dugas M, Wagner W, Berdel WE, Rosenbauer F, Muller-Tidow C (2016) Increased DNA methylation of Dnmt3b targets impairs leukemogenesis. Blood 127(12):1575–1586. https://doi.org/10.1182/blood-2015-07-655928
Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A (2009) Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324(5929):930–935. https://doi.org/10.1126/science.1170116
Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y (2011) Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333(6047):1300–1303. https://doi.org/10.1126/science.1210597
Gama-Sosa MA, Slagel VA, Trewyn RW, Oxenhandler R, Kuo KC, Gehrke CW, Ehrlich M (1983) The 5-methylcytosine content of DNA from human tumors. Nucleic Acids Res 11(19):6883–6894
Esteller M (2000) Epigenetic lesions causing genetic lesions in human cancer: promoter hypermethylation of DNA repair genes. Eur J Cancer 36(18):2294–2300
Guryanova OA, Shank K, Spitzer B, Luciani L, Koche RP, Garrett-Bakelman FE, Ganzel C, Durham BH, Mohanty A, Hoermann G, Rivera SA, Chramiec AG, Pronier E, Bastian L, Keller MD, Tovbin D, Loizou E, Weinstein AR, Gonzalez AR, Lieu YK, Rowe JM, Pastore F, McKenney AS, Krivtsov AV, Sperr WR, Cross JR, Mason CE, Tallman MS, Arcila ME, Abdel-Wahab O, Armstrong SA, Kubicek S, Staber PB, Gonen M, Paietta EM, Melnick AM, Nimer SD, Mukherjee S, Levine RL (2016) DNMT3A mutations promote anthracycline resistance in acute myeloid leukemia via impaired nucleosome remodeling. Nat Med 22(12):1488–1495. https://doi.org/10.1038/nm.4210
Sorm F, Vesely J (1964) The activity of a new antimetabolite, 5-Azacytidine, against lymphoid Leukaemia in Ak mice. Neoplasma 11:123–130
Vesely J, Sorm F (1965) The Cytologic and the metabolic effects of a new antileukemic analogue 5-Azacytidine in Normal mice followed autoradiographically with tritium. Neoplasma 12:3–9
Sorm F, Vesely J (1968) Effect of 5-aza-2′-deoxycytidine against leukemic and hemopoietic tissues in AKR mice. Neoplasma 15(4):339–343
Sorm F, Piskala A, Cihak A, Vesely J (1964) 5-Azacytidine, a new, highly effective cancerostatic. Experientia 20(4):202–203
Sato T, Issa JJ, Kropf P (2017) DNA Hypomethylating drugs in Cancer therapy. Cold Spring Harb Perspect Med 7(5). https://doi.org/10.1101/cshperspect.a026948
Momparler RL (2005) Pharmacology of 5-Aza-2′-deoxycytidine (decitabine). Semin Hematol 42(3 Suppl 2):S9–S16
Attadia V (1993) Effects of 5-aza-2′-deoxycytidine on differentiation and oncogene expression in the human monoblastic leukemia cell line U-937. Leukemia 7(Suppl 1):9–16
de Vos D (2005) Epigenetic drugs: a longstanding story. Semin Oncol 32(5):437–442. https://doi.org/10.1053/j.seminoncol.2005.07.025
Nieto M, Demolis P, Behanzin E, Moreau A, Hudson I, Flores B, Stemplewski H, Salmonson T, Gisselbrecht C, Bowen D, Pignatti F (2016) The European medicines agency review of decitabine (Dacogen) for the treatment of adult patients with acute myeloid leukemia: summary of the scientific assessment of the Committee for Medicinal Products for human use. Oncologist 21(6):692–700. https://doi.org/10.1634/theoncologist.2015-0298
Fandy TE, Jiemjit A, Thakar M, Rhoden P, Suarez L, Gore SD (2014) Decitabine induces delayed reactive oxygen species (ROS) accumulation in leukemia cells and induces the expression of ROS generating enzymes. Clin Cancer Res 20(5):1249–1258. https://doi.org/10.1158/1078-0432.CCR-13-1453
Kantarjian HM, Thomas XG, Dmoszynska A, Wierzbowska A, Mazur G, Mayer J, Gau JP, Chou WC, Buckstein R, Cermak J, Kuo CY, Oriol A, Ravandi F, Faderl S, Delaunay J, Lysak D, Minden M, Arthur C (2012) Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J Clin Oncol 30(21):2670–2677. https://doi.org/10.1200/JCO.2011.38.9429
Welch JS, Petti AA, Miller CA, Fronick CC, O'Laughlin M, Fulton RS, Wilson RK, Baty JD, Duncavage EJ, Tandon B, Lee YS, Wartman LD, Uy GL, Ghobadi A, Tomasson MH, Pusic I, Romee R, Fehniger TA, Stockerl-Goldstein KE, Vij R, Oh ST, Abboud CN, Cashen AF, Schroeder MA, Jacoby MA, Heath SE, Luber K, Janke MR, Hantel A, Khan N, Sukhanova MJ, Knoebel RW, Stock W, Graubert TA, Walter MJ, Westervelt P, Link DC, DiPersio JF, Ley TJ (2016) TP53 and Decitabine in acute myeloid leukemia and Myelodysplastic syndromes. N Engl J Med 375(21):2023–2036. https://doi.org/10.1056/NEJMoa1605949
Blum W, Sanford BL, Klisovic R, DeAngelo DJ, Uy G, Powell BL, Stock W, Baer MR, Kolitz JE, Wang ES, Hoke E, Mrozek K, Kohlschmidt J, Bloomfield CD, Geyer S, Marcucci G, Stone RM, Larson RA (2017) Maintenance therapy with decitabine in younger adults with acute myeloid leukemia in first remission: a phase 2 cancer and leukemia group B study (CALGB 10503). Leukemia 31(1):34–39. https://doi.org/10.1038/leu.2016.252
He PF, Zhou JD, Yao DM, Ma JC, Wen XM, Zhang ZH, Lian XY, Xu ZJ, Qian J, Lin J (2017) Efficacy and safety of decitabine in treatment of elderly patients with acute myeloid leukemia: a systematic review and meta-analysis. Oncotarget 8(25):41498–41507. https://doi.org/10.18632/oncotarget.17241
Li LH, Olin EJ, Buskirk HH, Reineke LM (1970) Cytotoxicity and mode of action of 5-azacytidine on L1210 leukemia. Cancer Res 30(11):2760–2769
Lu LJ, Randerath K (1980) Mechanism of 5-azacytidine-induced transfer RNA cytosine-5-methyltransferase deficiency. Cancer Res 40(8 Pt 1):2701–2705
Schaefer M, Hagemann S, Hanna K, Lyko F (2009) Azacytidine inhibits RNA methylation at DNMT2 target sites in human cancer cell lines. Cancer Res 69(20):8127–8132. https://doi.org/10.1158/0008-5472.CAN-09-0458
Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Finelli C, Giagounidis A, Schoch R, Gattermann N, Sanz G, List A, Gore SD, Seymour JF, Bennett JM, Byrd J, Backstrom J, Zimmerman L, McKenzie D, Beach C, Silverman LR (2009) Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol 10(3):223–232. https://doi.org/10.1016/S1470-2045(09)70003-8
Dombret H, Seymour JF, Butrym A, Wierzbowska A, Selleslag D, Jang JH, Kumar R, Cavenagh J, Schuh AC, Candoni A, Recher C, Sandhu I, Bernal del Castillo T, Al-Ali HK, Martinelli G, Falantes J, Noppeney R, Stone RM, Minden MD, McIntyre H, Songer S, Lucy LM, Beach CL, Dohner H (2015) International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood 126(3):291–299. https://doi.org/10.1182/blood-2015-01-621664
Bogenberger JM, Kornblau SM, Pierceall WE, Lena R, Chow D, Shi CX, Mantei J, Ahmann G, Gonzales IM, Choudhary A, Valdez R, Camoriano J, Fauble V, Tiedemann RE, Qiu YH, Coombes KR, Cardone M, Braggio E, Yin H, Azorsa DO, Mesa RA, Stewart AK, Tibes R (2014) BCL-2 family proteins as 5-Azacytidine-sensitizing targets and determinants of response in myeloid malignancies. Leukemia 28(8):1657–1665. https://doi.org/10.1038/leu.2014.44
DiNardo CD, Pratz K, Pullarkat V, Jonas BA, Arellano M, Becker PS, Frankfurt O, Konopleva M, Wei AH, Kantarjian HM, Xu T, Hong WJ, Chyla B, Potluri J, Pollyea DA, Letai A (2019) Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 133(1):7–17. https://doi.org/10.1182/blood-2018-08-868752
Griffiths EA, Choy G, Redkar S, Taverna P, Azab M, Karpf AR (2013) SGI-110: DNA Methyltransferase inhibitor Oncolytic. Drugs Future 38(8):535–543
Issa JJ, Roboz G, Rizzieri D, Jabbour E, Stock W, O'Connell C, Yee K, Tibes R, Griffiths EA, Walsh K, Daver N, Chung W, Naim S, Taverna P, Oganesian A, Hao Y, Lowder JN, Azab M, Kantarjian H (2015) Safety and tolerability of guadecitabine (SGI-110) in patients with myelodysplastic syndrome and acute myeloid leukaemia: a multicentre, randomised, dose-escalation phase 1 study. Lancet Oncol 16(9):1099–1110. https://doi.org/10.1016/S1470-2045(15)00038-8
Kantarjian HM, Roboz GJ, Kropf PL, Yee KWL, O'Connell CL, Tibes R, Walsh KJ, Podoltsev NA, Griffiths EA, Jabbour E, Garcia-Manero G, Rizzieri D, Stock W, Savona MR, Rosenblat TL, Berdeja JG, Ravandi F, Rock EP, Hao Y, Azab M, Issa JJ (2017) Guadecitabine (SGI-110) in treatment-naive patients with acute myeloid leukaemia: phase 2 results from a multicentre, randomised, phase 1/2 trial. Lancet Oncol 18(10):1317–1326. https://doi.org/10.1016/S1470-2045(17)30576-4
Fenaux P, Gobbi M, Kropf PL, Mayer J, Roboz GJ, Döhner H, Krauter J, Döhner K, Robak T, Kantarjian H, Novak J, Jedrzejczak WW, Thomas X, Ojeda-Uribe M, Miyazaki Y, Min YH, Brandwein J, Gercheva-Kyuchukova L, Demeter J, Griffiths E, Yee K, Azab M, Issa JP theligible for intensive chemotherapy (IC). EHA Library Fenaux P. 267462; S879
Kats LM, Vervoort SJ, Cole R, Rogers AJ, Gregory GP, Vidacs E, Li J, Nagaraja R, Yen KE, Johnstone RW (2017) A pharmacogenomic approach validates AG-221 as an effective and on-target therapy in IDH2 mutant AML. Leukemia 31(6):1466–1470. https://doi.org/10.1038/leu.2017.84
Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, Tallman MS, Sun Z, Wolniak K, Peeters JK, Liu W, Choe SE, Fantin VR, Paietta E, Lowenberg B, Licht JD, Godley LA, Delwel R, Valk PJ, Thompson CB, Levine RL, Melnick A (2010) Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18(6):553–567. https://doi.org/10.1016/j.ccr.2010.11.015
Enasidenib Approved for AML, but Best Uses Unclear (2017) Cancer Discov 7(10):OF4. https://doi.org/10.1158/2159-8290.CD-NB2017-117
Stein EM, DiNardo CD, Pollyea DA, Fathi AT, Roboz GJ, Altman JK, Stone RM, DeAngelo DJ, Levine RL, Flinn IW, Kantarjian HM, Collins R, Patel MR, Frankel AE, Stein A, Sekeres MA, Swords RT, Medeiros BC, Willekens C, Vyas P, Tosolini A, Xu Q, Knight RD, Yen KE, Agresta S, de Botton S, Tallman MS (2017) Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 130(6):722–731. https://doi.org/10.1182/blood-2017-04-779405
Amatangelo MD, Quek L, Shih A, Stein EM, Roshal M, David MD, Marteyn B, Farnoud NR, de Botton S, Bernard OA, Wu B, Yen KE, Tallman MS, Papaemmanuil E, Penard-Lacronique V, Thakurta A, Vyas P, Levine RL (2017) Enasidenib induces acute myeloid leukemia cell differentiation to promote clinical response. Blood 130(6):732–741. https://doi.org/10.1182/blood-2017-04-779447
DiNardo CD, Stein EM, de Botton S, Roboz GJ, Altman JK, Mims AS, Swords R, Collins RH, Mannis GN, Pollyea DA, Donnellan W, Fathi AT, Pigneux A, Erba HP, Prince GT, Stein AS, Uy GL, Foran JM, Traer E, Stuart RK, Arellano ML, Slack JL, Sekeres MA, Willekens C, Choe S, Wang H, Zhang V, Yen KE, Kapsalis SM, Yang H, Dai D, Fan B, Goldwasser M, Liu H, Agresta S, Wu B, Attar EC, Tallman MS, Stone RM, Kantarjian HM (2018) Durable remissions with Ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med 378(25):2386–2398. https://doi.org/10.1056/NEJMoa1716984
Norsworthy KJ, Luo L, Hsu V, Gudi R, Dorff SE, Przepiorka D, Deisseroth A, Shen YL, Sheth CM, Charlab R, Williams GM, Goldberg KB, Farrell AT, Pazdur R (2019) FDA approval summary: ivosidenib for relapsed or refractory acute myeloid leukemia with an isocitrate dehydrogenase-1 mutation. Clin Cancer Res. https://doi.org/10.1158/1078-0432.CCR-18-3749
Acknowledgments
The authors would like to thank Prof Fernando Luiz Affonso Fonseca for valuable discussions.
Funding
Mariana Lazarini gratefully acknowledges funding support from São Paulo Research Foundation, research grant 17/19674–2.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Bruna Contieri declares she has no conflicts of interest. Bruno Kosa Lino Duarte declares he has no conflicts of interest. Mariana Lazarini declares she has no conflicts of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Contieri, B., Duarte, B.K.L. & Lazarini, M. Updates on DNA methylation modifiers in acute myeloid leukemia. Ann Hematol 99, 693–701 (2020). https://doi.org/10.1007/s00277-020-03938-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-020-03938-2