
Abstract
Green clay and chrysoprase samples from the Ni-laterite deposit in Szklary, Poland, were studied by X-ray diffraction (XRD), electron microprobe analysis (EMPA), Raman, infrared and UV–VIS–NIR absorption spectroscopies. The clay samples proved to be Ni-rich “kerolite” and “pimelite”, and as these minerals are currently discredited, their nature was studied in more detail by high-temperature X-ray diffraction and thermal analysis coupled with mass spectrometry. These methods showed that “kerolite” and “pimelite” contain interlayer water, i.e. they differ from the non-hydrated talc–willemseite mineral series. Subsequently, the XRD and EMPA analyses of Szklary chrysoprase indicated that chrysoprase’s colouring agent is “pimelite”, which was further corroborated by Raman and UV–VIS–NIR spectroscopies. In addition, the Szklary chrysoprase, which has been quarried since the Middle Ages and used in jewellery and wall decorations (e.g. in the Prague Cathedral), was described from the gemmological point of view.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
“Pimelite” is a nickel-bearing clay mineral, but its precise definition remains rather controversial. It is currently not approved as a valid mineral species by the International Mineralogical Association—it was discredited in 2006, during the mass discreditation of almost 130 minerals in order to create an updated version of the mineral database, when no re-examination of type materials was undertaken due to the high number of discredited mineral species (Burke 2006). In addition, there has always been a discussion about the nature of the mineral—whether it belongs to expandable or non-expandable clay minerals.
The most comprehensive work on “pimelite” has been performed by Brindley and his colleagues (Brindley and Pham 1973; Brindley and Maksimović 1974, Brindley et al. 1979). They state that “pimelite” belongs to the 2:1 trioctahedral silicates with generally non-expandable talc-like structure and basal spacing at 1.0 nm. With the corresponding magnesium-rich mineral “kerolite” (also discredited), they form the Ni–Mg “pimelite–kerolite” series—the disordered and hydrated equivalent of willemseite–talc series. This seems to be the most approved “pimelite” definition (Moore and Reynolds 1997; Weiss and Kužvart 2005). However, some authors (Faust 1966) have described the structure of “pimelite” as expandable, belonging to the smectite group, but have not presented any conclusive data.
The essential problem of “pimelite’s” characterisation is its potential content of interlayer water, which would make it the hydrated equivalent of willemseite. Brindley et al. (1979) performed thermogravimetric analysis of “pimelite” and attributed the gradual weight loss between 110 and 730 °C to molecular water. However, they did not support it by the analysis of evolved gases. Miller et al. (1991) employed differential thermal analysis (DTA) for the study of “kerolite” under elevated pressures. They supposed that the observed broad peak on the DTA curve of “kerolite” is related to several thermal events and suggested the presence of interlayer water based on the enthalpy of “kerolite’s” dehydration, but they did not present any direct evidence.
“Pimelite” is typically formed in Ni-laterites, the soil layers derived from the weathering of ultramafic rocks under strongly oxidising conditions. In Europe, important deposits of this type are located in Greece and three adjacent countries in the Balkan Peninsula—Albania, Macedonia and Serbia (Berger et al. 2011). From the historical point of view, the most significant Ni-laterite deposit is located in Szklary, Lower Silesia, Poland. It has been exploited for nickel since the end of the 19th century, but its importance lies in the production of gem-quality chrysoprase, precious apple-green variety of chalcedony (Niśkiewicz 1982). Chrysoprase has been quarried there since the Middle Ages—its polished slabs are documented, e.g. in the Chapel of St. Wenceslaus in the Prague Cathedral at Prague Castle, Czech Republic, which was constructed in the end of the 14th century (Kostílková and Chotěbor 1999). In this period of time, Silesia was part of the Bohemian Kingdom, the historical predecessor of today’s Czech Republic.
The colouring agent of chrysoprase has also been a subject of long-lasting debates. There is a general agreement that the colour is caused by inclusions of Ni-rich mineral; Sachanbiński et al. (2001) reviewed the topic and stated two main theories—the colour is caused by the presence of either bunsenite (cubic NiO), or layered Ni-rich silicates. While Dyrek et al. (2001) excluded the possibility of the presence of dispersed bunsenite in chrysoprase based on UV–VIS–NIR spectroscopy, Sachanbiński et al. (2001) used transmission electron microscopy coupled with energy-dispersive spectrometry for the analysis of chrysoprase from Szklary, Poland, as well as Sarykul Boldy, Kazakhstan, and found Ni–Mg silicate aggregates with approx. 1.0 ± 0.7 nm spacing. Such structural data are not very specific; nevertheless, the authors attributed them (in combination with observed textures and chemical composition) to Ni-rich “kerolite” and minor amount of “pimelite”. Graetsch (2011) analysed chrysoprase samples from Haneti, Tanzania, using absorption spectroscopy and X-ray diffraction (XRD) and came to the conclusion that this chrysoprase is coloured by ca 0.5 wt % of gaspeite [hexagonal (Ni, Mg, Fe) CO3]. However, the performed XRD analysis has one significant drawback: the measured range covers only 15°–100° 2 Theta and therefore excludes the low-angle area diagnostic for the presence of clay minerals, i.e. also “kerolite” and “pimelite”.
In order to clarify whether “pimelite” is a distinct mineral species, we have decided to study Szklary “pimelite” reference samples by electron microprobe analysis (EMPA), and especially high-temperature X-ray diffraction (HT-XRD), differential thermal analysis (DTA) and thermal gravimetry (TG) coupled with mass spectrometry. The main focus was placed on the potential presence of water in the interlayer. To obtain further evidence regarding the colouring agent of Szklary chrysoprase, the chrysoprase samples were analysed by X-ray diffraction with special attention to the low-angle area. In addition, Raman, Fourier transform infrared (FTIR) as well as optical absorption spectroscopy was applied on both “pimelite” and chrysoprase samples to study the suggested presence of “pimelite” in chrysoprase. Finally, the Szklary chrysoprase material was described also from the gemmological point of view.
Materials and methods
The reference samples of green clay minerals were collected during a field trip to Szklary, Poland, which took place in October 2014. They were found on spoil heaps in the form of bluish green veins in the Mg-rich matrix, mostly containing smectite and serpentine group minerals together with quartz and talc (Fig. 1a). Ten samples were obtained in total. In the laboratory, they were carefully separated from the matrix and powdered for the SEM–EDS and XRD analyses. One of the samples was embedded in epoxy resin and cut in cross-section for the elemental EMPA analysis and Raman microspectroscopy (Fig. 1b). The occurrence of high-quality chrysoprase on the Szklary locality is much scarcer; therefore, the studied samples were obtained from a reputable mineral dealer (Minerály Jiří Špalek). Three cabochons were cut and polished from the chrysoprase raw; two of them are depicted in Fig. 1c. One part of the chrysoprase raw material was embedded in epoxy resin, cut and polished for the EMPA analysis.

(Micro-)photographs of the studied samples: a vein of the green clay mineral, b representative polished cross-section of the green clay mineral, c two chrysoprase cabochons (0.52 and 6.30 ct, i.e. 0.103 and 1.261 g)
Gemmological analysis
For the gemmological study of chrysoprase, the following equipment was employed: Axio Imager A.2 light microscope (Zeiss) coupled with digital microscope camera Olympus DP 75 and UV unit (365 nm) for visual observation, photographic documentation and UV luminescence assessment; Duplex II refractometer (GIA) for the measurement of refractive index; Sartorius TE64 precision balance with hydrostatic unit for the determination of hydrostatic specific gravity; and Chelsea colour filter (GIA) for the study of optical properties.
Scanning electron microscopy–energy-dispersive spectroscopy (SEM–EDS)
The powdered green clay mineral sample was studied by Jeol JSM6510 scanning electron microscope equipped with energy-dispersive spectrometer INCA (Oxford Instruments) with SDD detector allowing detection of elements heavier than Be (Z > 4) at resolution of 125 eV. Measurements were carried out in a low-vacuum mode under 30 Pa and accelerating voltage of 25 kV; backscattered electrons were detected. Low-vacuum mode allowed analysis of the sample without conductive coating of its surface.
The polished cross-section was analysed by TESCAN VEGA 3 XMU scanning electron microscope equipped with energy-dispersive system Quantax Bruker with SDD detector under following measurement conditions: accelerating voltage 15 kV, working distance 20 mm, BSE mode.
Electron microprobe analysis (EMPA)
The elemental analysis was performed on electron microprobe analyser CAMECA SX-100 equipped with four crystal spectrometers (with the possibility to quantify elements from B to U), back-scattered and secondary electrons detectors. For the calibration, the following standards were employed: diopside (for the measurement of Ca, Si, Mg), jadeite (Al, Na), nickel (Ni), haematite (Fe), rhodonite (Mn), sanidine (K), Mn Cr spinel (Cr) and cobalt (Co). The accelerating voltage was 15 kV, current 10 nA, beam diameter 5 µm and a counting time of 10, 20 or 30 s depending on the expected concentration of the element in the sample. The structural formulae were calculated on the O10(OH)2 basis.
X-ray diffraction (XRD)
Diffraction patterns were collected on PANalytical X’Pert PRO diffractometer equipped with conventional X-ray tube (CoKα radiation, 40 kV, 30 mA, line focus) and a multichannel detector X’Celerator with an anti-scatter shield. X-ray patterns were measured in the range of 4°–85° 2 Theta with the step of 0.0167° and 550 s counting per step (total time of the scan: approx. 6 h), using conventional Bragg–Brentano geometry. XRD patterns were not pre-treated before interpretation as no background correction was needed. Qualitative analysis was performed using HighScore Plus software package (PANalytical, The Netherlands, version 3.0.5) and JCPDS PDF-2 database (Release 2004).
The oriented specimen was prepared according to Moore and Reynolds (1997). It was air-dried and analysed by XRD in the range of 2°–40° 2 Theta with a step of 0.0167° and 100 s counting per step providing a scan of about 30 min. All other experimental parameters were kept the same as in the analysis of randomly oriented samples. Then, the oriented specimen underwent ethylene glycol solvation for 5 h at 70 °C. Immediately after the solvation, the specimen was re-analysed by XRD under the same experimental conditions as the air-dried specimen.
High-temperature X-ray diffraction (HT-XRD)
High-temperature X-ray diffraction patterns were collected using the same diffractometer and setting as for the normal room-temperature measurements (see above) together with high-temperature chamber (HTK 16, Anton Paar, Graz, Austria). X-ray patterns were measured at 25 °C and between 30 and 300 °C with a step of 10 °C in the range of 6.5°–36° 2 Theta (step of 0.0334° and 80 s counting per step yielding a scan of ca 10 min) using conventional Bragg–Brentano geometry. The measurements were performed while the sample was heated to the designated temperature, i.e. in situ.
Thermogravimetry (TG) and differential thermal analysis (DTA) coupled with mass spectrometry
The measurement was performed on SetSys Evolution (SETARAM) coupled with quadrupole mass spectrometer QMG700 (PFEIFFER) and took place under inert atmosphere (argon, 60 mL/min). The powdered sample (10 mg) was heated in an open α-Al2O3 crucible, from room temperature to 1000 °C with a step of 5 °C/min. Gaseous products were analysed in MID mode (multiple ion detection) as an intensity A of selected fragments, namely m/z = 16, 17, 18, 44 and 64.
Raman spectroscopy
Micro-Raman spectra of the samples were obtained on a Thermo Scientific DXR Raman Microscope with Peltier-cooled CCD detector interfaced to an Olympus microscope (10× and 50× objective lens) in the 50–3400 cm−1 spectral region with 4 cm−1 resolution. The spectrometer was calibrated by software-controlled calibration procedure using multiple neon emission lines (wavelength calibration), multiple polystyrene Raman bands (laser wavenumber calibration) and standardised white light sources (intensity calibration). The spectra were collected using four laser wavelengths (445, 532, 633 and 780 nm) with the following typical analytical conditions: 64 scans, 2 s exposure time; chrysoprase was generally analysed with full laser power, green clay samples often required restricted laser power due to occurring fluorescence signal (e.g. 2 mW with the 532 nm excitation wavelength).
Fourier transform infrared (FTIR) spectrometry
FTIR spectra were obtained using Thermo Scientific Nicolet NEXUS 670 FTIR in transmission mode. The samples were powdered and prepared in KBr pellets—the amount of the samples for the analysis ranged from 0.8 to 1.3 mg/300 mg KBr. The acquisition was set on 64 scans in the range of 4000–400 cm−1, resolution being 4 cm−1.
UV–VIS–NIR absorption spectroscopy
The UV–VIS–NIR spectroscopy was performed on UV–VIS spectrophotometer Lambda 35 from PerkinElmer Instruments using an integrating sphere, in the range of 250–1100 nm. Due to high noise in the UV part, the spectra are presented in the range of 380–1100 nm.
Results and discussion
“Pimelite” characterisation
As a first step, green clay mineral samples collected in Szklary were measured by XRD. It enabled the selection of five samples from one vein, which proved to be mineralogically consistent with the 0.99 nm d-spacing indicating the talc-type structure. To ensure sufficient amount of the sample for the analyses, they were mixed into one bulk powdered sample and one part of the vein was cut and polished in cross-section.
Subsequently, the cross-section (Fig. 1b) was analysed by SEM–EDS. The image in back-scattered electron (BSE) mode showed the distribution of phases in the vein (Fig. 2), and the EDS analysis indicated the presence of SiO2 phase and two different Ni–Mg silicates. After that, the sample was subjected to EMPA analysis. Twenty spot analyses confirmed that there are indeed two Ni–Mg silicate phases, and the analyses were highly consistent among each type. Analyses of each phase are summarised in Table 1. They correspond to the following structural formulae: Ni1.04Mg2.02Si3.97O10(OH)2·nH2O (Ni-rich “kerolite”) and Ni2.73Mg0.39Si3.94O10(OH)2·nH2O (“pimelite”). The atomic ratios of Si/(Si + Mg + Ni) slightly oscillated around 0.56; such ratio is by 0.05 higher than the one stated by Cathelineau et al. (2015), but in full agreement with the data stated by Brindley et al. (1979). The atomic ratio of Ni/(Ni + Mg) ranges from 0.29 to 0.37 in case of Ni-rich “kerolite” and from 0.87 to 0.89 in case of “pimelite”.

SEM image in BSE mode of the polished cross-section indicating the distribution of phases in the vein (white “pimelite”, lighter grey Ni-rich “kerolite”, darker grey SiO2 phase)
Even though the Ni-rich “kerolite” and “pimelite” are easily discernible on the BSE image (Fig. 2), their visual appearance (Fig. 1b) is almost the same, which prevented their separation in powdered sample used for the following bulk analyses.
The XRD analysis of the powdered sample showed a pattern of a single 2:1 clay mineral with d-spacing at 0.99 nm mixed with a small amount of quartz (Fig. 3). Such d-spacing is higher than that of talc (0.934 nm) as well as willemseite (0.940 nm) (Ross et al. 1968; Shameli et al. 2010; De Waal 1970; Villanova-de-Benavent et al. 2014), which corresponds with the idea of the presence of interlayer water in “kerolite” and “pimelite”. Brindley et al. (1979) mentioned the problem of obtaining sufficiently pure “kerolite”/“pimelite” samples for the analysis. It has been encountered, e.g. by Dubińska et al. (2000), who presented XRD patterns of Ni-rich clay minerals from Szklary containing (in most cases) not only the d-spacing at 1.01 or 1.02 nm, but also a smectite-like d-spacing at 1.24 and 1.26 nm, which they ascribed to the admixture of stevensite [monoclinic (Ca,Na) x Mg3−x (Si4O10)(OH)2]. To ascertain that the studied sample is not mixed with stevensite or any other expandable clay mineral, an oriented air-dried specimen was prepared and analysed by XRD, then subjected to ethylene glycol solvation and, again, analysed by XRD. The respective patterns are depicted in Fig. 3. The XRD pattern after solvation clearly shows that no expandable clay minerals are present; the d-spacing did not increase.

XRD patterns of the studied bulk green clay mineral sample—0.99 nm “pimelite” and Ni-rich “kerolite” together with a small admixture of quartz (Q): randomly oriented, oriented (air-dried) and oriented specimen after ethylene glycol solvation
The elemental and structural data indicated that the samples are composed of “kerolite” and “pimelite” and showed their significant structural disorder. Since these minerals have been discredited, further analyses were performed to assess whether the samples contain interlayer water and, therefore, represent distinct mineral species. The powdered sample was measured by HT-XRD. Figure 4 depicts the XRD patterns before and after the high-temperature measurement. Even though the difference is small, it can be seen that the main diffraction line of quartz remained exactly on its position, while the 0.99 nm line moved to the right to 0.98 nm due to the loss of water from the interlayer space. This effect is even more visible on Fig. 5, where the main quartz and the 0.99 nm line are depicted in each heating step of the measurement. With increasing temperature, the quartz line gradually moves to the left due to thermal expansion of the unit cell, while in case of the 0.99 nm line, this process is compensated by the loss of interlayer water and resulting shrinkage of the structure, i.e. the line stays on its position and after cool down moves to the 0.98 nm. Similar process can be observed also in Fig. 3, this time caused by the evaporation of interlayer water due to prolonged exposition to increased temperature (70 °C) during ethylene glycol solvation.

XRD patterns of the bulk “kerolite”/“pimelite” sample before and after heating to 300 °C. The heat treatment altered only the position of the “kerolite”/“pimelite” basal lines. Q quartz, b. basal line, n.b. non-basal line of the “kerolite”/“pimelite” sample

HT-XRD analysis of the bulk “kerolite”/“pimelite” sample: 0.99 nm basal “kerolite”/“pimelite” line (b.) and the main quartz line (Q) depicted in each heating step
To further confirm the presence of interlayer water, the powdered sample was subjected to thermal analysis. Figure 6 depicts the two thermogravimetric and DTA curves together with the curves of evolved gases; they indicate four steps of weight loss, with a total amount of 8.6 wt%. The first step represents loss of surface (hygroscopic) water with the fastest reaction at 73 °C and the highest endothermic energy at 90°. The second and the most important step relates to the loss of interlayer water (maximum at 157 °C, endothermic peak at 173 °C). The third step represents gradual weight decrease accompanied by a small release of CO2 (probably caused by decomposition of an admixture of carbonates indicated by the presence of small amount of calcium in the EMPA analyses, see Table 1; another possibility is the release of CO2 from fluid inclusions or CO2 trapped by clay minerals). Finally, the fourth step indicates rapid dehydroxylation with the fastest progress at 815 °C. At this phase, the MS indicated not only the production of hydroxyl groups (fragment 17), but also of water (fragment 18), which seems to be produced by rapid recombination of unstable hydroxyls. Brindley et al. (1979) performed thermogravimetric analyses on several samples of “pimelite” as well as “kerolite” and observed loss of hygroscopic water followed by gradual weight decrease and dehydroxylation, but they did not describe a step corresponding to the loss of interlayer water. Miller et al. (1991) indicated that “kerolite” samples seem to contain a small amount of interlayer water based on the reaction’s enthalpy; however, this is the first study that brings unambiguous data based on HT-XRD and TG/DTA analysis proving that “pimelite” and “kerolite” samples contain interlayer water and that they truly differ from non-hydrated willemseite and talc.

Thermal analysis of the bulk “kerolite”/“pimelite” sample. TG, DTG and DTA curves are in the upper part, while the MS analysis of evolved gases is presented below (fragment 17 indicates hydroxyl groups, fragment 18 water and fragment 44 CO2)
Regarding the actual amount of interlayer water in the minerals, the situation becomes slightly more complicated. Brindley et al. (1979) state that, on average, their “kerolite”/“pimelite” samples contain 0.89 H2O per formula unit. They ascribe the weight loss observable in the thermal analysis in the range of 110–730 °C solely to the loss of water. However, our measurement indicates that the (interlayer) water is released only up to ca 200 °C, and the continuing weight loss up to 730 °C should rather be ascribed to a small release of CO2. Therefore, instead of 4.21 wt% of weight loss attributable to water (phase II and III, 120–750 °C, see Fig. 6), the calculation should be made only with the weight loss in the phase II (120–200 °C, Fig. 6), i.e. only 0.26 wt%. This is in agreement with the assumption of Miller et al. (1991), who expected only a small amount of interlayer water based on the obtained DTA data. Our calculations are further complicated by the fact that there is more than one mineral phase present in the powdered bulk sample (“kerolite” and “pimelite”). Its SEM–EDS analysis indicated that the average ratio of Ni and Mg in the bulk sample is approx. 1:1. Taking into account this approximative mineral composition with an idealised formula of Ni1.5Mg1.5Si4O10(OH)2·nH2O, the amount of the water per formula unit equals to 0.06 H2O. It is not an exact procedure, but as the amount of water is small, the influence of the Ni–Mg average composition on the resulting absolute number of water is almost negligible. The results correspond with the HT-XRD measurements, which showed only a small change to the d-spacing after heating (0.01 nm) indicating only a minor amount of water in the interlayer.
To improve the characterisation of “kerolite”/“pimelite” samples from Szklary, we have recorded their Raman, infrared and UV–VIS–NIR absorption spectra (Fig. 7). The Ni-rich “kerolite” and “pimelite” in the polished cross-section were measured by four Raman excitation wavelengths (445, 532, 633 and 780 nm). All measurements yielded a considerable amount of fluorescence, but the best results were obtained using the 780 nm laser. After careful baseline correction, the Raman spectrum of Ni-rich “kerolite” is presented in Fig. 7a, while the measurement of “pimelite” resulted in only three main bands on similar position as for the Ni-rich “kerolite” (674, 188 and 107 cm−1). The spectrum of “pimelite” was not subjected to baseline correction as its lower quality posed a risk of misinterpretation of noise for actual signal. The infrared spectrum (Fig. 7b) was recorded in transmission mode, i.e. using the bulk powdered sample. The spectrum is strikingly similar to the one of synthetic Mg52Ni48 talc published by Wilkins and Ito (1967), except the presence of prominent water-related bands and the lack of one of the stretching OH vibrations, which is probably covered by the band of water. The UV–VIS–NIR absorption spectrum (Fig. 7c) was also obtained on the bulk sample, and according to Sachanbiński et al. (2001), these absorption bands are related to octahedrally coordinated Ni2+ ions.

Spectra of the “kerolite”/“pimelite” sample: a Raman spectra (780 nm) of Ni-rich “kerolite” (baseline corrected) and “pimelite” (no baseline correction) together with the band positions, b FTIR spectrum of the bulk “kerolite”/“pimelite” sample, c UV–VIS–NIR absorption spectrum of the bulk “kerolite”/“pimelite”
Szklary chrysoprase characterisation
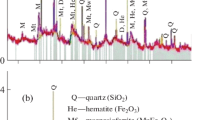
To study the colouring agent of Szklary chrysoprase, the chrysoprase sample was first analysed by XRD with special attention to the low-angle area. Its XRD pattern is depicted in Fig. 8 together with the pattern of “kerolite”/“pimelite” sample described in the previous section. The chrysoprase pattern is dominated by lines of quartz and moganite. In addition, there is also a line corresponding to 0.99 nm d-spacing, which is in very good agreement with the studied “kerolite”/“pimelite” (Fig. 8). Interestingly, similar diffraction line (at approx. 8.7° 2 Theta, CuKα, corresponding to 1.016 nm d-spacing) has been observed also by Hatipoğlu and Yardımcı (2014) in case of chrysoprase from Biga, Çanakkale region in Turkey. Even though the authors left it unassigned, it also indicates the presence of layered silicate with approximate d-spacing at 1.0 nm.

XRD patterns of Szklary chrysoprase and “kerolite”/“pimelite” sample. The dashed line indicates the main diffraction line of “kerolite”/“pimelite”. Q quartz, M moganite, K/P “kerolite”/“pimelite”
As the XRD data cannot provide information on whether the chrysoprase sample contains “kerolite” or “pimelite”, it has been subjected to elemental EMPA analysis to observe its Mg/Ni ratio. The analysed elements were the same as in case of the “kerolite”/“pimelite” sample (Table 1). The chrysoprase contained only negligible amounts of Na, K, Ca, Fe, Mn, Cr, Al and Co (from 0 to 0.0X wt%). The content of magnesium was also very small; five individual measurements gave values ranging around 0.05 wt%. Therefore, with the value of nickel being around 1.5 wt%, it is clear that these samples of Szklary chrysoprase are coloured by “pimelite”—the Ni-rich member of the “kerolite”–“pimelite” series.
To support these results, Szklary chrysoprase was analysed also by Raman, FTIR and UV–VIS–NIR spectroscopies. The Raman spectrum (Fig. 9a) represents a typical chrysoprase spectrum with bands attributable to two SiO2 phases, quartz and moganite (Kingma and Hemley 1994). However, there are two additional bands, one at 674 cm−1 and a shoulder positioned at approx. 188 cm−1 (see inset with resolved bands in Fig. 9a). These are in very good agreement with the two high-intensity bands of both the Ni-rich “kerolite” and “pimelite” analysed in the vein cross-section. The FTIR spectrum (Fig. 9b) again shows the presence of quartz and moganite, but there are no discernible bands of “kerolite”/“pimelite”. The chrysoprase UV–VIS–NIR spectrum (Fig. 9c) shows the absorptions related to octahedrally coordinated Ni2+ ions (Sachanbiński et al. 2001), which agree with the results obtained on the bulk “kerolite”/“pimelite” sample (Fig. 7c).

Spectra of chrysoprase: a Raman spectra (780 nm) of chrysoprase (no baseline correction) together with Ni-rich “kerolite” (baseline corrected); the main bands of “kerolite” are on the same position as “pimelite”; the bands of part of the chrysoprase spectrum were resolved (see the inset), b FTIR spectrum of chrysoprase, c UV–VIS–NIR absorption spectrum of chrysoprase
Finally, Szklary chrysoprase samples were subjected to gemmological analyses. The samples in this study have a very slightly bluish green to green colour. Their refraction index is 1.54, specific gravity is 2.63, and UV fluorescence (365 nm) is none to very weak blue, which is in agreement with the published data for chrysoprase (Shigley et al. 2009). The Chelsea filter reaction [which is used for the detection of chromium in the sample either as artificial colourant or as a rare natural colouring agent in chrome chalcedony (Petrová et al. 2012; Hyršl 1999)] was green, indicating none or negligible amount of chromium in agreement with the EMPA analysis.
Conclusions
Based on the elemental and XRD analyses, green clay samples from Szklary were identified as Ni-rich “kerolite” and “pimelite”. The results of their HT-XRD and thermal analyses proved for the first time that these minerals contain a small amount of interlayer water, and therefore, they differ from the non-hydrated talc and willemseite and represent distinct mineral species.
The long-debated colouring agent of Szklary chrysoprase, a gemstone originating from a historic site of unrivalled importance, was confirmed to be “pimelite” in the studied samples based on the XRD measurements with special attention to the low-angle area and the EMPA elemental analysis.
References
Berger VI, Singer DA, Bliss JD, Moring BC (2011) Ni–Co laterite deposits of the world—a database and grade and tonnage models. U.S. Geological Survey Open-File Report 2011-1058. http://pubs.usgs.gov/of/2011/1058/. Accessed 4 Mar 2016
Brindley GW, Maksimović Z (1974) The nature and nomenclature of hydrous nickel-containing silicates. Clay Miner 10:271–277
Brindley GW, Pham TH (1973) The nature of garnierites—I. Structures, chemical compositions and color characteristics. Clays Clay Miner 21:27–40
Brindley GW, Bish DL, Wan Hsien-Ming (1979) Compositions, structures, and properties of nickel-containing minerals in the kerolite–pimelite series. Am Mineral 64:615–625
Burke EAJ (2006) A mass discreditation of GQN minerals. Can Mineral 44:1557–1560
Cathelineau M, Caumon M-C, Massei F, Brie D, Harlaux M (2015) Raman spectra of Ni–Mg kerolite: effect of Ni–Mg substitution on O–H stretching vibrations. J Raman Spectrosc 46:933–940
De Waal SA (1970) Nickel minerals from Barberton, South Africa: III. Willemseite, a nickel-rich talc. Am Mineral 55:31–42
Dubińska E, Sakharov BA, Kaproń G, Bylina P, Kozubowski JA (2000) Layer silicates from Szklary (Lower Silesia): from ocean floor metamorphism to continental chemical weathering. Geol Sudet 33:85–105
Dyrek K, Sojka Z, Żabiński W, Bozon-Verduraz F (2001) On the problem of the occurrence of dispersed bunsenite (NiO) in chrysoprases. Mineral Pol 32:3–7
Faust GT (1966) The hydrous nickel-magnesium silicates—the garnierite group. Am Mineral 51:279–298
Graetsch HA (2011) Microstructure and origin of colour of chrysoprase from Haneti (Tanzania). Neues Jahrb Miner Abh 188:111–117
Hatipoğlu M, Yardımcı Y (2014) Optical and cathodoluminescence investigations of the green microcrystalline (chrysoprase) quartz. J Lumin Appl 1:87–104
Hyršl J (1999) Chrome chalcedony, a review. J Gemmol 26:364–370
JCPDS PDF-2 database (2004) International Centre for Diffraction Data, Newtown Square, PA, U.S.A. release 54
Kingma KJ, Hemley RJ (1994) Raman spectroscopic study of crystalline silica. Am Mineral 79:269–273
Kostílková M, Chotěbor P (1999) The chapel of St. Wenceslaus, Prague Castle Administration, Prague (in Czech)
Miller AK, Guggenheim S, van Groos AFK (1991) Bond energy of adsorbed and interlayer water: kerolite dehydration at elevated pressures. Clays Clay Miner 39:127–130
Moore DM, Reynolds RC Jr (1997) X-ray diffraction and the identification and analysis of clay minerals, vol 2. Oxford University Press, Oxford
Niśkiewicz J (1982) Geological setting of the occurrence of chrysoprase and related gemstones in the Szklary Massif, Lower Silesia. Geol Sudet 17:125–139 (in Polish with English summary)
Petrová Z, Jehlička J, Čapoun T, Hanus R, Trojek T, Goliáš V (2012) Gemstones and noble metals adorning the sceptre of the Faculty of Science of Charles University in Prague: integrated analysis by Raman and XRF handheld instruments. J Raman Spectrosc 43:1275–1280
Ross M, Smith WL, Ashton WH (1968) Triclinic talc and associated amphiboles from Gouverneuer mining district, New York. Am Mineral 53:751–769
Sachanbiński M, Janeczek J, Platonov A, Rietmeijer JM (2001) The origin of colour of chrysoprase from Szklary (Poland) and Sarykul Boldy (Kazakhstan). Neues Jahrb Miner Abh 177:61–76
Shameli K, Ahmad MB, Yunus WZW, Ibrahim NA, Darroudi M (2010) Synthesis and characterization of silver/talc nanocomposites using the wet chemical reduction method. Int J Nanomed 5:743–751
Shigley JE, Laurs BM, Renfro ND (2009) Chrysoprase and prase opal from Haneti, Central Tanzania. Gems Gemol 45:271–279
Villanova-de-Benavent C, Tredoux M, Aiglsperger T, Proenza J (2014) Ni–Mg-phyllosilicates from Bon Accord, Barberton, South Africa: new data on willemseite and nimite. 21st General meeting of the IMA (IMA2014), Gauteng, South Africa, September 1–5, Abstract Volume, p 212
Weiss Z, Kužvart M (2005) Clay minerals: their nanostructure and application. Charles University in Prague, Karolinum Press, Prague (in Czech)
Wilkins RWT, Ito J (1967) Infrared spectra of some synthetic talcs. Am Mineral 52:1649–1661
Acknowledgments
The authors would like to express their thanks to Monika Palkovská (Institute of Inorganic Chemistry of the CAS) for the performance of the thermal analysis, Irena Matulková (Faculty of Science, Charles University in Prague) for her help during Raman spectroscopic measurements and Zuzana Korbelová (Geological Institute of the CAS) for the EMPA analyses. The authors are also grateful to Jaroslava Jabůrková (Geological Institute of the CAS), who cut and polished the chrysoprase cabochons. Vlasta Vašutová and Veronika Kohútová (Academy of Fine Arts in Prague) are acknowledged for their versatile cooperation. This research was supported by the Czech Science Foundation, project no. 14-22984S and by Institutional Budgets of Research Institutions (RVO 61388980 and 60461446).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Čermáková, Z., Hradil, D., Bezdička, P. et al. New data on “kerolite–pimelite” series and the colouring agent of Szklary chrysoprase, Poland. Phys Chem Minerals 44, 193–202 (2017). https://doi.org/10.1007/s00269-016-0848-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00269-016-0848-z