
Abstract
The crystal structure of hydroxide perovskite Ga(OH)3, the mineral söhngeite, has been determined for a natural sample by single-crystal XRD in space group P42/nmc to R 1 = 0.031, wR 2 = 0.071, GoF = 1.208, and for comparison also in space group P42/n to R 1 = 0.031, wR 2 = 0.073, GoF = 1.076. Unit cell parameters are a = 7.4546(2) Å, c = 7.3915(2) Å, V = 410.75(2) Å3. The two structures are very similar and both have tilt system a + a + c −. The approximate positions of all H atoms in each structure have been refined. In the P42/nmc structure all five H sites are half-occupied, whereas in the P42/n structure four sites are half-occupied and one is fully occupied. The presence of five non-equivalent OH groups in söhngeite is confirmed by single-crystal Raman spectroscopy, but does not allow a choice between these two space groups to be made. There is only a single very weak violator of the c-glide of P42/nmc and the two refined structures are essentially the same, but are significantly different from that of the original description in which orthorhombic space group Pmn21 was reported with corresponding tilt system a 0 a 0 c +. It is argued here that such a structure is very implausible for a hydroxide perovskite. On heating söhngeite to 423 K, transformation to a cubic structure with \(Im\bar{3}\) symmetry (a + a + a +) of the aristotype occurs. This cubic phase was recovered on cooling to 293 K without back-transformation to the tetragonal polymorph. As there is no continuous group/subgroup pathway from P42/nmc (or P42/n) to \(Im\bar{3}\), the transformation must be first-order, which is consistent with the large hysteresis observed. The change from the tetragonal to cubic structures involves a change in tilt system a + a + c − → a + a + a +, with a significant reconfiguration of hydrogen-bonding topology. The very different tilt systems and hydrogen-bonding configurations of the two polymorphs are responsible for hysteresis and metastable preservation of the cubic phase at 293 K. As the Ga(OH)6 octahedra of the low- and high-T polymorphs are very similar it is inferred that the transformation is driven by proton behaviour, presumably involving proton re-ordering.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hydroxide perovskites are frameworks composed of corner-linked octahedra in which all O atoms form OH groups and, unlike perovskites, there is no A cation. Extensive hydrogen bonding, coupled with collapse of the framework around the empty A site, is responsible for highly tilted octahedra. Transformational behaviour in hydroxide perovskites is largely unexplored. The simple topology of hydroxide perovskites allows the structural role of hydrogen bonding to be evaluated.
There are two general stoichiometries: BB′(OH)6 and B(OH)3 (B = B′). “Double hydroxide perovskites” are analogous to double perovskites in that they have a framework composed of alternating B and B′ octahedra in which the cations are different (Howard et al. 2003). “Single hydroxide perovskites” have a single cation type, e.g. In(OH)3. Natural examples of double hydroxide perovskites include burtite CaSn(OH)6, vismirnovite ZnSn(OH)6, schönfliesite MgSn(OH)6, natanite FeSn(OH)6, mushistonite (Cu,Zn)Sn(OH)6, wickmannite and tetrawickmanite MnSn(OH)6, and stottite FeGe(OH)6. Natural single hydroxide perovskites include dzahlindite In(OH)3, bernalite Fe(OH)3 and söhngeite Ga(OH)3. Synthetic hydroxide perovskites have also been produced, such as MgSi(OH)6 (Wunder et al. 2011, 2012; Welch and Wunder 2012) and δ-Al(OH)3 (Matsui et al. 2011). Hydroxide perovskites studied so far occur as cubic, tetragonal, orthorhombic and monoclinic varieties. Polymorphism occurs in MnSn(OH)6 in the forms of wickmannite (cubic, \(Pn\bar{3}\)) and tetrawickmanite (tetragonal, P42/n). The reader is referred to Mitchell (2002) for a thorough review of the diversity of natural and synthetic hydroxide perovskites. Information relating to the synthesis of stannate hydroxide perovskites is given by Kramer et al. (2010).
Zero tilts of octahedra are very unlikely in hydroxide perovskites, due to (a) the empty A-site, and (b) the formation of strong hydrogen bonds across each quartet of octahedra bordering the A-site cavity (Fig. 1). In double hydroxide perovskites, in which the octahedral framework has heterovalent cations alternating in a fully-ordered motif, mirror planes can only occur by bisecting octahedra. However, such mirror planes lead to extremely distorted octahedra, as can be seen in the reported structures of natanite FeSn(OH)6 (\(Pn\bar{3}m\), Strunz and Contag 1960) and CuSn(OH)6 (P42/nnm, Morgernstern-Badarau 1976), which are wrong—the correct space groups are likely to be \(Pn\bar{3}\) and P42/n, respectively, both of which have non-zero tilts.

The crystal structure of söhngeite. a A quartet of Ga(OH)3 octahedra of the P42/nmc structure. H5 atoms of the ring of four pairs of ½-occupied sites are highlighted in blue. The H5···H5 distance of each pair is 0.8 Å. Each oxygen atom receives a hydrogen bond. b A quartet of Ga(OH)3 octahedra of the P42/n structure. The ring consists of four fully-occupied H5 sites. O–H5···O hydrogen bonds are shown as dotted red lines. Each oxygen atom receives a hydrogen bond. c Polyhedral representation of the P42/nmc söhngeite structure projected on to (001) with the set of eight ½-occupied H5 sites of the ring highlighted in light blue. All other coloured H atoms belong to the crankshafts and are ½-occupied (see text). d Polyhedral representation of the P42/n söhngeite structure projected on to (001) with the set of four fully-occupied H5 sites of the ring highlighted in light blue. All other coloured H atoms belong to the crankshafts and are ½-occupied (see text). e Polyhedral representation of the structure of söhngeite proposed by Scott (1971) projected on to (001) and showing the untitled Ga(OH)6 octahedra (see text). Hydrogen atoms were not located. (f) Polyhedral representation of the structure of söhngeite proposed by Scott (1971) projected on to (100) and showing the large axial distortion of Ga(OH)6 octahedra (see text)
Scott (1971) reported the structure of Tsumeb söhngeite as being a perovskite-like with orthorhombic space group Pmn21 and lattice vectors [2, 0, 0] [0, 2, 0] [0, 0, 2] relative to the \(Im\bar{3}\) aristotype; unit cell parameters are a 7.4865 Å, b 7.4379 Å, c 7.4963 Å, V 417.42 Å3 (unspecified uncertainties on these values). This topology has tilt system a 0 a 0 c +, with two zero tilts and is, as we discuss in this paper, very unlikely for a hydroxide perovskite. We distinguish that structure from a Pmn21 perovskite with lattice vectors [0, 0, 2] [1, 1, 0] [\(\bar{1}\), 1, 0] relative to the axes of the aristotype, which has a different tilt system a − a − c + and is a plausible hydroxide perovskite topology. These questionable features of Scott’s structure led us to reinvestigate this phase as part of wider study of the structural chemistry of hydroxide perovskites. In this paper we report the determination of the crystal structure of söhngeite from the type locality at Tsumeb (specimen BM 1975, 398) and we also describe its transformation to a high-temperature cubic polymorph.
Experimental methods and samples
Samples
Two crystals were taken from hand specimen BM1975, 398 of the Natural History Museum’s collection from the type locality at Tsumeb, South West Africa. The structures of both were determined by single-crystal XRD. Crystal 1 (0.13 × 0.11 × 0.05 mm) was retained as a co-type, and Crystal 2 (0.22 × 0.15 × 0.04 mm) was used in the heating experiment described below. The latter crystal transformed irreversibly to a cubic polymorph.
Electron microprobe microanalysis
After the single-crystal XRD experiment Crystal 2 was mounted on a glass slide with a clean, smooth crystal surface parallel to the slide surface and carbon-coated for wavelength-dispersive analysis using a Cameca SX100 electron microprobe (20 kV, 20 nA, graphite monochromator). A 0.01 mm beam diameter was used with an on-peak counting time of 20 s and background counting time of 10 s. Element standards, X-ray lines and spectrometer crystals used were: corundum (AlKα, LTAP), scandium metal (ScKα, LPET), yttrium gallium garnet (GaKα, LLIF), fayalite (FeKα, LLIF), MnTiO3 perovskite (MnKα, LLIF), indium phosphide (InLα, LPET). Mn, In and Sc were below detection limits. The average composition of seven spot analyses (excluding H2O) is: 0.9 (±0.7) wt% Al2O3, 0.6 (±0.2) wt% Fe2O3, 79.4 (±2.0) wt% Ga2O3, total oxides (excluding H2O) 80.9 (±1.6) wt%. The corresponding average chemical formula normalised to three oxygens is Ga0.97Al0.02Fe 3+0.01 (OH)3, being essentially of end-member composition. The ideal H2O content of Ga(OH)3 is 22.4 wt%, and is in good agreement with the H2O deficiency of the average empirical anhydrous analysis.
Single-crystal X-ray diffraction
Each of the two sohngeite crystals described above was mounted on a non-diffracting amorphous carbon fibre (0.01 mm diameter), itself glued to a glass fibre support. Single-crystal X-ray diffraction data were collected using a XcaliburE® four-circle diffractometer equipped with an EoS® area detector (®Rigaku Oxford Diffraction) operated with graphite-monochromatised MoKα radiation at 45 kV and 40 mA. Data collection strategies for the crystals were based upon 25-min pre-experiments. In all experiments a whole sphere of data was collected to θ = 35° with 99.7–100 % completeness. Reflection intensities were corrected for Lorentz-polarization effects and absorption (empirical multi-scan) and converted to structure factors using the program CrysalisPro® (®Rigaku Oxford Diffraction).
The structures of both söhngeite crystals were determined at 293 K. In a second experiment Crystal 2 was heated from 293 to 423 K using a stream dry nitrogen gas from a Cryojet® (®Rigaku Oxford Diffraction) at a rate of 10 K/min and allowed to equilibrate at 423 K for 105 min before commencing data collection. It was evident from the 20-min pre-experiment that this crystal had transformed to a cubic-I phase during the equilibration period. A 1° frame-width and 30-s frame-time were used. The 423 K experiment lasted 19 h.
Finally, a return-to-ambient dataset for Crystal 2 was collected using the same experimental strategy as for the 423 K experiment. As we shall show, the crystal structure at 423 K is cubic \(Im\bar{3}\) and was recovered on cooling to 293 K without back-transformation.
Raman spectroscopy
Unpolarized Raman spectra at various crystal-to-laser beam orientations have been recorded for the Crystal 2 plate mounted on a 0.4 mm diameter culet of a low fluorescence Raman diamond. Ambient spectra were collected in 180° back-scattering geometry over the range 100–4000 cm−1. The instrument used was a Labram HR800 (Horiba Jobin–Yvon) spectrometer on beamline I15 at Diamond Light Source (UK). It was equipped with 1200 g grating and a CCD detector. The spectra were excited by the 473 nm line of a 50 mW Cobalt Blues TM laser focused down to a 0.01 mm spot on the sample and collected through a 0.05 mm confocal aperture. The intrinsic resolution of the spectrometer is <1 cm−1 and calibrations are accurate to ±1 cm−1. The frequency of each Raman band was obtained by fitting Voigtian line profiles using a least-squares algorithm.
Results
Crystal structure determination
Information relating to all data collections and structure refinements is summarised in Tables 1 and 2; atom coordinates and equivalent displacement parameters are given in Tables 3, 4 and 5; polyhedral parameters are given in Tables 6 and 7. CIF files for all refinements and lists of structure factors are deposited with the journal. Structures were solved and refined using the program SHELX (Sheldrick 2008) within the WinGX program suite (Farrugia 1999). Neutral atomic scattering factors for Ga, Fe, Sn and O were taken from International Tables for Crystallography, Volume C (Wilson 1992).
Data collection and refinement of Crystals 1 and 2 at 293 K
Systematic absences are consistent with space groups P42/nmc and P42/n. There was only a single weak “unobserved” violator of the c-glide of P42/nmc [{113}, I/σ(I) = 3.4]. However, the structure was refined in both space groups. Refinement in P42/n required 〈110〉 merohedral twinning for which the refined value of the batch scale factor (BASF, Sheldrick 2008) was 0.49(1). This value might be taken to imply that space group P42/nmc is the correct choice (the twinning would correspond to a diad axis of this space group), but we have found that a BASF value near 0.5 is frequently obtained when refining 〈110〉 merohedral twinning in double hydroxide perovskites, for which 〈110〉 mirror symmetry cannot occur, i.e. in these structures higher symmetry is not implied by BASF ~0.5. The very subtle differences between P42/nmc and P42/n refined structures are discussed below. Both structures have tilt system a + a + c −. As we also discuss below, the ambient Raman spectrum of tetragonal söhngeite is compatible with both space groups.
Data collection and refinement of Crystal 2 at 423 and 293 K
Diffraction patterns of söhngeite at 423 K are consistent with cubic space group \(Im\bar{3}\), the aristotype topology with corresponding tilt system a + a + a +. Both ½-occupied non-equivalent hydrogen positions were located and refined isotropically (see below). In both refinements of the cubic phase (at 423 K and return-to-ambient) 〈110〉 merohedral twinning was evident from residual electron density maps. Merohedral twinning using this twin law was refined. However, unlike the original tetragonal structure, the BASF values were 0.852(4) and 0.873(4) for 423 and 293 K structures, respectively. At 293 K the unit–cell volumes of the tetragonal and cubic phases differ by 1 %, with the former being smaller.
Description of the crystal structures of tetragonal and cubic polymorphs
The structural features of tetragonal and cubic söhngeite are shown in Figs. 1, 2 and 3.

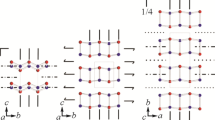
Hydrogen bonding topology of the O–H···O crankshafts of P42/nmc söhngeite (upper two diagrams) and P42/n söhngeite (lower two diagrams). In both structures each H site (H1–4) is ½-occupied and each oxygen atom receives a hydrogen bond. The crankshaft of the P42/nmc structure lies in a mirror plane which constrains O–H bond orientations

Crystal structure of the cubic (\(Im\bar{3}\)) polymorph of söhngeite at 423 K. H1 and H2 atoms are shown as grey and light blue spheres, respectively. Both H sites are ½-occupied. A ring of H(1,2) sites is highlighted by red circles
Ga(OH)6 octahedra
Ga–O bond distances, quadratic elongation (λ) and mean bond-angle variance 〈σ 2〉 of Ga octahedra for P42/nmc, P42/n and \(Im\bar{3}\) structures are given in Table 5 and 6; λ and 〈σ 2〉 values were calculated using the formula of Robinson et al. (1971). Differences between the two non-equivalent octahedra of the P42/n structure are minor, the Ga(2)(OH)6 octahedron being 3 % larger than the Ga(1)(OH)6 octahedron. The single octahedron of the P42/nmc structure is clearly an average of those of the P42/n structure. There is no detectable expansion of the octahedron on heating to 423 K.
The octahedra of P42/nmc and \(Im\bar{3}\) structures at 293 and 423 K, respectively, are almost identical. The octahedron of the cubic structure is unchanged on cooling from 423 to 293 K. There is no significant difference in Ga–O–Ga angles between tetragonal and cubic structures (Tables 6, 7). The octahedral tilts for the two tetragonal structures are 16° [100, 010] and 14° [001]. The octahedral tilt for the cubic structure is 15° [100, 010, 001] at 293 and 423 K. Hence, while there is a change in tilt phase along [001], Ga–O–Ga angles are retained with negligible deformation and there is a very minor change of tilt angle (1°) across the transformation.
Hydrogen positions in söhngeite
Recognition of plausible H sites in difference-Fourier maps of hydroxide perovskites is aided by the distinctive oxygen arrays that arise from highly tilted framework octahedra which allow obvious O–H···O bridges to be identified. The rhombus of oxygens associated with a quartet of octahedra (Fig. 1a, b) has one short O···O distance at ~2.7–2.8 Å and one much longer distance at ~4 Å. It is known from neutron powder diffraction studies of burtite CaSn(OH)6 and schönfliesite MgSn(OH)6 (Basciano and Peterson 1998) and synthetic In(OH)3 (Mullica et al. 1979), that a O–H···O hydrogen bond forms across the shorter O···O distance. Hence, if a weak residual peak in the difference-Fourier map of an X-ray-determined structure lies at a position 0.8–0.9 Å from a donor oxygen and points towards a plausible oxygen acceptor (i.e. across the short O···O distance), then refinement of its position can be tested. In the case of hydroxide perovskites there is a further consideration: some H sites are half-occupied. Lafuente et al. (2015) showed that tetrawickmanite MnSn(OH)6, space group P42/n, has five non-equivalent H sites, four of which are half-occupied. We have found the five analogous sites in stottite FeGe(OH)6 (P42/n) which correlate with the five OH stretching peaks of its ambient Raman spectrum (Kleppe et al. 2012). The half-occupied H sites are associated with the O(H)···O(H)··· crankshaft (Fig. 2), whereas the fully occupied H site is associated with the isolated four-membered ring.
One of us (MDW, unpublished data) has located the two analogous non-equivalent half-occupied sites in wickmanite and schönfliesite MgSn(OH)6 (both \(Pn\bar{3}\)) using single-crystal XRD. The close correspondence between the approximate positions of these sites determined by XRD and those determined by neutron powder diffraction (Basciano and Peterson 1998) leaves little room for doubt about the validity of the XRD refinements.
It has been possible to determine the approximate positions of all five non-equivalent H atoms in P42/nmc and P42/n structures of söhngeite by SCXRD. Space group P42/nmc imposes 〈100〉 mirror symmetry on the four-membered O–H···O ring and splits the H site into a pair of mirror-related ½-occupied sites. This arrangement differs from that of P42/n, which has no mirrors and thereby allows ordering of H at a single ring site, as in tetrawickmanite (Lafuente et al. 2015). The configurations of the O–H···O crankshafts of both structures are shown in Fig. 2. The only clear difference is in their H positions, which in P42/nmc are constrained to lie within a mirror plane, whereas this is not a constraint in P42/n. Although all five H atoms of each structure were located and their positions refined, their half occupancy prevented meaningful refinement of U iso values, and so H positions for both structures should be seen as being approximate. Apart from the split/unsplit H ring site the only significant topological difference between the two tetragonal structures is that the single Ga site of P42/nmc becomes two non-equivalent sites in P42/n. Hence, there are no indicators that would allow a clear choice to be made between these two space groups for söhngeite. Consequently, a choice between the two space groups cannot be made based upon single-crystal XRD data. Both half-occupied non-equivalent H sites in the cubic structure were located and their positions refined.
The cubic \(Im\bar{3}\) structure of sohngeite is shown in Fig. 3. It has three equal in-phase tilts (a + a + a +) that result in isolated rings of O–H···O hydrogen-bonded linkages (a ring is highlighted in Fig. 3). Both non-equivalent hydrogen atoms were located in difference-Fourier maps and their positions refined keeping U iso fixed at 0.04 Å2. There are no O–H···O crankshafts. Hence, the tetragonal → cubic transformation involves disruption of the crankshaft topology. Each oxygen atom of the four-membered O–H···O ring is (in the average structure represented by \(Im\bar{3}\)) bonded to two H atoms, each with half-occupancy, as also reported for synthetic cubic In(OH)3 (\(Im\bar{3}\)) by Mullica et al. (1979).
Raman spectrum
The ambient Raman spectra of tetragonal (unheated) söhngeite is shown in Fig. 4. The low-wavenumber region 150–1000 cm−1 has one intense band at 327 cm−1 and five much weaker bands at 185, 256, 273, 455 and 923 cm−1. The spectrum in the OH-stretching region 2800–3500 cm−1 is best fitted with a five-bands with maxima at 3000, 3100, 3189, 3240 and 3334 cm−1. A four-peak fit (3000, 3101, 3226, 3335 cm−1) is less satisfactory (Fig. 4). It results in a significant discrepancy between the width of the 3226 cm−1 band (FWHM ~ 110 cm−1) compared with FWHM values of 75–80 cm−1 for the other three bands. The 3226 cm−1 band has a significant unfitted residual shoulder on the its low-wavenumber side that corresponds to the extra band at 3189 cm−1 of the five-band fit.

a Ambient single-crystal Raman spectrum of söhngeite. The peak positions of the most intense modes are labelled. b Single-crystal Raman spectrum of söhngeite in the OH-stretching region showing a four-peak fit in blue and a five-peak fit in red. Peak positions are labelled and the numbers in brackets give the FWHM of each peak. The weak and broad peaks on the extreme high and low wavenumber sides of the spectrum fit instrumental background
The five-band Raman spectrum is consistent with both space groups. A four-band fit is consistent with neither. If H atoms were all at fully-occupied sites in both structures we would expect three O–H bands for each space group as they both have three non-equivalent oxygen atoms. Both structure refinements require ½-occupancy of the crankshaft H positions (H 1–4), with a doubling of the number of non-equivalent H sites (giving a total of five sites), as has been reported for tetrawickmanite by Lafuente et al. (2015) and also observed by us for stottite (MDW, unpublished data). The ambient Raman spectrum of stottite, space group P42/n has five OH bands (Kleppe et al. 2012).
Discussion
Options for tetragonal space groups in hydroxide perovskites are constrained by three important topological features: (a) the empty A site; (b) the formation of O–H···O linkages; (c) the avoidance of 〈110〉 mirrors. The first two constraints exclude space groups of structures having one or more zero tilts, as O–H···O bridges form strong links between octahedra and enhance tilting. The third constraint arises because 〈110〉 mirrors must bisect an octahedron and this leads to highly distorted octahedra, as can be seen in the published structures of natanite FeSn(OH)6 reported as having space group \(Pn\bar{3}m\) (Strunz and Contag 1960) and of CuSn(OH)6, space group P42/nnm (Morgernstern-Badarau 1976). These two reported structures also have mirror symmetry—a feature that, as we argued above, is very unlikely for hydroxide perovskites. The correct space group of natanite is likely to be \(Pn\bar{3}\) (as in wickmanite, burtite and schönfliesite), and the most likely space group for CuSn(OH)6 is P42/n. In single hydroxide perovskites, such as In(OH)3 (dzahlindite) and söhngeite, 〈100〉 mirror planes pass through oxygen atoms, with octahedra either side being equivalent. In double hydroxide perovskites such as wickmanite MnSn(OH)6 (\(Pn\bar{3}\)) and stottite FeGe(OH)6 (P42/n), 〈100〉 mirror symmetry is not possible because adjacent octahedra are non-equivalent and have different cations; as for single hydroxide perovskites 〈110〉 mirrors lead to gross distortions of octahedra.
The ambient structure of söhngeite reported here is tetragonal with space group P42/nmc or P42/n. Refinements in both space groups are of comparably good quality. The ambient Raman spectrum has five O–H stretching peaks and is consistent with the five non-equivalent H atoms of both space groups. The distinction between the two structures lies with a pair of ½-occupied equivalent H sites of the four-membered ring in P42/nmc compared with a single fully-occupied site in P42/n. The differences between the two non-equivalent Ga(OH)6 octahedra of P42/n are very minor.
The tetragonal structure(s) for söhngeite reported here differ significantly from that obtained by Scott (1971) who reported space group Pmn21 for a (2, 0, 0) (0, 2, 0) (0, 0, 2) cell. Scott’s structure (Fig. 1e, f) is highly implausible for two significant reasons: (a) it has tilt system a 0 a 0 c + with zero tilts; (b) the shortest next-nearest neighbour O···O distances are ~3.7 Å and are far too long to form the necessary O–H···O bridges. A further anomaly is that while Scott’s structure could have a single tilt (about [001]), the atom coordinates reported result in a topology with no tilt.
The cubic \(Im\bar{3}\) aristotype formed at 423 K was recovered metastably at ambient conditions without back-transformation. The hysteresis observed in söhngeite is consistent with a change in tilt system, i.e. P42/nmc or P42/n → \(Im\bar{3}\), for which the corresponding change in tilt system is a + a + c − → a + a + a +, and is associated with a significant reconstruction of the hydrogen-bonding topology, namely the conversion of O–H···O–H···O crankshafts into four-membered rings.
At 293 K the unit cell volume of the cubic structure is 1 % larger than that of the tetragonal polymorph. Group/sub-group relations for perovskites (Howard and Stokes 2005) indicate that there is no continuous pathway for transformation from P42/nmc or P42/n to \(Im\bar{3}\). Available continuous transformations with symmetry increase for the two tetragonal structures involve space groups having zero tilts: I4/mmm (a 0 b + b +) and I4/mcm (a 0 a 0 c −) for P42/nmc, and P42/nnm (a 0 b + b +) and I4/m (a 0 a 0 c −) for P42/n (Howard and Stokes 2005, Figs. 9 and 11). Thus, the observed transformation to the \(Im\bar{3}\) aristotype is essentially reconstructive in character as it involves the breakage and reformation of hydrogen bonds (with attendant hysteresis).
Kleppe et al. (2012) reported Raman spectra of stottite FeGe(OH)6 that showed significant hysteresis on decompression from a high-pressure (>11 GPa) P42/n structure to an ambient structure with inferred space group P2/n (a + a + c −). On decompression, the two structures coexisted from at P ≤ 10 GPa and the final spectrum of the fully decompressed crystal was the same as its original ambient spectrum. However, the observed hysteresis on decompression and the coexistence of two structures suggest that a change in tilt system is involved (viz söhngeite), whereas both proposed structures have the same tilt system (a + a + c −) and hydrogen-bonding topology. Also, the very recent discovery that ½-occupied split H sites can occur in hydroxide perovskites (Lafuente et al. 2015) would seem to require re-evaluation of stottite Raman spectra.
The occurrence of tetragonal and cubic polymorphs in söhngeite is analogous to that in the MnSn(OH)6 minerals tetrawickmanite (P42/n) and wickmanite (\(Pn\bar{3}\)), although these are double hydroxide perovskites with a very different composition. The occurrence of tetrawickmanite and wickmanite (presumably metastably) at the Earth’s surface is consistent with significant hysteresis, as found for söhngeite.
The interesting question arises as to the role of hydrogen bonding in driving the tetragonal → cubic transformations in Ga(OH)3 and MnSn(OH)6. Is it reactive or causative? Given that the geometrical parameters of the Ga(OH)6 polyhedra at 293 and 423 K are almost identical and Ga–O–Ga angles are unchanged despite a switch in tilt phase (− to +) along [001], it seems likely that the transition is driven by the behaviour of protons, i.e. site re-ordering. If this is the case, then it is the H atoms of the crankshafts, H(1–4), that are involved, as it is the crankshafts that become four-membered rings.
References
Basciano L, Peterson RL (1998) Description of schoenfliesite, MgSn(OH)6, and roxbyite, Cu1.72S, from a 1375 BC shipwreck, and Rietveld neutron-diffraction refeinement of synthetic schoenfliesite, wickmanite, MnSn(OH)6, and burtite CaSn(OH)6. Can Mineral 36:1203–1210
Farrugia LJ (1999) WinGX: an integrated system of Windows Programs for the solution, refinement and analysis of single-crystal X-ray diffraction data. J Appl Crystallogr 32:837–838
Howard CJ, Stokes H (2005) Structures and phase transitions in perovskites—a group-theoretical approach. Acta Crystallogr A A61:93–111
Howard CJ, Kennedy BJ, Woodward PM (2003) Ordered double perovskites—a group-theoretical analysis. Acta Crystallogr A B59:463–471
Kleppe AK, Welch MD, Crichton WA, Jephcoat AP (2012) Phase transitions in hydroxide perovskites: a Raman spectroscopic study of stottite FeGe(OH)6 to 21 GPa. Mineral Mag 76:949–962
Kramer JW, Kelly B, Manivannan V (2010) Synthesis of MSn(OH)6 (where M = Mg, Ca, Zn, Mn, or Cu) materials at room temperature. Cent Eur J Chem 8:65–89
Lafuente B, Yang H, Downs RT (2015) Crystal structure of tetrawickmanite Mn2+Sn4+(OH)6. Acta Crystallogr A E71:234–237
Matsui M, Komatsu K, Ikeda E, Sano-Furukawa A, Gotou H, Yagi T (2011) The crystal structure of δ-Al(OH)3: neutron diffraction measurements and ab initio calculations. Am Mineral 96:854–859
Mitchell RH (2002) Perovskites modern and ancient. Almaz Press, Thunder Bay
Morgernstern-Badarau I (1976) Effet Jahn–Teller et structure cristalline de l’hydroxyde CuSn(OH)6. J Solid State Chem 17:399–406
Mullica DF, Beall GW, Milligan WO (1979) The crystal structure of cubic In(OH)3 by X-ray and neutron diffraction methods. J Inorg Nucl Chem 41:277–282
Robinson K, Gibbs GV, Ribbe PH (1971) Quadratic elongation: a quantitative measure of distortion in coordination polyhedra. Science 172:567–570
Scott JD (1971) Crystal structure of a new mineral, söhngeite. Am Mineral 56:355
Sheldrick GM (2008) A short history of SHELX. Acta Crystallogr A A64:112–122
Strunz H, Contag B (1960) Hexahydroxostannate Fe, Mn Co, Mg, Ca[Sn(OH)6] und deren kristallstrukur. Acta Crystallogr A 13:601
Welch MD, Wunder B (2012) A single-crystal X-ray diffraction study of the 3.65 Å phase MgSi(OH)6, a high-pressure hydroxide perovskite. Phys Chem Miner 39:693–697
Wilson AJC (ed) (1992) International tables for crystallography, vol C. Kluwer Academic Publishers, Dordrecht
Wunder B, Wirth R, Koch-Muller M (2011) The 3.65 Å-phase in the system MgO–SiO2–H2O: synthesis, composition, and structure. Am Mineral 96:1207–1214
Wunder B, Jahn S, Koch-Müller M, Speziale S (2012) The 3.65 Å phase, MgSi(OH)6: structural insights from DFT calculations and T-dependent IR spectroscopy. Am Mineral 97:1043–1048
Acknowledgments
MDW thanks Roy Kristiansen for support of his research on hydroxide perovskites by the provision of high-quality samples. We thank John Spratt (NHM) for carrying out electron microprobe analysis. František Laufek and an anonymous reviewer are thanked for their helpful comments on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Welch, M.D., Kleppe, A.K. Polymorphism of the hydroxide perovskite Ga(OH)3 and possible proton-driven transformational behaviour. Phys Chem Minerals 43, 515–526 (2016). https://doi.org/10.1007/s00269-016-0812-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00269-016-0812-y