
Abstract
Approximately 10% of patients with polycythemia vera (PV) transform to acute leukemia (blast phase) at 10 years after initial diagnosis of PV. The bone marrow pathologic, cytogenetic, and molecular features of blast phase have not been well characterized. In this study, we reviewed 422 PV patients over a period of 11 years and identified 58 patients who developed acute myeloid leukemia (blast phase) during the course of disease. We found that blast phase of PV was characterized by overt myelodysplasia (n = 51, 88%); moderate to severe myelofibrosis (33 of 45, 73%); an abnormal karyotype (n = 51, 88%) that was often complex karyotype (n = 42, 72%); and gene mutations involving TP53 (55%), TET2 (27%), and DNMT3A (25%). Patients with blast phase of PV had an aggressive clinical course, with a median overall survival of 4 months after onset of blast phase. Eleven patients had close follow-up from polycythemic phase to blast phase: Four patients showed dysplastic changes in the polycythemic phase, and three of them transformed to blast phase without a “middle phase” of post-PV myelofibrosis.We conclude that blast phase of PV is characterized by myelodysplasia, moderate to severe fibrosis, a high frequency of an abnormal and often complex karyotype, and frequentTP53mutation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Polycythemia vera (PV) is the second most common myeloproliferative neoplasm (MPN) [1] characterized by increased red blood cell (RBC) production, somatic gain-of-function mutation of JAK2,and panmyelosis in the bone marrow (BM) [1,2,3].The natural course of PV usually includes pre-polycythemic phase, polycythemic phase (PP), and post-polycythemic myelofibrosis (post-PV MF) phase. The natural progression of PV also includes a low incidence of evolution to a myelodysplastic and/or blast phase (BP) [3]. In earlier reports, within 10 years after initial diagnosis of PV, 2.3~14.4% of patients have transformed to BP or acute myeloid leukemia (AML) with blasts ≥ 20% in peripheral blood (PB) and/or BM [4,5,6,7,8]. Risk factors for BP transformation reported by others include longer disease duration, older age, treatments with P32 or alkylating agents, high leukocyte count, BM myelofibrosis, splenomegaly, an abnormal karyotype, and somatic mutations involving TP53, SRSF2, RUNX1, and IKZF1 [4, 5, 7,8,9,10,11,12,13,14]. Progression to BP is invariably associated with a poor response to treatment and shorter overall survival; the median survival after onset of BP is 3 to 6 months [15, 16].
In the revised World Health Organization (WHO) classification of tumors of hematopoietic and lymphoid tissue [3], the diagnostic criteria for PV were modified substantially. These changes include lowering the hemoglobin or hematocrit thresholds of diagnosis for PV (> 16.5 g/dL or > 49% for men and > 16.0 g/dL or > 48% for women) and upgrading BM morphology (hypercellularity for age with trilineage growth) from a minor to major criterion. Until now, the pathologic features of the blast phase of PV have been reported rarely [13, 17]. Others have shown that patients with higher grade myelofibrosis (≥ MF-1) in the polycythemic phase (PP) have a higher risk of disease progression and transformation to AML [14, 18]. Morphologic features of a myelodysplasia also have been observed in a subset of PV patients during the course of disease [8], and morphologically, these changes may resemble a myelodysplastic syndrome (MDS) or MDS/MPN. However, little is known regarding the potential prognostic impact or biology of myelodysplasia or myelofibrosis in blast phase of PV. It is not clear whether development of morphologic evidence of dysplasia is associated with transformation of PV to BP.
Most cases of acute leukemia that arise in patients with PV are AML. In the current WHO classification [3], though blast phase was used to describe this stage, various terms have been used in BM diagnosis, such as blast phase of PV, AML evolving from PV, secondary AML, or simply AML, not otherwise specified. It is clinically important to render a consistent and biologically accurate diagnosis for the BP of PV, and having a better understanding of the clinicopathologic, cytogenetic, and molecular genetic features of this neoplasm is therefore required for this purpose [19].
In this study, we retrospectively reviewed 422 PV patients and identified 58 patients who developed AML (blast phase) during the course of disease. We summarize the clinicopathologic, cytogenetic, and molecular genetic features of blast phase of PV. In addition, 11 patients underwent multiple bone marrow examinations from the polycythemic phase to blast phase. This subset of patients allowed us to better study the dynamic changes during the course of PV and analyze for possible factors that contribute to transformation into blast phase.
Materials and methods
Patients
We searched the archives of The University of Texas MD Anderson Cancer Center for patients with PV who were diagnosed and/or managed at this institution from January 1, 2005 through June 30, 2016. A detailed chart review was conducted for all patients and the following data were collected: clinical presentation, treatment history, clinical outcomes, laboratory data, and pathological findings. Blasts were counted on PB smears based on a 200-cell differential count and on BM smears based on a 500-cell differential count. Blast phase was defined as ≥ 20% myeloblasts in PB or BM or both. This study was approved by the Institutional Review Board of MDACC.
Histological assessment
Wright-Giemsa stained PB and BM aspirate smears and H&E stained bone marrow aspirate clot and core biopsy tissue sections were reviewed. The PB smear blast percentage and BM cellularity, blast percentage, and morphologic dysplasia were evaluated. Granulocytic dysplasia includes hypogranulation, nuclear hypolobation, or hypersegmentation; erythroid dysplasia includes nuclear budding or hyperlobation, ring sideroblasts, or cytoplasmic vacuolization; and megakaryocytic dysplasia includes micromegakaryocytes, nuclear hypolobation, widely separated nuclear lobes, or atypical clusters. In this study, only “overt dysplasia” was considered if dysplasia involved two or more lineages or > 50% cells if only one lineage was involved. Myelofibrosis was evaluated by reticulin and trichrome stains performed on the BM core biopsy in all cases with core biopsy available. The grade of myelofibrosis was based on theEuropean Consensus on grading of BM fibrosis [20].
Cytogenetics
Conventional chromosomal analysis was performed on all patients, and G-banded metaphases were prepared from unstimulated 24- and 48-h BM aspirate cultures using standard techniques. Twentymetaphases were analyzed, and the results were reported using the 2016 International System for Human Cytogenetics Nomenclature (ISCN 2016) [21]. A complex karyotype was defined as ≥ 3 chromosomal abnormalities.
Mutational analysis
Molecular analysis was performed in most patients as a part of the routine clinical work-up.JAK2 V617F and codon 12 mutations were evaluated in all patients at time of initial diagnosis and in most patients in BP. In addition, NPM1 (exon 12),KIT (exons 8 and 17), CEBPA, FLT3 (internal tandem duplication and D835 point mutation), and IDH1 and IDH2 mutations were analyzed at the time of BP transformation. Next-generation sequencing (NGS) for somatic mutations was performed in 11 patients at the time of BP progression as described previously [22]. To identify pathways involved in BP progression, genes were grouped by functional classification as defined by The Cancer Genome Atlas Research Network [23].
Statistical analysis
Frequencies and percentages were calculated for categorical variables, and medians (range) were calculated for continuous variables. Overall survival was calculated from the date of transformation to BP to the date of death or censored at the date of last follow-up for surviving patients. p value of < 0.05 was considered statistically significant.
Results
Patients and clinical features
From a group of 422 patients with PV, 58 (14%) patients progressed to AML and formed the study group. There were 30 men and 28 women, with a median age 67 years (range, 32–82 years) at the time of onset of BP. The median interval from initial diagnosis of PV to onset of BP was 109 months (range, 9–283 months). The first BM biopsy was performed at a median of 95 months (range, 0–167 months) after initial diagnosis of PV. At the time of first BM evaluation, 11 patients had PP, 13 had post-PV MF, 6 had accelerated phase (AP), and 28 had BP (Fig. 1a). For the 28 patients who showed BP in the first BM specimen available for our review, all these patients had an established diagnosis of PV at another hospital before they were referred to our institution. For the other 30 patients, the median interval from first BM evaluation to BP was 53 months (range, 4–137) in patients with PP, 12.5 months (range, 1–48) in patients with post-PV MF, and 7 months (range, 1–24) in patients with AP.

a Disease stage of polycythemia vera at the first bone marrow evaluation and the number of patients who progressed to blast phases by the end of follow-up.b The risk of acute leukemia transformation is significantly higher in patients with myelodysplasia comparing to that in patients without myelodysplasia at polycythemic phase (20 vs. 5.6%, p = 0.0453)
Prior to development of BP, 30 patients had splenomegaly, 26 had constitutional symptoms (weight loss, night sweats, and/or fever), and 10 patients had a history of thrombotic events. Prior to onset of BP, 3 patients were treated by phlebotomy only and 53 patients received various therapeutic agents, including hydroxyurea (n = 44), ruxolitinib (n = 5), interferon alpha (n = 3), aspirin or coumadin or Plavix (n = 4), and P32 (n = 1). The therapeutic history was unclear in 3 patients.
Laboratory and pathological findings
At time of onset of BP, the complete blood count showed a median hemoglobin level of 9.4 g/dL (range, 6.8–11.1; normal 14–18 g/dL for men and 12–16 g/dL for women), leukocyte count 9.7 × 109/L (range, 4.8–118.3; normal 4–11 × 109/L), platelet count 44 × 109/L (range, 7–724; normal, 150–440 × 109/L), and blast percentage of 18% (range, 8–78%) (Table 1).
BM blast counts ranged from 14 to 70% (median 30%). In the cases with blast counts < 20% in BM, blast counts were ≥ 20% in the PB.Based on WHO classification criteria [3], the acute leukemia was classified as follows: AML with myelodysplasia-related changes (AML-MRC) (n = 48), AML with inv(3)(q21.3q26.2)/MECOM rearrangement (n = 3), AML with maturation (n = 3), AML with t(9;11)(p22;q23)/MLLT3-MLL rearrangement (n = 1),AML with minimal differentiation (n = 1), acute myelomonocytic leukemia (AMML, n = 1), andacute erythroid leukemia (n = 1).
Overt dysplasia was observed in 51 (88%) patients including 16 with trilineage, 24 with bilineage, and 11 with unilineage dysplasia. Granulocytic dysplasia was observed in 34 patients, erythroid dysplasia in 34 patients, and megakaryocytic dysplasia in 39 patients.There were 7 patients without overt dysplasia; 2 had a normal karyotype, 3 had single karyotypic abnormality (+ 13, + 11, t(5;10)(q33;q21), respectively), and 2 had trisomy 1q in a complex karyotype. None of these 7 patients showed abnormalities involving chromosomes of 5, 7, or 17.
Reticulin and trichrome stains were performed on BM biopsy specimens of 45 (76%) patients to evaluate the grade of myelofibrosis. Moderate to severe myelofibrosis (MF-2 or MF-3) was detected in 33 (73%) patients, MF-1 in 7 (16%) patients, and MF-0 in 5 (11%) patients. Among the group of 12 patients who had MF-0/1, 8 patients had BP involving the first BM specimen reviewed by us at our institution; 2 patients showed 3 distinct stages including PP, post-PV MF (with MF-2), and BP; and 2 patients transformed to BP directly from PP stage. All 12 patients showed overt dysplasia at time of BP.
We evaluated the association between myelodysplasia developing in polycythemic phase and the risk of transformation to BP. As shown in Fig.1b, of 271 patients who had a BM specimen that showed the PP stage of PV, 146 patients had follow-up BM evaluation.Twenty of these 146 (14%) patients showed dysplastic changes and 4 (20%) subsequently transformed to BP. Of note, none of these 4 patients had blast counts ≥ 5% in PB or BM at the time when morphologic dysplasia was detected. By contrast, of the other 126 patients in whom the BM did not show dysplastic changes, 7 (5.6%) transformed to BP. The difference between these two groups was significant (p = 0.0453). In addition, these 20 patients with dysplasia showed a higher frequency of abnormal karyotype (9/20 vs. 28/126, p = 0.0492), an inferior overall survival (median 126 vs. 169 months from the first BM diagnosis, p = 0.0332), but comparable value in hemoglobin level, white blood cell, and platelet counts compared to the 126 patients without dysplasia.
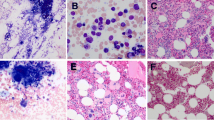
Sequential BM biopsies from PP to BP were available in 11 patients. In polycythemic phase, overt dysplasia was detected in three patients, moderate fibrosis (MF-2) in two patients, overt dysplasia plus MF-2 in one patient, and the remaining five patients did not show dysplasia or myelofibrosis.Of the four patients (including the one with MF-2) who showed overt dysplasia, all had an abnormal karyotype at the first kayotyping analysis;three of four did not show a distinct stage of post-PV MF and progressed to BP from PP stage directly (Fig. 2, patient A). Of the two patients with MF-2 without dysplasia at first BM biopsy, both cases progressed to post-PV MF and then BP (Fig.2, patient B).

Sequential bone marrow morphologic evolution from polycythemic phase to blast phase. a Bone marrow core biopsy (× 40),b reticulum stain (× 40), and c bone marrow aspirate smear (× 100). Case A showed overt myelodysplasia but no or mild myelofibrosis in polycythemic phase and all follow-up bone marrow biopsies. Bone marrow 1: polycythemic phase (the first biopsy, 0 months); bone marrow 2: accelerated phase with 12% blasts (biopsy at 2 years);bone marrow 3: blast phase with 38% blasts (biopsy at 2 years plus 4 months). Case B showed moderate to marked myelofibrosis in the polycythemic phase and all follow-up bone marrow biopsies, and myelodysplasia was only observed at blast phase. Bone marrow 1: polycythemic phase (the first biopsy, 0 months); bone marrow 2: post-polycythemic myelofibrosis (biopsy at 5 years plus 2 months); bone marrow 3: blast phase with 66% of blasts (biopsy at 6 years plus 6 months).Cells with morphologic dysplasia are marked with arrow head
Cytogenetic analysis
For the 58 patients in the study, 6 (10%) had a normal karyotype and 52 (90%) had an abnormal karyotype at the onset of BP. The latter included a complex karyotype (n = 42, 72%), double abnormalities (n = 4), and a single abnormality (n = 6).The most common abnormalities included − 7/del7q (n = 26), − 5/del5q (n = 21), − 17/del17p (n = 14), + 21 (n = 10), trisomy 1q (n = 10), and − 18 (n = 9); all of these abnormalities were parts of complex karyotype. Three patients showed inv(3)(q21q26), one as sole abnormality and the other two as part of a complex karyotype. One patient showed t(9;11)(p22;q23).
For 19 patients with available data, the karyotypes before and after onset of BP were compared (Table 2). Prior to BP, 11 patients had a normal karyotype, 1 had isolated del(20q), and 7 had a complex karyotype. At the onset of BP, 10 patients gained new clones; 4 showed clonal evolution; and in 5 patients, the karyotype was unchanged.
Molecular mutations
All 58 patients showed a JAK2 mutation prior to BP transformation. At the time of BP, 54 patients underwent molecular studies to assess for gene mutations, either by the next-generation sequencing (NGS, n = 11) or quantitative PCR of selected genes (n = 43). JAK2 mutation status at BP stage was available for 41 patients: 36 (88%) patients had JAK2 mutation, including 35 with V617F (with a median allele burden of 64.1%) and 1 had exon 12 mutation; 2 (5%) patients did not have detectable JAK2 mutation; and 3 patients had JAK2 V617F mutations, but the mutant JAK2 was not consistent with AML status; there was a high allelic burden of mutant JAK2 when AML was in remission, or there was a low mutant JAK2 allele burden when the blast count was high.
Other molecular mutations were also detected in a subset of patients, including TP53 (6/11, 55%), TET2 (3/11, 27%), DNMT3A (3/12, 25%), IDH1 (4/20, 20%), RUNX1 (2/11, 18%), PTPN11 (2/11, 18%),EZH2 (2/11, 18%), NRAS (2/42, 5%), KIT (1/23, 4.3%), FLT3 (1/48, 2%), and NPM1 (1/11, 9%) (Table 3).
We were unable to compare the mutation profile before and after BP transformation in the same group of patients because there were only a few patients that underwent NGS analysis more than once during the course of disease. For this reason, we compared the mutation frequencies among the patients in BP and 83 patients in PP/post-PV MF stage and who never transformed to BP during the follow-up (Table 3). In general, mutations were more frequent in BP than those in the PP or post-PV MF phases, especially for TP53 (55 vs. 1.2%, p < 0.0001), DNMT3A (25 vs. 4%, p = 0.0410), PTPN11 (18 vs. 0%, p = 0.0126), IDH1 (20 vs. 5%, p = 0.0440), EZH2 (18 vs. 1.2%, p = 0.0353), and RUNX1 (18 vs. 1.2%, p = 0.0353). Among 11 patients who had NGS at time of BP (28 gene panel), all patients had at least one mutated gene in addition toJAK2 mutation, and 5 patients showed more than 4 gene mutations.
Treatments and outcomes
After BP transformation, 22 patients were enrolled in clinical trials: 17 on a protocol that included hypomethylating agents and 5 on a protocol that included ruxolitinib. In addition, 12 patients received standard induction chemotherapy (“7 + 3 regimen”); 1 patient received FLAG-IDA; 10 patients had initial monotherapy with a hypomethylating agent; and 12 patients went to palliative care. Three patients also received allogeneic stem cell transplant. Two patients were lost to follow-up and their treatments are unknown. At time of the last follow-up, 53 (91%) patients were dead with a median overall survival of 4 months (Fig. 3). The overall survival was not significantly different among patients with MF 0/1 and patients with MF 2/3 (6.5 vs. 4 months, p = 0.6237).

Overall survival from blast phase transformation, with a median overall survival of 4 months
Discussion
In this study, we have shown that patients with blast phase of PV have a high frequency of myelodysplasia, moderate to severe BM fibrosis, an abnormal and often complex karyotype, and gene mutations, particularly TP53.
During the polycythemic phase of PV, patients have an increased red blood cell (RBC) mass, and the BM is typically characterized by hypercellularity and panmyelosis. When the disease progresses to post-PV MF phase, BM cellularity and erythropoiesis decrease, leading to a decreased RBC mass. In the blast phase of PV, patients develop acute myeloid leukemia [17, 24, 25]. Based on the presence of overt dysplasia and a complex karyotype, most patients in this cohort belong to the category of AML-MRC as defined in the WHO classification. The high frequency of an abnormal karyotype or a complex karyotype, TP53 mutation, and overt dysplasia indicates that the BP of PV is usually a high-grade AML similar to therapy-related AML [26]. In our cohort, only 1 of 58 patients developed acute erythroid leukemia, which is similar to the study by Passamonti et al.[17], in which only 1 out of 23 patients developed erythroid leukemia. However, in a study by Weinfeld et al.[13], 3 out of 8 patients developed erythroid leukemia.
In de novo AML, reticulin fibrosis is observed frequently, but usually is present to a mild or moderate degree, with the exception of acute megakaryoblastic leukemia in which fibrosis is often severe [27]. In this cohort, 33 (73%) patients showed moderate to severe fibrosis (MF-2/3). The interval from initial diagnosis to BP was slightly shorter in 12 (27%) patients with MF 0/1 compared with patients with MF 2/3 (median 94 vs. 129 months, p = 0.1054). Interestingly, all 12 patients with MF 0/1 showed dysplastic changes. In a previous study, we showed that a higher grade myelofibrosis (≥ MF-1) in PP stage was associated with a higher frequency of disease progression and inferior overall survival [14].
Morphologic dysplasia has been studied infrequently in PV patients. Hydroxyurea could cause erythrocytes megaloblastoid changes, which may mimic dysplastic changes. However, hydroxyurea often affects erythrocytes only, granulocytes and megakaryotypes are often spared. Besides, the effect of hydroxyurea is often transient, erythrocytes morphology restores to normal after stopping the use of hydroxyurea.Development of MDS-type PV (morphologically resembling MDS/MPN) has been reported to be related to exposure to high-dose P32 and alkylator agents [8], but only one patient in this study cohort received P32 treatment. In this cohort, of 11 patients who had PP phase at time of initial BM evaluation, 4 showed overt dysplasia and all 4 patients had an abnormal karyotype. Of interest, 3 of 4 patients progressed to BP without going through a “middle phase” of post-PV MF. We found that patients with myelodysplasia detected at polycythemic phase had a significantly higher risk for BP transformation, higher frequency of abnormal karyotype, and inferior survival, compared to patients without myelodysplasia. These findings indicate that overt dysplasia or MDS-like BM changes that develop during the course of PV are adverse features, similar to significant BM fibrosis and should be considered as one form of progression that may herald impending BP. Morphologic dysplasia was a prominent feature in most patients at time of BP, and development of myelodysplasia was closely correlated with chromosomal abnormalities, especially abnormalities involving chromosomes 5, 7, and 17.
In contrast to the low frequency of an abnormal karyotype in PP, 90% of patients in BP had an abnormal karyotype and about 70% of patients showed a complex karyotype, in line with previous studies [14, 17, 28]. The most commonly detected chromosomal abnormalities in BP were 7/del(7q), − 5/del(5q), and − 17/del(17p); these abnormalities were detected infrequently in PP of PV. Clonal evolution or acquisition of new clone(s) was very common in PV, in almost 75% of patients during BP transformation in this cohort, which supports the concept that acquisition of cytogenetic abnormalities plays a critical role in progression of PV to BP [14]. Interestingly, 4 patients acquired recurrent translocations, 3 with inv(3)/MECOM rearrangement and 1 with t(9;11)/MLLT3-MLL rearrangement; these abnormalities have not been reported in PV previously. A complex karyotype is a high risk factor in PV and AML [14, 29].
A higher mutant JAK2 V617F allele burden has been associated with pruritus and post-PV MF in PV patients but does not appear to affect transformation to BP [30]. In this cohort, JAK2 mutation was detected persistently at BP stage in 88% patients; the other 12% of patients had no detectable JAK2 or a low percentage of mutated JAK2 while blast counts were high. These findings suggest that the myeloblastic clone in BP is derived from the original PV clone in most patients. This is different from the report by Theocharides et al. that included 17 patients with JAK2-positive MPN (7 PV, 7 essential thrombocythemia, and 2 primary myelofibroses). In their study, 5 (29%) patients developed JAK2-positive AML, 9 (53%) developed JAK2-negative AML, and 3 (18%) were uninformative [31].These results suggest that JAK2-negative BP may arise from a cell ancestral to the JAK2 mutation or represents another independent clone arising from a different progenitor [31]. This phenomenon also has been observed in patients with chronic myeloid leukemia [32].
In this cohort, 55% of PV patients showed TP53 mutation and 24% of patients showed del(17p)/−17 with TP53 deletion. TP53 mutation therefore seems to be detected more frequently in BP of PV than in patients with therapy related-AML (33.3%) or de novo AML (~ 14.5%) [33].Loss of TP53 is sufficient to induce leukemic transformation in JAK2 V617F-driven PV in a murine model [34]. The presence of TP53 mutation at time of diagnosis of PV has been associated with BP progression [35,36,37]. TP53 mutation is also often associated with a poor response to treatment and shorter overall survival [38]. All of these data suggest that acquisition of TP53 mutation plays an important role for BP transformation and an aggressive disease course in PV patients. Other than TP53 mutation, DNMT3A, IDH1, PTPN11, EZH2,and RUNX1 were also more frequently mutated in BP, and these mutated genes also may have prognostic significance [36].
In conclusion, the blast phase of PV is characterized by frequent morphologic evidence of dysplasia, myelofibrosis, an abnormal/complex karyotype, frequent gene mutations, and an aggressive clinical course.
References
Titmarsh GJ, Duncombe AS, McMullin MF, O’Rorke M, Mesa R, de Vocht F, Horan S, Fritschi L, Clarke M, Anderson LA (2014) How common are myeloproliferative neoplasms? A systematic review and meta-analysis. Am J Hematol 89(6):581–587. https://doi.org/10.1002/ajh.23690
Tefferi A, Barbui T (2017) Polycythemia vera and essential thrombocythemia: 2017 update on diagnosis, risk-stratification, and management. Am J Hematol 92(1):94–108. https://doi.org/10.1002/ajh.24607
Swerdlow SH, Campo E, Harris NL et al (eds) (2017) WHO classification of tumors of haematopoietic and lymphoid tissue (Revised 4th edition). IARC, Lyon
Tefferi A, Guglielmelli P, Larson DR, Finke C, Wassie EA, Pieri L, Gangat N, Fjerza R, Belachew AA, Lasho TL, Ketterling RP, Hanson CA, Rambaldi A, Finazzi G, Thiele J, Barbui T, Pardanani A, Vannucchi AM (2014) Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood 124(16):2507–2513; quiz 615. https://doi.org/10.1182/blood-2014-05-579136
Tefferi A, Rumi E, Finazzi G, Gisslinger H, Vannucchi AM, Rodeghiero F, Randi ML, Vaidya R, Cazzola M, Rambaldi A, Gisslinger B, Pieri L, Ruggeri M, Bertozzi I, Sulai NH, Casetti I, Carobbio A, Jeryczynski G, Larson DR, Müllauer L, Pardanani A, Thiele J, Passamonti F, Barbui T (2013) Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia 27(9):1874–1881. https://doi.org/10.1038/leu.2013.163
Finazzi G, Caruso V, Marchioli R, Capnist G, Chisesi T, Finelli C, Gugliotta L, Landolfi R, Kutti J, Gisslinger H, Marilus R, Patrono C, Pogliani EM, Randi ML, Villegas A, Tognoni G, Barbui T, ECLAP Investigators (2005) Acute leukemia in polycythemia vera: an analysis of 1638 patients enrolled in a prospective observational study. Blood 105(7):2664–2670. https://doi.org/10.1182/blood-2004-09-3426
Cerquozzi S, Tefferi A (2015) Blast transformation and fibrotic progression in polycythemia vera and essential thrombocythemia: a literature review of incidence and risk factors. Blood Cancer J 5(11):e366. https://doi.org/10.1038/bcj.2015.95
Bjorkholm M, Derolf AR, Hultcrantz M et al (2011) Treatment-related risk factors for transformation to acute myeloid leukemia and myelodysplastic syndromes in myeloproliferative neoplasms. J Clin Oncol 29(17):2410–2415. https://doi.org/10.1200/JCO.2011.34.7542
Stein BL, Oh ST, Berenzon D et al (2015) Polycythemia vera: an appraisal of the biology and management 10 years after the discovery of JAK2 V617F. J Clin Oncol 33(33):3953–3960. https://doi.org/10.1200/JCO.2015.61.6474
Sever M, Quintas-Cardama A, Pierce S et al (2013) Significance of cytogenetic abnormalities in patients with polycythemia vera. Leuk Lymphoma 54(12):2667–2670. https://doi.org/10.3109/10428194.2013.784970
Gangat N, Strand J, Li CY, Wu W, Pardanani A, Tefferi A (2007) Leucocytosis in polycythaemia vera predicts both inferior survival and leukaemic transformation. Br J Haematol 138(3):354–358. https://doi.org/10.1111/j.1365-2141.2007.06674.x
Marchioli R, Finazzi G, Landolfi R, Kutti J, Gisslinger H, Patrono C, Marilus R, Villegas A, Tognoni G, Barbui T (2005) Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J Clin Oncol 23(10):2224–2232. https://doi.org/10.1200/JCO.2005.07.062
Weinfeld A, Westin J, Ridell B, Swolin B (1977) Polycythaemia vera terminating in acute leukaemia. A clinical, cytogenetic and morphologic study in 8 patients treated with alkylating agents. Scand J Haematol 19(3):255–272
Tang G, Hidalgo Lopez JE, Wang SA et al (2017) Characteristics and clinical significance of cytogenetic abnormalities in polycythemia vera. Haematologica. available from: URL:www.haematologica.org/content/early/2017/05/02/haematol.2017.165795
Tam CS, Nussenzveig RM, Popat U, Bueso-Ramos CE, Thomas DA, Cortes JA, Champlin RE, Ciurea SE, Manshouri T, Pierce SM, Kantarjian HM, Verstovsek S (2008) The natural history and treatment outcome of blast phase BCR-ABL−myeloproliferative neoplasms. Blood 112(5):1628–1637. https://doi.org/10.1182/blood-2008-02-138230
Rampal R, Mascarenhas J (2014) Pathogenesis and management of acute myeloid leukemia that has evolved from a myeloproliferative neoplasm. Curr Opin Hematol 21(2):65–71. https://doi.org/10.1097/MOH.0000000000000017
Passamonti F, Rumi E, Arcaini L, Castagnola C, Lunghi M, Bernasconi P, Giovanni Della Porta M, Columbo N, Pascutto C, Cazzola M, Lazzarino M (2005) Leukemic transformation of polycythemia vera: a single center study of 23 patients. Cancer 104(5):1032–1036. https://doi.org/10.1002/cncr.21297
Barbui T, Thiele J, Passamonti F, Rumi E, Boveri E, Randi ML, Bertozzi I, Marino F, Vannucchi AM, Pieri L, Rotunno G, Gisslinger H, Gisslinger B, Mullauer L, Finazzi G, Carobbio A, Gianatti A, Ruggeri M, Nichele I, D'Amore E, Rambaldi A, Tefferi A (2012) Initial bone marrow reticulin fibrosis in polycythemia vera exerts an impact on clinical outcome. Blood 119(10):2239–2241. https://doi.org/10.1182/blood-2011-11-393819
Czader M, Orazi A (2015) Acute myeloid leukemia and other types of disease progression in myeloproliferative neoplasms. Am J Clin Pathol 144(2):188–206. https://doi.org/10.1309/AJCPZQK40JOZZZCC
Thiele J, Kvasnicka HM, Facchetti F, Franco V, van der Walt J, Orazi A (2005) European consensus on grading bone marrow fibrosis and assessment of cellularity. Haematologica 90(8):1128–1132
Simons A, Shaffer LG, Hastings RJ (2013) Cytogenetic nomenclature: changes in the ISCN 2013 compared to the 2009 edition. Cytogenet Genome Res 141(1):1–6. https://doi.org/10.1159/000353118
Kanagal-Shamanna R, Luthra R, Yin CC, Patel KP, Takahashi K, Lu X, Lee J, Zhao C, Stingo F, Zuo Z, Routbort MJ, Singh RR, Fox P, Ravandi F, Garcia-Manero G, Medeiros LJ, Bueso-Ramos CE (2016) Myeloid neoplasms with isolated isochromosome 17q demonstrate a high frequency of mutations in SETBP1, SRSF2, ASXL1 and NRAS. Oncotarget 7(12):14251–14258. https://doi.org/10.18632/oncotarget.7350
Cancer Genome Atlas Research N (2013) Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 368(22):2059–2074. https://doi.org/10.1056/NEJMoa1301689
Camos M, Cervantes F, Montoto S et al (1999) Acute lymphoid leukemia following polycythemia vera. Leuk Lymphoma 32(3–4):395–398. https://doi.org/10.3109/10428199909167404
Gawel WB, Helbig G, Boral K, Kyrcz-Krzemien S (2016) Acute lymphoblastic leukemia transformation in polycythemia vera: arare phenomenon. Indian J Hematol Blood Transfus 32(S1):62–65. https://doi.org/10.1007/s12288-016-0673-z
Bjorkholm M, Hultcrantz M, Derolf AR (2014) Leukemic transformation in myeloproliferative neoplasms: therapy-related or unrelated? Best Pract Res Clin Haematol 27(2):141–153. https://doi.org/10.1016/j.beha.2014.07.003
Manoharan A, Horsley R, Pitney WR (1979) The reticulin content of bone marrow in acute leukaemia in adults. Br J Haematol 43(2):185–190. https://doi.org/10.1111/j.1365-2141.1979.tb03740.x
Swolin B, Rodjer S, Westin J (2008) Therapy-related patterns of cytogenetic abnormalities in acute myeloid leukemia and myelodysplastic syndrome post polycythemia vera: single center experience and review of literature. Ann Hematol 87(6):467–474. https://doi.org/10.1007/s00277-008-0461-4
Rollig C, Bornhauser M, Thiede C et al (2011) Long-term prognosis of acute myeloid leukemia according to the new genetic risk classification of the European LeukemiaNet recommendations: evaluation of the proposed reporting system. J Clin Oncol 29(20):2758–2765. https://doi.org/10.1200/JCO.2010.32.8500
Passamonti F, Rumi E, Pietra D, Elena C, Boveri E, Arcaini L, Roncoroni E, Astori C, Merli M, Boggi S, Pascutto C, Lazzarino M, Cazzola M (2010) A prospective study of 338 patients with polycythemia vera: the impact of JAK2 (V617F) allele burden and leukocytosis on fibrotic or leukemic disease transformation and vascular complications. Leukemia 24(9):1574–1579. https://doi.org/10.1038/leu.2010.148
Theocharides A, Boissinot M, Girodon F, Garand R, Teo SS, Lippert E, Talmant P, Tichelli A, Hermouet S, Skoda RC (2007) Leukemic blasts in transformed JAK2-V617F-positive myeloproliferative disorders are frequently negative for the JAK2-V617F mutation. Blood 110(1):375–379. https://doi.org/10.1182/blood-2006-12-062125
Kovitz C, Kantarjian H, Garcia-Manero G, Abruzzo LV, Cortes J (2006) Myelodysplastic syndromes and acute leukemia developing after imatinib mesylate therapy for chronic myeloid leukemia. Blood 108(8):2811–2813. https://doi.org/10.1182/blood-2006-04-017400
Ok CY, Patel KP, Garcia-Manero G, Routbort MJ, Fu B, Tang G, Goswami M, Singh R, Kanagal-Shamanna R, Pierce SA, Young KH, Kantarjian HM, Medeiros LJ, Luthra R, Wang SA (2015) Mutational profiling of therapy-related myelodysplastic syndromes and acute myeloid leukemia by next generation sequencing, a comparison with de novo diseases. Leuk Res 39(3):348–354. https://doi.org/10.1016/j.leukres.2014.12.006
Tsuruta-Kishino T, Koya J, Kataoka K et al Loss of p53 induces leukemic transformation in a murine model of Jak2 V617F-driven polycythemia vera. Oncogene. available from: URL:www.nature.com/onc/journal/vaop/ncurrent/full/onc2016478a
Courtier F, Carbuccia N, Garnier S et al Genomic analysis of myeloproliferative neoplasms in chronic and acute phases. Haematologica. available from: URL:www.ncbi.nlm.nih.gov/pubmed/27742771
Lundberg P, Karow A, Nienhold R, Looser R, Hao-Shen H, Nissen I, Girsberger S, Lehmann T, Passweg J, Stern M, Beisel C, Kralovics R, Skoda RC (2014) Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood 123(14):2220–2228. https://doi.org/10.1182/blood-2013-11-537167
Harutyunyan A, Klampfl T, Cazzola M, Kralovics R (2011) p53 lesions in leukemic transformation. N Engl J Med 364(5):488–490. https://doi.org/10.1056/NEJMc1012718
Kadia TM, Jain P, Ravandi F et al TP53 mutations in newly diagnosed acute myeloid leukemia: clinicomolecular characteristics, response to therapy, and outcomes. Cancer. available from: URL:www.ncbi.nlm.nih.gov/pubmed/27463065
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
This study was approved by the Institutional Review Board of MDACC.
Conflict of interest
The authors declare that they have noconflict of interest.
Rights and permissions
About this article

Cite this article
Hidalgo López, J.E., Carballo-Zarate, A., Verstovsek, S. et al. Bone marrow findings in blast phase of polycythemia vera. Ann Hematol 97, 425–434 (2018). https://doi.org/10.1007/s00277-017-3211-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-017-3211-7