
Abstract
A vauxite mineral sample from Huanuni, Bolivia, was studied by XRD, TGA and Mössbauer spectroscopy. The XRD revealed the sample as having the typical triclinic structure of vauxite. The chemical formula was determined as (Fe0.88Mn0.01)Al1.99(PO4)2(OH)1.75(H2O)5.31, implying some Fe2+, OH− and H2O deficiencies. The TGA curve showed ca. 27% loss of weight over a temperature range from 80 to 400 °C, supposedly due to the loss of water and hydroxyl groups. For the first time, Mössbauer spectra for vauxite were collected over a wide temperature range between 9 and 310 K. No magnetic ordering was detected. The spectra could be successfully and consistently analyzed by a superposition of four doublet subspectra. On the basis of the relation between the center shift and the mean Fe-ligand distance on the one hand and the center shift values for the various doublets on the other hand, one doublet was assigned to Fe(2). For the other doublets, it is proposed that, as a result of the H2O deficiency in the structure of the present vauxite sample, vacancies are present in the second coordination spheres of some Fe(1) and that these vacancies affect the quadrupole splitting of the corresponding Fe(1) cations, thus causing three Fe(1) doublet components in the Mössbauer spectra. The temperature variations of center shift and quadrupole splitting of the various doublet contributions are presented and discussed.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Vauxite, ideally FeAl2(PO4)2(OH)2(H2O)6, is a secondary mineral derived from the alteration of apatite and belongs to the vauxite group of minerals with the triclinic space group \({\text{P}}\bar{1}\) (Baur and Rama Rao 1968). Closely related minerals are paravauxite, FeAl2(PO4)2(OH)2(H2O)8 (Baur 1969), with the same space group as vauxite, and metavauxite (Baur and Rama Rao 1967) with space group P 21/c, being the monoclinic dimorph of paravauxite.
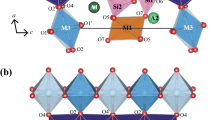
The structure of vauxite (see Fig. 1) was unraveled by Baur and Rama Rao (1968), and recently reviewed by Ventruti et al. (2016). It consists of [FeAl3(PO4)4(OH)4(OH2)2]5− units arranged in infinite triple chains along the c-axis, and connected to neighboring units in neighboring triple chains via vertex-linked Al coordination octahedra in the a-direction, and via Fe coordination octahedra in the b-direction. The triple chains themselves are interlinked by phosphate tetrahedra and each consist of a central chain of edge sharing coordination octahedra with central cations Fe and Al alternately. Within these triple chains, parallel to the central octahedral chains, are vertex-linked chains flanking them on two opposite sides, and consisting of phosphate tetrahedra alternating with coordination octahedra. Two types of Fe octahedral sites can be differentiated in the structure: one, Fe(1), links neighboring triple chains to each other and the other, Fe(2), is located within the central chains of the triple chains. Per formula unit, two water oxygen atoms belong to the first coordination sphere of Fe(1), two other water oxygen atoms are located in the first coordination spheres of Al cations and the remaining two water molecules are not coordinated to any cations, but are situated in channels that extend parallel to the [001]-axis through the structure (Baur and Rama Rao 1968).

Structure of vauxite (after Baur and Rama Rao 1968) viewed along the c-axis. The parallelogram indicates the unit cell
Experimental
The investigated vauxite sample, collected from the Huanuni mine, Bolivia, consists of a conglomerate of small blue crystals, which were pulverized in an agate mortar to produce fine powder samples for the various characterization techniques described in this section. A powder X-ray diffraction (XRD) pattern was recorded with a Shimadzu XRD-6000 diffractometer equipped with an iron tube and graphite monochromator. The scan was done between 7° and 70° (2θ) with a scanning speed of 0.5°/min. Silicon was added to the powder sample as an internal standard. Unit-cell parameters were calculated by means of the Jade+ software using least-squares refinement after subtracting the background and the Kα2 contribution and using intensity and angular weighting of the most intense diffraction peaks. Microprobe elemental analysis was carried out with a Jeol microscope, model JXA 8900R, equipped with a Noran EDS system and operating at (20 kV, 25 nA). A distinct well-developed crystallite, selected on the basis of the SEM observations, was probed at 10 different spots. Counting times were 10–100 s. Apatite, fayalite and synthetic corundum were used for the determinations of P, Fe and Al, respectively. Simultaneous thermo-gravimetric analysis (TGA) and differential thermal analysis (DTA) were performed in a Du Pont SDT2960 apparatus. The temperature ranged from 25 to 550 °C applying a constant flow of 100 ml/min of synthetic air and a heating rate of 20 °C/min. Masses of ~8 mg were evenly spread out in the pans to ensure a homogeneous thin layer of material.
Mössbauer spectra (MS) were collected in transmission geometry using 57Co(Rh) sources with active diameter of 5 mm and initial activity of ~75 mCi (2.78 GBq), provided by Gamma-Lab Development S.L. The time-mode spectrometer was composed of Wissel GmbH drive, detection and data-acquisition (CMCA-550) modules. The triangular source motion option was used. The spectrometer was experienced to exhibit excellent linearity. Counts were accumulated in 1024 channels until an off-resonance count rate of ~106 per channel had been reached. Fe foil was used for regular calibration and all center shift values quoted hereafter are relative to α-Fe at room temperature (RT). For all spectra the velocity (v) increment per channel was ~0.0165 mm/s and from the calibration spectra an instrumental line width of ~0.22 mm/s was observed for the innermost absorption lines of the α-Fe foil sextet. The absorber thickness was approximately 10 mg Fe per cm2. The temperature of the absorber was varied within the range 5–310 K using standard commercial cryogenic equipment (CF506 continuous flow cryostat with ITC4 temperature controller from Oxford Instruments.)
All MS down to 9 K were observed to be composed of a number of doublet components and no magnetic ordering appears to be present in the studied vauxite species at the applied temperatures. To avoid any possible disturbing effect of peak–depth asymmetry of the two absorption lines constituting a particular quadrupole doublet as a result of texture effects (i.e., preferred orientation of the crystallites’ axes with respect to the direction of the incident γ-rays), all MS were recorded under the so-called magic angle of 54.7° between the normal to the absorber plane and the γ-ray direction (Ericsson and Wäppling 1976; Greneche and Varret 1982). The spectra were fitted with a superposition of four discrete symmetric quadrupole doublets using in-house developed software based on the IMSL FORTRAN library optimizer routine ZXSSQ. For each discrete doublet the center shift δ, the quadrupole splitting ΔE Q, and the full width at half maximum Γ were adjustable parameters. However, as will be discussed in the next section, the fractional spectral areas RA of the four doublets had to be constraint to obtain consistent parameter values at all applied temperatures.
Results
The X-ray diffractogram of the powder is reproduced in Fig. 2. No phases other than vauxite could be detected. The unit-cell cell parameters were determined as a = 9.131(13) Å, b = 11.587(2) Å, c = 6.154(97) Å, α = 98.24(2)°, β = 92.14(7)°, γ = 108.19(3)° and V = 609.89 Å3, Z = 2.0, ρcalc = 2.4059 g cm−3, and are in excellent agreement with earlier reported values (Baur and Rama Rao 1968; Ventruti et al. 2016).

X-ray diffraction pattern of the present vauxite sample
From the EDX, the weight percentages of the compositional oxides in the present vauxite sample were determined as follows: MnO 0.11(3), FeO 15.21(53), Al2O3 24.41(63), P2O5 34.12(80). Fluorine content was not determined. Calculating the P and Al contents from these data, it is concluded that their % are nearly the same. Normalizing to 6 H2O molecules per formula unit, as is done by Ventruti et al. (2016) for their sample, yields (PO4)2.05 and Al2.04 per formula unit for the present sample. But this should mean that P occupies more tetrahedral sites than are available in the structure. Normalizing to (PO4)2, however, shows that P sites are fully occupied and as good as all the Al sites with Al, in accordance with the crystallographic structure as is determined by XRD. As consequence of this normalization, some deficiency of water has to be concluded, which should mean that some water sites in the structure are not occupied. This is not so surprising, because two of the 6 H2O molecules in the ideal vauxite structure are not coordinated to any cations. Consequently, these are not strongly bound in the structure and can be easily removed from the structure. From these considerations, assuming two phosphorous atoms per formula unit and requiring charge neutrality to derive the OH− content, (Fe0.88Mn0.01)Al1.99(PO4)2(OH)1.75(H2O)5.31 is proposed for the chemical composition of the presently investigated vauxite sample. Although the water content could be obtained by complementing the EDX results to 100%, the assessment of the water content in the chemical formula is based on the combined Mössbauer spectroscopic and thermo-gravimetric analyses (see further). Thus, the as-such proposed chemical formula implies the existence of significant Fe2+, H2O and OH− deficiencies with respect to the ideal composition. The TGA–DTA recording (Fig. 3) reveals a total mass loss of ca. 27.0% which is attributed to the release of water molecules. This value is in good agreement with the weight percent of H2O (26.6%) present in the structure as reflected in the above chemical formula.

DTA–TGA graph of vauxite
Selected Mössbauer spectra recorded at different temperatures are reproduced in Fig. 4. According to the crystallographic structure of vauxite, two ferrous doublet subspectra are expected to appear in its MS. From the low-temperature MS, however, it is obvious that at least three ferrous doublets need to be considered to properly and consistently fit the spectra. Numerous and meticulous trial fits have eventually inferred that excellent and consistent reproduction of the spectra for T ≤ 80 K could only be achieved using four symmetric doublet subspectra D1, D2, D3, and D4, of which the doublet with the lowest area fraction (D4) shows a line width close to the instrumental value as determined from the calibration spectra, i.e., 0.22 mm/s. Moreover, a weak Fe3+ absorption (<1%) at velocities between ca. 0.6 and 1.0 mm/s, barely noticeable on inspecting the raw experimental spectra, had to be considered. Because of irrelevance, its presence will be disregarded in what follows. The ratios of the respective area fractions of the four resolved ferrous components (at T ≤ 80 K) were found to be 0.444(4):0.246(2):0.231(2):0.079(1). As will be argued in the next section, it is believed that the doublet with the highest RA is due to Fe2+ cations located at Fe(2) sites, while the three other doublets are due to Fe2+ cations located at three differently coordinated Fe(1) sites.

Experimental (+) and calculated (black full line) MS at selected temperatures. Blue, red, green and purple full lines: calculated spectra for D1, D2, D3 and D4, respectively. Relative areas of D2, D3 and D4 were fixed at 0.246:0.231:0.079. Mössbauer parameters are in Table 1
In the next stage of the analysis process, the MS recorded at relatively higher temperatures (T > 80 K) were fitted by imposing fixed ratios for the fractional areas of the three Fe(1) doublets, i.e., 0.246:0.231:0.079. A further constraint consisted of the requirement that the line width parameter could not be lower than 0.22 mm/s. As illustrated in Fig. 4 (solid lines) this fitting procedure produced excellent reproductions of the experimental line shapes over the entire temperature range applied in this study. The adjusted parameter values for the distinct doublets at some selected temperatures are listed in Table 1. The temperature variations of the center shifts and quadrupole splittings of the various doublets are depicted in Figs. 5 and 6, respectively. Finally, it is worth mentioning that a fitting model using three ferrous doublets did not result in consistently varying parameter values as a function of temperature and that moreover the goodness-of-fit values obtained for each of the MS using such model were significantly higher as compared to the respective values obtained from the four-doublet fits (e.g., goodness-of-fit values 1761 compared to 1213 at 9 K and 2827 compared to 1948 at 80 K, for the three and four doublet fits, respectively).

Center shifts as function of temperature for the Fe in the present vauxite. •, ■, ▲ and ♦: experimental values for doublets D1, D2, D3 and D4, respectively. Corresponding colored solid lines are the related calculated temperature dependencies following the Debye model of the lattice vibrations as described in the text

Quadrupole splittings as function of temperature for the respective doublets. •, ■, ▲ and ♦: experimental values for doublets D1, D2, D3 and D4, respectively. Corresponding colored solid lines are the related calculated temperature dependencies calculated using the crystal field approximation as described in the text
Discussion
Assignment of the doublets
The assignment of the four ferrous doublets to specific crystallographic sites in the vauxite structure (see previous section) is not straightforward. It is well-documented that in the case of the 57Fe nucleus, a lower center shift generally implies a higher s-electron density at the nucleus, and hence a smaller mean Fe-ligand distance. Because the mean Fe-ligand distance in ideal vauxite is smallest for the Fe(2) atoms, the authors are inclined to attribute the doublet with the lowest δ value (D1) to Fe(2) atoms. From the relative area of this doublet, being 0.444, one then determines that 0.39 of the total of 0.88 iron per formula unit is located at the Fe(2) site, i.e., only ~78% of these Fe(2) sites would be occupied. The total relative area of the other three doublets (D2, D3, and D4) amounts to 0.556, implying that 0.49 Fe per formula unit is located at the other available site, i.e., the Fe(1) site. Hence, nearly all (98%) Fe(1) sites seem to be occupied by Fe2+ cations in the present vauxite sample. The center shifts of the three respective doublets are found to be equal within experimental error limits and the value is significantly higher than that of D1. This latter finding would infer that the mean Fe-ligand distance is comparatively somewhat higher for the Fe(1) sites than for the Fe(2) sites, which is in agreement with the crystallographic data reported by Baur and Rama Rao (1968). According to these authors the Fe(2) site, having four oxygen ions and two hydroxyl groups in its nearest coordination, is slightly distorted from octahedral symmetry. This distortion causes the 5 D energy level scheme of the central Fe2+ cation to show a relatively small splitting between the two lowest energy levels, and a much higher energy gap between the ground state and the third most energetic level. Consequently, the quadrupole splitting ΔE Q is expected to exhibit a relatively strong temperature dependence as is indeed observed for doublet D1 (see Fig. 5). The maximum at T ≈ 75 K that occurs for this ΔE Q(T) curve can be ascribed to the effect of spin–orbit interaction between the different levels (see next subsection).
The authors believe that the three other doublets D2, D3 and D4 are all due to Fe2+ cations that occupy Fe(1) sites. The finding that the adjusted values of their center shifts are identical within experimental error limits has prompted that opinion. In pure vauxite all Fe(1) sites are crystallographically identical with ideally two O2− ions and four H2O molecules in their first coordination sphere, and two non-coordinated H2O molecules situated at the channel sites in the vicinity of the Fe(1) octahedrons. It is suggested that doublet D2 can be attributed to Fe(1) sites showing this ideal nearest-neighbor configuration. Its quadrupole splitting exhibits a weaker temperature dependence as compared to the Fe(2) quadrupole splitting, implying a stronger distortion of the Fe(1) octahedron relative to the Fe(2) octahedron. It is further proposed that the two additional doublets D3 and D4 also arise from Fe2+ cations that are situated at Fe(1) sites which, however, show a chemical next-nearest neighbor environment that is different from the ideal one and that the presence of such sites is related to the observed deficiency of water molecules in the structure of the present vauxite sample. It is believed that this deficiency is the result of intra-channel water molecules being released from the ideal structure. It is thus suggested that doublet D3 is due to these Fe(1) sites for which one of the neighboring intra-channel sites per formula unit is vacant, and doublet D4 to Fe(1) sites for which two of the neighboring intra-channel sites per formula unit are vacant. The absence of one or two H2O molecules in the channel sites in the vicinity of the Fe(1) octahedrons is expected to have a minor influence upon the electronic state of the central Fe2+ cation at the Fe(1) site, and hence upon the hyperfine interaction parameters.
Temperature dependence of the center shift
It is well-known that the center shift (δ) consists of two contributions and can be expressed as δ = δ I + δ SOD. The intrinsic center shift δ I is determined by the s-electron density at the nucleus and is to first approximation invariant with temperature. In contrast, the second-order Doppler shift δ SOD is significantly dependent on the temperature of the absorber and can be expressed in terms of the Debye approximation of the lattice vibrations, yielding (Pound and Rebka 1960):
where c, k B and M are the velocity of light, the Boltzmann constant and the mass of the 57Fe nucleus, respectively. The quantity ΘM is the so-called characteristic Mössbauer temperature and is related to the well-known Debye temperature of the lattice. Its value can be determined from the experimental δ(T) data using a proper self-consistent iteration method. Applied to the δ(T) values of doublet D1 resolved from the MS of the present sample, values of ΘM,D1 = 320 ± 20 K and δ I = 1.38 ± 0.01 mm/s are obtained. For doublets D2, D3 and D4 the respective ΘM values (see Table 2) were found to be somewhat lower than 300 K. Figure 5 presents the experimental δ(T) values and the calculated curves (solid lines) following this method for the distinct Fe sites in the present vauxite. It is not clear whether the lower ΘM values for the Fe(1) sites reflect any structural feature concerning the Fe–O bonding at these sites related to the presence of the four water molecules in the first coordination sphere comparatively to the Fe(2) sites, or that they are merely the result of the constraints used in the analysis of the MS, and hence of the possibly less precise values of the adjusted δ values. Also, one must bear in mind that the limited temperature range of available experimental δ data will certainly affect the accuracy of the iterated ΘM values. However, the instability of the vauxite structure at temperatures above ca. 320 K as determined from the TGA–DTA analysis, does not allow extending the temperature interval of the MS measurements to higher temperatures. Finally, it should be noted that the result for ΘM,D1 is in line with a general tendency observed for Fe-bearing compounds, that Mössbauer temperatures for ferrous sites are commonly found to lie in the range 300–400 K, while for ferric sites values close to or exceeding 500 K are usually calculated (De Grave and Van Alboom 1991; Eeckhout and De Grave 2003a).
Temperature dependence of the quadrupole splitting
The quadrupole splitting ΔE Q as observed in a doublet spectrum is calculated by the relation (Ingalls 1964):
in which
In foregoing equation, e represents the proton charge, Q is the nuclear quadrupole moment and V zz (often denoted as eq) stands for the principal component of the diagonalized electric field gradient (EFG) tensor, being positive or negative depending on the local symmetry experienced by the probe 57Fe nuclei. η = |V xx − V yy |/V zz is the asymmetry parameter, being zero for axial symmetry. The quantities V ii are the diagonal elements of the EFG tensor organized so that V zz > V xx ≥ V yy.
In general, the components of the EFG, V ij (i, j = x, y, or z) are each composed of a valence term and a lattice term, i.e.:
R and γ∞ being the Sternheimer shielding and anti-shielding factors, respectively (Sternheimer 1972). The valence term (indicated by subscript val) reflects the contribution of the non-spherical charge distribution of the 3d-”valence” electrons of the Fe2+ ion, while the lattice term (indicated by subscript latt) originates from the charge distribution of the neighboring ions in the crystalline lattice.
The valence contribution typically being the largest one for Fe2+, largely explains the temperature dependence of ΔE Q by the Boltzmann population of the lower ferrous 5D energy levels, of which the relative positions are determined by the local crystal field, and therefore, by the corresponding ferrous site symmetry. Generally, the primary effect of the crystal field is to lift the fivefold spatial degeneracy of the 5 D state. In the case of a cubic field, the five orbital levels are split in a higher orbital doublet (E g) and a lower orbital triplet (T 2g). Further distortion of the crystallographic site can lift the orbital degeneracy of the 5 D orbital energy level scheme fully, giving rise to five separate orbital levels, depending on the site symmetry. Spin–orbit interaction further lifts the fivefold spin degeneracy of the orbital levels, giving rise to 25 levels in the most extreme case. Qualitatively one can say that a relative strong decrease of the quadrupole splitting as function of temperature occurs when the distortion of the local octahedral symmetry of the ferrous sites is relatively small, yielding a relatively small orbital-splitting of the lower 5D energy levels. On the other hand, a strong distortion of the local octahedral ferrous coordination typically causes a larger gap between the lower 5D levels by which the decrease of the quadrupole splitting as function of temperature will be smaller.
From these considerations and judging the experimental ΔE Q values as function of T in Fig. 6, D2, D3 and D4 concern Fe2+ in relative stronger distorted sites than for D1. For D1, here an additional feature can be observed for the ΔE Q values as function of T. When the splitting of the two lowest 5D orbital levels is relatively small (≤ca. 500 cm−1), also spin–orbit interaction between the lowest energy levels becomes visible in the course of the quadrupole splitting as function of T (Eeckhout and De Grave 2003b). This effect yields an increase of ΔE Q(T) at low temperatures when temperature increases, yielding a maximum at a certain T, which then is followed by a decrease of ΔE Q when T further increases.
A more quantitative evaluation of ΔE Q(T) can be obtained by, e.g., a crystal field determination of the relative positions of the ferrous 5D levels as described in Van Alboom et al. (2009, 2011, 2015). To calculate the valence contributions and the lattice contributions to the EFG components, all the ions are considered as point charges. Spin–orbit coupling also can be taken into account. In calculating the lattice contributions, the charge numbers (Z) of the ions in the lattice summations were based on the chemical formula of vauxite (\(Z_{{{\text{Fe}}^{2 + } }} = + 2\), \(Z_{{{\text{Al}}^{3 + } }} = + 3\), \(Z_{{{\text{P}}^{5 + } }} = + 5\), \(Z_{{{\text{O}}^{2 - } }} = - 2\), \(Z_{{{\text{H}}_{2} {\text{O}}}} = 0\), \(Z_{{{\text{OH}}^{ - } }} = - 1\)). The calculation of the valence contribution to ΔE Q(T) is based on the positions and the charges of the oxygens in the first coordination sphere of the Fe2+ probe. In the octahedral coordination of an Fe(1) ion four of the six oxygens are bounded in water molecules. In comparison with the other two oxygens, it is believed that the influence of these water oxygen atoms on the EFG is somewhat toned through the H ions in the water molecules. To take into account this effect in the calculation of the valence contribution to the EFG, the charge of these oxygens was somewhat reduced in comparison with the other two oxygens. An analogous method already explained successfully the course of the ferrous quadrupole splitting as function of temperature in szomolnokite (Van Alboom et al. 2009) and in eosphorite (Van Alboom et al. 2015). Therefore, as for these minerals, in the crystal field calculation of the valence contribution to the EFG of the Fe atoms in the present vauxite, effective charges of −1.0e for the water oxygen atoms in the first coordination sphere of Fe(1) were used, and analogously −1.5e for the hydroxyl oxygen atoms in the first coordination sphere of Fe(2), respectively. The relative positions of the oxygens and other ions in all these calculations were taken from the crystallographic data of vauxite determined by Baur and Rama Rao (1968).
It is noticed that the calculations were done for the ideal vauxite structure and that the deficiency of Fe2+ or OH− for example, as indicated in the chemical formula of the present vauxite, was not taken into account.
By fitting the as-developed theoretical model to the experimental ΔE Q(T) curve for the present vauxite sample, five parameters have to be evaluated: 〈r 2〉, 〈r 4〉, λ, ΔE 0 and C lat. 〈r 2〉 and 〈r 4〉 (and analogously 〈r −3〉, see further) are quantum mechanical expectation values for r 2 and r 4 of the radial part of the 5D wave functions. λ is the spin–orbit interaction constant and ΔE 0 is a proportionality factor that is commonly used in EFG calculations and that arises from the practice of expressing the valence contributions to the EFG components in units \(\frac{4}{7}e \langle r^{ - 3}\rangle\). In that respect, ΔE 0 can be considered as the free-ion zero-Kelvin axial valence term and is given by:
Analogously, C lat is a proportionality coefficient in the lattice contribution ΔE Q, and is given by:
Values for the various constants used in these calculations and formulas have previously been calculated for free ions from theoretical models and can be found in literature: Q = 0.15 barn (Lauer et al. 1979), 〈r 2〉 = 2.295 a.u. and 〈r 4〉 = 14.0 a.u. (Zhao and Du 1983), 〈r −3〉 = 4.93 a.u. (Freeman and Watson 1963), R = 0.12 and γ∞ = −10.97 (Sternheimer 1972; Lauer et al. 1979), λ = −103 cm−1 (Ballhausen 1962). Note that 1 a.u. refers to a length dimension equals 5.29 × 10−11 m. With these theoretical values, ΔE 0 = 3.76 mm/s and C lat = 2.69 10−3 nm3 mm/s were calculated. In real cases, however, ΔE 0 can be (strongly) reduced from its theoretical value due to covalence effects. Also C lat can differ from its free ion value in real lattices. The same is valid for 〈r 2〉, 〈r 4〉 and λ. Hence, all these constants were considered as parameters to be adjusted to the experimental ΔE Q(T) curve. The results of this procedure are indicated in Table 3 for the respective Fe sites in the vauxite sample, with the exception of D4-Fe(1). For this doublet, it is meant that the overlap with the other doublets makes it impossible to obtain a reliable temperature dependence of ΔE Q that could be described by the model. Table 3 also gives the corresponding calculated positions of the 5D orbital levels relative to the ground state, Δ i (i = 1,…,4), respectively. Only for D1, spin–orbit interaction was taken into account. For the other doublets the influence was negligible and not indicated. The full lines in Fig. 6 represent the corresponding theoretical ΔE Q(T) curves. As Fig. 6 shows, the agreement between experimental and calculated ΔE Q(T) is quite acceptable.
Generally, the contribution of the higher E g levels can be neglected in the calculation of ΔE Q(T), as is the case for D1-Fe(2) (Ingalls 1964). The reason for this is the typically relative high value of the so-called crystal field splitting 10Dq for Fe2+, as is the case in distorted octahedral O2− co-ordinations, a parameter that among other things is related to 〈r 4〉. Typically, this parameter cannot be determined accurately from the adjustment to the experimental ΔE Q(T), and therefore, was constraint on its theoretical free ion value in the case of D1-Fe(2). The strong reduction of the values for ΔE 0 and 〈r 2〉 in comparison with the free ion values indicate a strong covalence of the Fe–O bonds for Fe(2), a phenomenon that inherently cannot be accurately taken into account by a point-charge calculation. Also the reduction of λ is due to covalency effects.
Comparing the ΔE Q(T) dependencies of D1, D2 and D3, one observes that ΔE Q,D3 varies less as function of T than ΔE Q,D1 and ΔE Q,D2 do. To describe ΔE Q(T) accurately for the D2-Fe(1) and D3-Fe(1) doublets, also 〈r 4〉 had to be adjusted. The lower value of 〈r 4〉 shifts the fourth orbital level to values where it may also contribute to the valence terms of the EFG. It is meant that this effect is induced by the water molecules in the Fe(1) coordination. For D3-Fe(1) all the orbital level splittings are somewhat larger than for D2-Fe(1), causing a less varying ΔE Q as function of T for D3-Fe(1) with respect to D2-Fe(1). It is believed that the differences in the adjusted parameters for D2 and D3 are due to the deficiency of water molecules in the second coordination spheres of the Fe(1) site which cause small variations in the local charge distribution around these Fe(1) and these in turn cause changes in the ferrous 5D level scheme and as consequence influence the valence contribution to the EFG differently and finally also ΔE Q(T). Although these effects cannot be reproduced as such within the as-developed crystal field model, the differences in the adjusted parameters, therefore, reflects the differences caused by the presence or the absence of water molecules in the second coordination spheres of Fe(1).
Conclusion
Based on the results of EDX and TGA on the one hand, and the interpretation of the Mössbauer spectra as function of temperature on the other hand, the chemical formula for the present vauxite is determined, showing water deficiency in the structure. From the temperature dependences of the center shifts, values of ΘM,D1 = 320 ± 20 K and δ I = 1.38 ± 0.01 mm/s are determined for Fe(2) in agreement with values at ferrous sites in other Fe2+-bearing minerals. However, the value of ΘM for Fe(1) is somewhat lower than those values. The temperature dependences of the quadrupole splittings are satisfactorily reproduced based on crystal field calculations of the 5 D orbital energy level schemes of the corresponding ferrous sites. The maximum in the Fe(2) ΔE Q(T) curve is attributed to spin–orbit interaction. 5 D energy level splittings of the second and third orbital level with respect to the lowest orbital level are ca. 410 and 2040 cm−1 for Fe(2) and ca. 620 and 1120 cm−1 for Fe(1), respectively.
References
Ballhausen CJ (1962) Introduction to ligand field theory. McGraw-Hill Book Company Inc, New-York
Baur WH (1969) The crystal structure of paravauxite, Fe2+Al2(PO4)2(OH)2(OH2)6.2H2O. Neues Jahrbuch fur Mineralogie, Monatshefte:430-433
Baur WH, Rama Rao B (1967) The crystal structure of metavauxite. Die Naturwissenschaften 54:561
Baur WH, Rama Rao B (1968) The crystal structure and the chemical composition of vauxite. Am Miner 53:1025–1028
De Grave E, Van Alboom A (1991) Evaluation of ferrous and ferric Mössbauer fractions. Phys Chem Miner 18:337–342
Eeckhout SG, De Grave E (2003a) Evaluation of ferrous and ferric Mössbauer fractions: Part II. Phys Chem Miner 30:142–146
Eeckhout SG, De Grave E (2003b) Fe-57 Mössbauer-effect studies of Ca-rich, Fe-bearing clinopyroxenes: Part I. Paramagnetic spectra of magnesian hedenbergite. Am Miner 88:1129–1137
Ericsson T, Wäppling R (1976) Texture effects in 3/2 → 1/2 Mössbauer spectra. J Phys Colloq C6(37):719–723
Freeman AJ, Watson RE (1963) Antishielding of magnetic and electric hyperfine interactions in open shell ions. Phys Rev 131:2566–2573
Greneche JM, Varret F (1982) On the texture problem in Mossbauer spectroscopy. J Phys C Solid State Phys 15:5333–5344
Ingalls R (1964) Electric-field gradient tensor in ferrous compounds. Phys Rev 133:787–795
Lauer S, Marathe VR, Trautwein A (1979) Sternheimer shielding using various approximations. Phys Rev A 19:1852–1861
Pound RV, Rebka GA Jr (1960) Variation with temperature of the energy of recoil free gamma rays from solids. Phys Rev Lett 4:274–277
Sternheimer RM (1972) Quadrupole shielding and antishielding factors for several atomic ground states. Phys Rev A 6:1702–1709
Van Alboom A, De Resende VG, De Grave E, Gomez JM (2009) Hyperfine interactions in szomolnokite (FeSO4·H2O). J Mol Struct 924–926:448–456
Van Alboom A, De Grave E, Wohlfahrt-Mehrens M (2011) Temperature dependence of the Fe2+ Mössbauer parameters in triphylite (LiFePO4). Am Miner 96(2–3):408–416
Van Alboom A, De Resende VG, da Costa GM, De Grave E (2015) Mössbauer spectroscopic study of natural eosphorite, (Mn, Fe)AlPO4(OH)2H2O. Am Miner 100(2–3):580–587
Ventruti G, Schingaro E, Monno A, Lacalamita M, Della Ventura G, Bellatreccia F, Cuocci C, Rossi M, Capitelli F (2016) Structure refinement and vibrational spectroscopy of vauxite from the type locality, Llallagua (Bolivia). Can Mineral 54:163–176
Zhao M, Du M (1983) Two-center transitions in the anti-ferromagnetic salt FeCO3. Phys Rev B 28(11):6481–6484
Acknowledgements
This work was funded by the Fund for Scientific Research—Flanders, Belgium.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Van Alboom, A., da Costa, G.M. & De Grave, E. Deficiency of water molecules in the crystallographic structure of vauxite. Phys Chem Minerals 45, 249–257 (2018). https://doi.org/10.1007/s00269-017-0913-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00269-017-0913-2