
Abstract
The crystal structures of synthetic K-dravite [XKYMg Z3 Al T6 Si6O18(BO3) V3 (OH) W3 (OH)], dravite [XNaYMg Z3 Al T6 Si6O18(BO3) V3 (OH) W3 (OH)], oxy-uvite [XCaYMg Z3 Al T6 Si6O18(BO3) V3 (OH) W3 O], and magnesio-foitite [X☐Y(Mg2Al)ZAl T6 Si6O18(BO3) V3 (OH) W3 (OH)] are investigated by polarized Raman spectroscopy, single-crystal structure refinement (SREF), and powder X-ray diffraction. The use of compositionally simple tourmalines characterized by electron microprobe analysis facilitates the determination of site occupancy in the SREF and band assignment in the Raman spectra. The synthesized K-dravite, oxy-uvite, and magnesio-foitite have significant Mg–Al disorder between their octahedral sites indicated by their respective average 〈Y–O〉 and 〈Z–O〉 bond lengths. The Y- and Z-site compositions of oxy-uvite (YMg1.52Al1.48(10) and ZAl4.90Mg1.10(15)) and magnesio-foitite (YAl1.62Mg1.38(18) and ZAl4.92Mg1.08(24)) are refined from the electron densities at each site. The Mg–Al ratio of the Y and Z sites is also determined from the relative integrated peak intensities of the Raman bands in the O–H stretching vibrational range (3250–3850 cm−1), producing values in good agreement with the SREF data. The unit cell volume of tourmaline increases from magnesio-foitite (1558.4(3) Å3) to dravite (1569.5(4)–1571.7(3) Å3) to oxy-uvite (1572.4(2) Å3) to K-dravite (1588.1(2) Å3), mainly due to lengthening of the crystallographic c-axis. The increase in the size of the X-site coordination polyhedron from dravite (Na) to K-dravite (K) is accommodated locally in the crystal structure, resulting in the shortening of the neighboring O1–H1 bond. In oxy-uvite, Ca2+ is locally associated with a deprotonated W (O1) site, whereas vacant X sites are neighbored by protonated W (O1) sites. Increasing the size of the X-site-occupying ion does not detectably affect bonding between the other sites; however, the higher charge of Ca and the deprotonated W (O1) site in oxy-uvite are correlated to changes in the lattice vibration Raman spectrum (100–1200 cm−1), particularly for bands assigned to the T 6O18 ring. The Raman spectrum of magnesio-foitite shows significant deviations from those of K-dravite, dravite, and oxy-uvite in both the lattice and O–H stretching vibrational ranges (100–1200 and 3250–3850 cm−1, respectively). The vacant X site is correlated with long- and short-range changes in the crystal structure, i.e., deformation of the T 6O18 ring and lengthening of the O1–H1 and O3–H3 bonds. However, X-site vacancies in K-dravite, dravite, and oxy-uvite result only in the lengthening of the neighboring O1–H1 bond and do not result in identifiable changes in the lattice-bonding environment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The flexible crystal structure of tourmaline supergroup minerals (XY3Z6T6O18(BO3)3V3W; Hamburger and Buerger 1948; Hawthorne and Henry 1999) allows them to incorporate a large variety of elements, leading to the growing list of 31 endmember species accepted by the International Mineralogical Association (Henry et al. 2011). Although almost all the alkali and alkaline earth metals have been measured in tourmaline, the X site is principally occupied by Na and Ca, or left vacant (☐). K is the third most common X-site-occupying ion and is typically considered a minor or trace element (Henry and Dutrow 1996). However, in K-rich environments at ultrahigh pressure and temperature, K-dominant Mg-Al tourmaline can form as the endmember maruyamaite \(\left[ {^{\text{X}} {\text{K}}^{\text{Y}} \left( {{\text{Mg}}_{ 2} {\text{Al}}} \right)^{\text{Z}} {\text{Al}}_{ 6}^{\text{T}} {\text{Si}}_{ 6} {\text{O}}_{ 1 8} \left( {{\text{BO}}_{ 3} } \right)_{ 3}^{\text{V}} \left( {\text{OH}} \right)_{ 3}^{\text{W}} {\text{O}}} \right]\) (Lussier et al. 2014) or as K-dravite \(\left[ {^{\text{X}} {\text{K}}^{\text{Y}} {\text{Mg}}_{ 3}^{\text{Z}} {\text{Al}}_{ 6}^{\text{T}} {\text{Si}}_{ 6} {\text{O}}_{ 1 8} \left( {{\text{BO}}_{ 3} } \right)_{ 3}^{\text{V}} \left( {\text{OH}} \right)_{ 3}^{\text{W}} {\text{OH}}} \right]\) (Henry et al. 2011; Berryman et al. 2014, 2015). K-dominant Fe-Mg tourmaline has also been identified at the type locality of povondraite \([^{\text{x}} {\text{Na}}^{\text{Y}} {\text{Fe}}^{3 + }_{3} \;^{\text{Z}} ({\text{Mg}}_{2} {\text{Fe}}^{3 + }_{4} )^{\text{T}} {\text{Si}}_{6} {\text{O}}_{18} ({\text{BO}}_{3} )_{3} ({\text{OH}})_{3} ]\) in Bolivia (Grice et al. 1993; Žáček et al. 2000). The K+ ion is significantly larger (1.55 Å) than the more common X-site-occupying cations, Na+ (1.24 Å) and Ca2+ (1.18 Å) (all in ninefold coordination; Shannon 1976). With increasing size of its occupant, the X-site coordination polyhedron is expected to expand (Ertl and Tillmanns 2012). This raises the question of how the range in size of the X site is accommodated in tourmaline, and more generally, what is the relationship between the X-site-occupying ion and the rest of the crystal structure.
X-ray diffraction and Raman spectroscopy are powerful tools for investigating long- and short-range features of crystal structures. Single-crystal structure refinement from X-ray diffraction data of a compositionally well-characterized tourmaline provides information about average site occupancies and bond lengths. Raman spectroscopy provides further information about the bonding environment around different crystallographic sites, including qualitative information about the O–H bond lengths. Recent Raman spectroscopy studies have largely focussed on the assignment of the high wavenumber bands (3250–3850 cm−1) of tourmalines of various compositions (e.g., Gonzalez-Carreño et al. 1988; Skogby et al. 2012; Fantini et al. 2014). The low wavenumber lattice vibration Raman bands (100–1200 cm−1) in tourmaline are associated with cation–oxygen bonds (mainly B–O, T–O, Y–O, Z–O, e.g., Mihailova et al. 1996; Gasharova et al. 1997; McKeown 2008; Zhao et al. 2012; Fantini et al. 2014). The high wavenumber bands (3250–3850 cm−1) are associated with O–H stretching and are influenced by substitution at the X, Y, and Z sites, as well as by deformation of the T 6O18 ring. For example, the presence of significant IVB (>0.33(6) pfu; Kutzschbach et al. in review) can be detected and quantified in the Raman spectra of B-rich olenites from the O–H stretching vibrations. Similarly, shifts in the position and intensity of these bands provide insight into the structural effect of the X-site-occupying cation on its local crystal environment.
To date, almost all Raman spectroscopy studies have been conducted on natural tourmaline crystals, which are solid solutions of various endmember species. The presence of multiple site substitutions in these tourmalines leads to complicated Raman spectra, making it difficult to unambiguously assign bands to particular bonding environments. The investigation of compositionally restricted, synthetic tourmalines facilitates band assignment.
To better understand how the tourmaline structure accommodates variations in the composition of the X site, we characterized synthetic K-dravite, dravite, oxy-uvite, and magnesio-foitite by electron microprobe, single-crystal (except dravite) and powder X-ray diffraction, and oriented Raman spectroscopy. Each of these synthetic tourmalines has a different X-site composition [K, Na, Ca, or vacancy (☐)], and comparison of their combined crystal structure data allows us to better understand the X-site occupancy’s influence on tourmaline’s crystal structure.
Tourmaline’s crystal structure
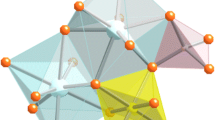
Tourmaline crystallizes in the trigonal space group R3m (Buerger and Parrish 1937). The cyclosilicate crystal structure (Fig. 1) consists of rings of six corner-sharing TO4 tetrahedra, occupied primarily by Si and more rarely by Al and/or B. The bases of the tetrahedra face the c +-crystallographic axis direction, making tourmaline a polar crystal. The ninefold-coordinated X site lies directly above the T 6O18 ring, coordinated with the inner corners of the tetrahedra. Below the ring, the apex of each tetrahedron shares a corner with one of three edge-sharing Y-site octahedra and one of six Z-site octahedra, which surround the Y-site trimer. The three trigonally-coordinated B atoms share corners with both the Y and Z octahedra. The W and V sites, defined as O1 and O3, respectively, can contain OH−, O2−, or, in the case of the W site, F−. The single W (O1) site is found at the center of the T 6O18 ring, coordinated with the three Y sites. The O1–H1 bond is aligned with the c +-axis, with H1 orientated toward the X site. Asymmetry in the occupation of the coordinated Y sites can cause the O1–H1 bond to diverge from complete parallelism with the c-axis (Gatta et al. 2014). Very weak hydrogen bonds exist between H1 and O4 and O5 (0.026 and 0.047 vu, respectively; Gatta et al. 2014), oxygen sites in the T 6O18 ring. The three V (O3) sites are found around the Y-site trimer, between two Z-site octahedra. Each V (O3) site is coordinated with one Y and two Z sites. The O3–H3 bond is orientated with H3 pointing in the c −-crystallographic axis direction. There is weak, but significant (0.114 vu; Gatta et al. 2012, 2014) hydrogen bonding between H3 and O5, an oxygen atom coordinated with both the T site and the X site. As a result, these hydroxyl groups may be expected to point slightly toward the O5 site and not be completely parallel to the c-axis (Gatta et al. 2014).

Schematic representation of tourmaline’s crystal structure viewed normal (A) and parallel (B) to the c-axis. The X site (in purple, occupied by Na, Ca, K, or left vacant) is located just above the tetrahedral ring (in dark blue) and is bonded to the inner corners of each tetrahedra (T) and trigonally-coordinated B group (in green). The O1 (W) site (in red) is coordinated to three Y sites (in yellow). The O1–H1 bond is orientated with the hydrogen (in light blue) pointing toward the X site. The O3 (V) sites (in red) are each coordinated to one Y site (in yellow) and two Z sites (in orange). The O3–H3 bond is orientated in the c − direction with H3 sharing a H bond with the O5 site (dashed circle) in the tetrahedral ring
Experimental methods
K-dravite crystals synthesized and described by Berryman et al. (2015) (EB13) were used in this study. In addition, four tourmaline-synthesis experiments were done to produce dravite (EB28 and EB29), oxy-uvite (CN11), and magnesio-foitite (MF2) using experimental procedures modified after Berryman et al. (2014, 2015). At pressures of 0.4 GPa and below, tourmaline crystals synthesized in one-chamber capsules tend to be small (<5 μm) (e.g., Berryman et al. 2015), making it difficult to select and orientate single crystals for Raman spectroscopy. For this reason, tourmaline synthesized at these pressures (MF2 and CN11) applied the two-chamber method of von Goerne et al. (1999), which promotes large crystal growth. All tourmalines were synthesized from a homogeneous oxide mixture of MgO, γ-Al2O3, H3BO3, ± SiO2 (as quartz) and an excess amount of fluid. Quartz was included in the oxide mixture for syntheses using the one-chamber method, but not for those using the two-chamber method, in which it was added separately (see below). The details of each tourmaline synthesis, including the atomic proportions of each oxide mixture and the fluid composition, are provided in Table 1.
One-chamber synthesis method
The fluid and oxide mixture were placed in clean gold capsules (13 mm length, 3 mm outer diameter, 0.25 mm wall thickness), welded shut, and left in an oven at 100 °C overnight. The capsules were then placed in sets of two in a steel furnace assembly with sodium chloride as a pressure medium and loaded into the piston-cylinder apparatus. Pressure was calibrated according to the quartz–coesite transition (Mirwald and Massonne 1980), which is accurate within 50 MPa. Temperature was measured with a Ni–CrNi thermocouple (accurate within 10 °C) placed into the assembly close to the centers of the capsules. Following pressurization, the system was heated to the target temperature and monitored throughout the experiment; pressure was maintained within 50 MPa. At the end of the experiment, the sample was quenched isobarically to a temperature below 200 °C in less than 15 s and pressure slowly released. The capsule was removed from the assembly, cleaned, reweighed, and opened. The solid material was removed and prepared for subsequent analysis.
Two-chamber synthesis method
One end of a clean gold capsule (30 mm length, 4 mm diameter, and 0.25 mm wall thickness) was loaded with the fluid and oxide mixture. The center of the capsule was pinched closed, thereby forming two separate chambers. SiO2 (as quartz) was added to the second chamber and the capsule welded shut. The loaded capsule was stored at 100 °C overnight, reweighed, and placed in a standard, cold-seal hydrothermal apparatus. Pressure was maintained within 10 MPa, and temperature was measured with a Ni–CrNi thermocouple placed within the wall of the autoclave. At the end of the experiment, the sample was isobarically quenched by cooling the autoclave with compressed air to less than 100 °C in 5 min and then depressurized. The capsule was removed, cleaned, reweighed, and opened. The solid product was removed and prepared for subsequent analysis.
Electron microprobe analysis (EMPA)
A portion of the solid product of each experiment was mounted in an epoxy puck, polished, and carbon-coated. The compositions of the synthetic tourmalines, including the previously characterized sample EB13 (cf. Berryman et al. 2015), were determined by EMPA on a JEOL Hyperprobe JXA-8500F equipped with a thermal field-emission cathode and five wavelength-dispersive spectrometers. The operating conditions were as follows: 10.0 kV accelerating voltage, 10 nA beam current, 4 μm beam diameter, 30 s counting time for all elements (except B, which had a 80-s counting time). The standards used were schorl (B), jadeite (Na), wollastonite (Ca), periclase (Mg), pyrope (Al), orthoclase (K and Si), and tugtupite (Cl). Data reduction was done with a φ(ρZ) correction scheme (CITZAF; Armstrong 1995). Under these conditions, relative analytical errors and detection limits (1σ) are 2.5 % and 1800 ppm for B, 2.0 % and 110 ppm for Na, 0.85 % and 120 ppm for Mg, 0.5 % and 150 ppm for Al, 0.9 % and 250 ppm for Si, 10 % and 70 ppm for Cl, 2.1 % and 150 ppm for K, 1.5 % and 210 ppm for Ca, respectively.
Powder and single-crystal X-ray diffraction (XRD)
A finely ground portion of the solid product of each experiment was glued between two boPET foils and mounted in a transmission XRD sample holder. Powder XRD patterns of all tourmalines were collected on a STOE Stadi P diffractometer equipped with a position-sensitive detector under the following conditions: Cu Κα1 radiation; 0.01° steps in the 5–125° 2θ range; 40 kV accelerating voltage; 40 mA beam current; Ge (111) primary monochromator. Phase proportions in the product assemblage and the average unit cell dimensions of each tourmaline were determined by Rietveld refinement of the diffraction patterns using the GSAS software package (Larson and Von Dreele 1987). Peak intensities were extracted using the default Rietveld method, and the peak profiles fit to the constant wavelength (CW) profile function 2 using silica standard parameters as initial coefficients. Initial structure models for tourmaline (dravite), coesite, boromuscovite, boralsilite, and pinnoite applied the structural parameters of Donnay and Buerger (1950), Geisinger et al. (1987), Zhukhlistov et al. (1973), Peacor et al. (1999), and Genkina and Malinovskii (1983), respectively. The X-site composition of each tourmaline was fixed using values determined by EMPA. The Rietveld refinements were then performed in the following order: zero-point correction, background subtraction, scale factor, lattice parameters, and Caglioti W for each phase. The structural parameters of tourmaline were further refined in the following order: Caglioti V, Lorentz X, atomic positions, isotropic temperature displacement factors (Uiso; constrained to be identical for all oxygen-bearing sites), and Lorentz Y.
Single-crystal XRD analyses were done at the Institut für Mineralogie und Kristallographie at the University of Vienna, Austria. The quality of different crystals was determined with a Bruker APEXII diffractometer equipped with a CCD area detector and an Incoatec Microfocus Source IμS (30 W, multilayer mirror, Mo-Kα radiation). The XRD pattern of a high-quality oxy-uvite crystal was recorded on the same Bruker APEXII diffractometer, whereas those of K-dravite (EB13) and magnesio-foitite (MF2) were recorded on a STOE StadiVari four-circle goniometer with a Dectris PILATUS 300-K pixel detector. Single-crystal XRD data were collected at room temperature, integrated, and corrected for Lorentz and polarization factors with an absorption correction by evaluation of partial multiscans. The structure was refined with SHELXL97 (Sheldrick 1997) using scattering factors for neutral atoms and a tourmaline starting model taken from Ertl et al. (2012). The H3 atom bonded to the O3 atom was located from a difference Fourier map and subsequently refined. Refinement was performed with anisotropic displacement parameters for all non-hydrogen atoms. Table 2 provides crystal data and details of the structure refinement. Site occupancies were refined according to expected variations in the tourmaline structure (Table 3).
Polarized Raman spectroscopy
Polarized micro-Raman spectra were collected at room temperature using either a HORIBA Jobin LabRAM HR800 UV–Vis (MF2) or HR800 Vis (EB13, EB28, EB29, CN11) spectrometer with a 1800 grooves/mm grating, a 100-μm pinhole in backscattering configuration, and a CCD detector (spectral resolution ~1 cm−1). The polarization analyzer had no observable effect on the collected spectra and was therefore not used. Tourmaline crystals were mounted on a glass slide and focused on using an optical microscope with a 50× (MF2, CN11) or 100× (EB13, EB28, EB29) objective and excited with a linearly polarized blue laser (488 or 473 nm wavelength and 30- or 12-mW laser power at sample surface for spectra collected using the HR800 UV–Vis or HR800 Vis, respectively). No damage to the crystal surfaces from the laser was observed. Raman spectra in the lattice (100–1200 cm−1) and O–H stretching (3250–3850 cm−1) vibrational ranges were generated from three (EB13, EB28, CN11) or five (EB29) 40-s accumulations, or four 50-s accumulations (MF2). Although multiple spectra were collected from different spots on each crystal to verify consistency, only representative spectra are reported here. As O–H bonds in tourmaline’s structure are largely parallel to the crystallographic c-axis, spectra were collected with the electric field vector of the polarized laser light (E) oriented parallel to the c-axis (e.g., Fig 2), with the exception of one spectrum collected with E perpendicular to the c-axis of EB13. The Raman spectra in the O–H stretching range were deconvoluted using PeakFit (Jandel Scientific) to identify hidden bands, their centers, and their integrated peak intensities. The coefficient of determination (R2) for all fitted spectra was >0.999.

Photograph of magnesio-foitite (MF2) synthesized using the two-chamber method of von Goerne et al. (2001). The crystal is 516 μm long. The arrow is aligned to the crystallographic c-axis
Results
All synthesis experiments produced tourmaline along with the additional phases listed in Table 1. Tourmaline crystals synthesized at 4.0 GPa (EB13, EB28, EB29) were up to 10 μm in length, whereas those synthesized using the two-chamber method (CN11, MF2) were up to ~600 μm in length (Fig. 2). All synthesized tourmalines were relatively homogeneous in composition (Fig. 3; Table 4), with those from EB28 showing the highest amount of inhomogeneity in the backscattered electron image (Fig. 3). In general, the total oxides measured by EMPA (Table 4) are lower than expected for tourmaline (~96–97 wt%, depending on composition) by ~2 wt%. This likely reflects the larger excitation volume of the electron beam relative to the crystal size. The minor reduction in total measured oxides is expected to be shared proportionally among the different oxides and therefore should mostly not affect the ratio of the elements determined by their normalization. The tourmaline compositions determined by EMPA were normalized both to 18 cations at the Y, Z, and T and B sites (18YZTB) and alternatively to 15 cations at the Y, Z, and T sites (15YZT) allowing the effect of the measured B concentrations on the normalized values to be evaluated. The quantities of O and OH at the V and W sites were determined by charge balance (Table 4). In the case of EB13, EB28, and EB29, the 18 YZTB normalized values give Si + B > 9, which cannot be accommodated in tourmaline’s structure. This likely reflects an analytical problem, particularly in the measurement of B resulting from the small crystal size of these samples. The larger crystal sizes of MF2 and CN11 correlate with 15 YZT and 18 YZTB normalized cation values with Si + B < 9, supporting this conclusion (Table 4). Normalization of the data to 15 YZT, assuming 3 B pfu, avoids this problem. The proportion of OH and O at the VW sites calculated using either the 15 YZT or 18 YZTB normalizations indicates that only oxy-uvite (CN11) has a largely deprotonated W site. However, in the case of EB13, EB28, EB29, and MF2, these normalization schemes result in significant vacancies at the VW sites (i.e., VW(OH) + VWO < 4). To overcome this problem, the values are additionally normalized to 31 oxygens pfu, assuming 3 B pfu and fully protonated VW sites (i.e., 4 OH pfu) for EB13, EB28, EB29, and MF2 (Table 4). The same normalization scheme was applied to CN11, but assuming a deprotonated W site (i.e., V(OH)3 and WO). Comparison of the different normalization schemes (Table 4) shows that only the proportion of tetrahedral B and the occupancy of the VW sites are significantly influenced by the different normalization schemes. The synthesized K-dravite (EB13) has an average of 0.68(6) [0.69(6)] K pfu based on the 18YZTB [15YZT or 31O] normalization. Tourmaline synthesized in the alkali-free system (MF2) is endmember magnesio-foitite in composition. In the Na-bearing system (EB28, EB29), dravite with variable Na content was synthesized. Dravite crystallized from a 3.99 m NaCl fluid (EB28) has an average of 0.81(3) [0.85(3) or 0.84(3)] Na pfu and that crystallized from a 3.00 m fluid (EB29) has 0.63(5) [0.66(5)] Na pfu based on the 18YZTB [15YZT or 31O] normalization (Table 4). The correlation of Na content in the dravitic tourmaline with Na concentration in the fluid is consistent with the findings of von Goerne et al. (2001) and Berryman et al. (2015). In the Ca system (CN11), oxy-uvite with 0.72(10) [0.73(10)] Ca pfu was synthesized based on the 18 YZTB and 15 YZT [31O] normalization schemes (Table 4). The EMPA for all tourmalines except oxy-uvite (CN11) show 0.22(20)–0.70(22) B in excess of 3 B pfu when normalized to 18 YZTB, with the two synthetic dravites (EB28 and EB29) having the highest amounts (Table 4; 0.70(22) and 0.67(19) excess B pfu, respectively).

Backscattered electron images of polished grain mounts of synthetic tourmaline (K-drv: K-dravite; Drv: dravite; Mg-f: magnesio-foitite; Oxy-uv: oxy-uvite). The lighter gray round spots in the center of the tourmaline aggregates in EB28 are caused by the electron beam during EMPA. The non-tourmaline phases in the dravite synthesis (EB29: coesite and boromuscovite) and magnesio-foitite synthesis (MF2: boralsilite and pinnoite) were identified by powder XRD
The average unit cell parameters obtained by powder XRD are in good agreement with those obtained by single-crystal XRD (Table 5). K-dravite (EB13) has the largest unit cell parameters, which are comparable to those reported for the K-dominant tourmaline maruyamaite (a = 15.955(10) Å, c = 7.227(4) Å, V = 1593.2(17) Å3; Lussier et al. 2014). The synthetic magnesio-foitite (MF2) has the smallest unit cell parameters of the investigated tourmalines, consistent with values obtained for synthetic (a = 15.902(1) Å, c = 7.1263(3) Å, V = 1560.52(12) Å3; von Goerne et al. 2001; Rosenberg and Foit 1979) and natural (a = 15.884(4) Å, c = 7.178(3) Å, V = 1568.0(6) Å3; Hawthorne et al. 1999) magnesio-foitite. The unit cell parameters of the two synthesized dravites (EB28 and EB29) are very similar despite their variable Na content, and are comparable to those of the synthesized oxy-uvite (CN11) (Table 5). The values are consistent with previously synthesized dravite and oxy-uvite [cf. a = 15.875(1) Å, c = 7.1790(2) Å, V = 1566.78(2) Å3 for dravite (von Goerne et al. 2001); a = 15.919(2) Å, c = 7.167(2) Å, V = 1572.97(4) Å3 for oxy-uvite (von Goerne and Franz 2000)]. The unit cell parameters of dravite and oxy-uvite are intermediate in value between those of K-dravite and magnesio-foitite (Table 5). The atom parameters and a selection of interatomic distances for synthetic K-dravite, oxy-uvite, and magnesio-foitite as determined by refinement of the single-crystal X-ray diffraction data (SREF) are given in Tables 3 and 6, respectively. The values obtained for magnesio-foitite are similar to values for synthetic foitite (Kahlenberg and Veličkov 2000), with the average 〈Y–O〉 distance being slightly smaller in magnesio-foitite (cf. 〈Y–O〉 = 1.992 and 2.020 Å for magnesio-foitite and foitite, respectively). The quality of the SREF of synthetic oxy-uvite (CN11) and magnesio-foitite (MF2) is significantly higher than that of K-dravite (EB13) (Table 2), likely due to the different size and quality of the investigated crystals. As a result, in the former two tourmalines, it was feasible to refine the occupancies of the Y and Z sites by Mg and Al from the electron densities at each respective site (Table 3). The presence of tetrahedral B was identified in the SREF of K-dravite (EB13) and, to a lesser extent, magnesio-foitite (MF2). No tetrahedral B was identified in the SREF of oxy-uvite, consistent with the 18 YZTB normalized EMPA (Table 4).
The polarized lattice vibration Raman spectra of synthetic K-dravite, dravite, and oxy-uvite share the same major bands (Fig. 4). The relative intensities of the bands are similar, with the exception of the band at 661 cm−1 having a higher relative intensity in the spectrum of oxy-uvite and that at 676 cm−1 having a higher relative intensity in the spectra of K-dravite and dravite. The lattice vibration Raman spectrum of magnesio-foitite is generally similar to those of the other tourmalines, but is distinguished by the bands at 228, 267, 311, and 688 cm−1 having higher and that at 676 cm−1 reduced relative intensities. The polarized Raman spectra in the O–H stretching region are more characteristic of each investigated tourmaline (Fig. 5). In particular, bands at 3769 and 3817 cm−1 are unique to K-dravite (EB13); bands at 3723, 3739–3740, and 3776–3778 cm−1 are unique to dravite; and the band at 3459 cm−1 is unique to magnesio-foitite (MF2) (Fig. 5, Tables 7, 8). All bands in the polarized Raman O–H stretching spectrum of oxy-uvite can be identified in the spectra of the other tourmalines.

Polarized lattice vibration Raman spectra for synthetic oxy-uvite (CN11), K-dravite (EB13), dravite (EB29), and magnesio-foitite (MF2). All spectra were collected with the electrical field vector of the linearly polarized laser light parallel to the crystallographic c-axis. The inverted triangles indicate the wavenumber of deviations between the spectra, whereby black symbols indicate a band more pronounced in magnesio-foitite; the white symbol indicates a band more pronounced in dravite and K-dravite; and the gray symbol indicates a band more pronounced in oxy-uvite

Polarized Raman spectra in the O–H stretching vibration range for synthetic K-dravite, dravite, magnesio-foitite, and oxy-uvite. The baseline-corrected spectra (black) were collected with the electrical field vector of the linearly polarized laser light parallel to the crystallographic c-axis, except for the second spectrum of K-dravite (EB13 marked with ┴), which was collected with the electrical field vector of the laser light polarized perpendicular to the crystallographic c-axis. The intensity scales for both EB13 spectra are identical. The spectra have been deconvoluted (red) with the PeakFit program to identify the underlying bands (green)
Discussion
Single-crystal XRD refinement (SREF)
K-dravite (EB13)
Comparison of the interatomic distances obtained from the SREF of synthetic K-dravite (EB13; Table 6) to those obtained for naturally occurring Na- and Ca-bearing tourmalines reveals significant expansion of the X-site polyhedron in accommodating the relatively large K+ ion. For example, whereas the average 〈X–O〉 distance determined for the synthesized K-dravite (EB13) was 2.737(12) Å, the largest 〈X–O〉 distance measured by Bloodaxe et al. (1999) was 2.696(5) Å, corresponding to dravitic tourmaline with an average X-site occupancy of 0.540(52) Na, 0.027(2) Ca, and 0.025(31) K pfu. The average 〈Y–O〉 bond length of 2.009(11) Å obtained for synthetic K-dravite (EB13) is relatively small compared to tourmaline with a Y site dominated by Mg (cf. 2.044 Å; Pertlik et al. 2003). This suggests that the smaller Y-site-occupying ion Al (cf. atomic radius of 0.535 Å for VIAl3+ with 0.72 Å for VIMg2+; Shannon 1967) is present, reducing the size of the Y-site coordination polyhedron. Moreover, comparison of the average 〈Z–O〉 bond length of synthetic K-dravite (1.931(8) Å) with values obtained for Mg- and Al-bearing tourmalines investigated by Bloodaxe et al. (1999) suggests the Z site is occupied by ~Al5Mg. Increasing amounts of Mg at the Z site would result in increasing 〈Z–O〉 bond lengths (Bloodaxe et al. 1999). Based on the measured 〈Y–O〉 and 〈Z–O〉 bond lengths, we expect significant Mg–Al disorder between the Y and Z sites in the synthetic K-dravite. The average 〈T–O〉 bond length of 1.615(6) Å is slightly smaller than, albeit within error of the bond length expected for a T site fully occupied by Si (~1.62 Å; MacDonald and Hawthorne 1995). Nevertheless, based on electron densities in the tourmaline structure, the single-crystal structure refinement suggests the presence of 0.6(1) tetrahedral B pfu, a value 3σ greater than that determined by EMPA. The occurrence of tetrahedral B is not uncommon for tourmalines crystallized at high temperature and pressure (Ertl et al. 2008, 2010); however, the 〈T–O〉 bond length and EMPA data suggest it is minor. Comparison of the position of H3 (x = 0.246(9)) and O3 (x = 0.2628(9)) in the atom parameters (Table 3) shows that the H3 atom is not perfectly in line with O3. It is slightly shifted toward the tetrahedral site, as expected from the presence of its hydrogen bond with O5 (Fig. 1).
Oxy-uvite (CN11)
In the structure of the ideal, ordered oxy-uvite endmember [XCaYMg Z3 Al T6 Si6O18(BO3) V3 (OH) W3 O)], Mg is found at the octahedral Y site and Al at the octahedral Z site. The synthesized oxy-uvite (CN11) has a lower Mg/Al ratio than the endmember (Table 4), indicating that some Al must occur at the Y site. In addition, the SREF revealed that in the investigated crystal, not all the Mg is restricted to the Y site. Instead, there is Mg–Al disorder between the two octahedral sites, which results in an average of 1.52(10) Mg and 1.48(10) Al pfu occurring at the Y site and 1.10(15) Mg and 4.90(15) Al pfu at the Z site (Table 3). The average 〈T–O〉 bond length of 1.619(1) is in agreement with a T site fully occupied by Si (MacDonald and Hawthorne 1995), as determined by EMPA. The absence of tetrahedral B is expected from the low synthesis pressure of 0.2 GPa.
Compared to K-dravite (EB13; Table 3), the O3–H3 bond in oxy-uvite (CN11) is slightly more parallel to the c-axis (x = 0.26024(6) for O3 and x = 0.257(2) for H3). This suggests that H bonding between H3 and O5 is less pronounced in oxy-uvite than in K-dravite.
Magnesio-foitite (MF2)
In the structure of the ideal, ordered magnesio-foitite endmember [X☐Y(Mg2Al)ZAl T6 Si6O18(BO3) V3 (OH) W3 (OH)], Mg is found at the octahedral Y site. Although the synthetic magnesio-foitite (MF2) has approximately endmember stoichiometry with ~2 Mg and 7 Al as determined by EMPA (Table 4), the SREF revealed that in the investigated crystal, an average of 1.38(18) Mg and 1.62(18) Al pfu occurs at the Y site and 1.08(24) Mg and 4.92(24) Al pfu at the Z site (Table 3). The presence of minor Na refined at the X site (~0.09(6) apfu; Table 3) likely reflects Na contamination in the synthesis procedure. This emphasizes the high preference of Na for tourmaline over the coexisting fluid, as even traces of Na in the fluid are incorporated into the crystallizing tourmaline. Small amounts of Na (0.01(2) apfu) were also detected by EMPA in the nominally Na-free magnesio-foitite crystals. The average 〈T–O〉 bond length of 1.616(1) Å is comparable to that of synthetic K-dravite (EB13, Table 6), but the amount of B refined at the T site based on electron densities is less (cf. 0.34(4) IVB in MF2 with 0.6(1) IVB in EB13), as expected from the lower synthesis pressure of 0.4 GPa (cf. 4.0 GPa for EB13; Berryman et al. 2015). The low amount of tetrahedral B refined for magnesio-foitite (MF2) is in agreement with the results of EMPA (Table 4) to a higher degree than for K-dravite (EB13), likely due to the higher quality of the former’s SREF (cf. R1 = 1.19 % for CN11 and 4.88 % for EB13).
Compared to K-dravite (EB13) and oxy-uvite (CN11; Table 3), the O3–H3 bond in magnesio-foitite (MF2) is the most parallel to the c-axis (x = 0.26152(8) for O3 and x = 0.262(2) for H3). This suggests that H bonding between H3 and O5 is least pronounced in magnesio-foitite (MF2).
Polarized Raman spectroscopy
Lattice vibration Raman spectra
In the lattice vibration Raman spectra, bands in the 200–300 cm−1 wavenumber range have been previously assigned to vibrations at the Y site (e.g., Gasharova et al. 1997). The increased intensity of the bands around 228 and 267 cm−1 in magnesio-foitite (MF2) compared to dravite (EB29), K-dravite (EB13), and oxy-uvite (CN11) may reflect the higher occupancy of the Y site by Al (Fig. 4). This is consistent with the lattice vibration Raman spectrum of synthetic olenite [XNaYAl Z3 Al6(TSi6O18)(BO3)3O3(OH)], which also has distinct bands around 228 and 267 cm−1 (Martin Kutzschbach, personal communication). Although the sharp band at 311 cm−1 in the spectrum of magnesio-foitite has not been previously assigned, its increased intensity for magnesio-foitite (as well as for synthetic olenite) compared to dravite and K-dravite may also reflect its association with Al at the Y site. The bands between 600 and 800 cm−1 have been associated with Si–O vibrations in the Si6O18 ring and are affected by substitutions at the neighboring Y and Z sites (Mihailova et al. 1996; Bosi and Lucchesi 2007; Fantini et al. 2014). Although the position and intensities of these bands are similar for K-dravite and dravite, they are distinct in the lattice vibration Raman spectra of magnesio-foitite (MF2) and oxy-uvite (CN11) (Fig. 4). The different shape of these bands in the spectrum of magnesio-foitite (MF2) likely reflects the effect of the vacant X site on the Si6O18 ring. In the case of oxy-uvite, the different shape likely results from the increased charge of Ca2+ as an X-site-occupying ion, or less likely, the effect of a deprotonated W (O1) site on the Si6O18 ring structure. Together, the variations in the lattice vibrations of MF2, and to a lesser extent CN11, show distinct differences in the structure of magnesio-foitite and oxy-uvite compared to K-dravite and dravite. In contrast, the lattice vibration Raman spectra of K-dravite and dravite are comparable, with no clear variations in major band positions or relative intensities. This observation suggests that substituting K for Na at dravite’s X site does not significantly influence the bonding environment around the other sites in the structure.
O–H stretching Raman spectra
The O–H stretching vibrations in the Raman spectra (Fig. 5) are more sensitive than the lattice vibrations (Fig. 4) to compositional variations in tourmaline. With the exception of magnesio-foitite, if the synthesized tourmaline crystals had their exact endmember compositions, whereby each site is occupied by a single element, we would expect two symmetrical bands in the O–H stretching region of the Raman spectra: one for each OH-occupied site, W (O1) and V (O3). In the case of magnesio-foitite, its ordered, endmember composition with YMg2Al would generate three bands in the O-H stretching region: one for the W (O1) site and two for the V (O3) site, corresponding to the latter's coordination with either YMg or YAl. Additional bands reflect variations in the local bonding environment around the different hydroxyl groups, with each band representing a different bonding environment. If either of the W (O1) or V (O3) sites is deprotonated [as is the case for W (O1) in oxy-uvite (XCaYMg Z3 Al T6 Si6O18(BO3) V3 (OH) W3 O)], its associated bands will be absent. The OH bond at the W (O1) site is sensitive to variations in the occupation of the three coordinated Y sites and the nearby X site (Fig. 1). The three OH bonds at the V (O3) sites are sensitive to variations in the occupation of the coordinated Z and Y sites. Variation in the occupancy of the T or X sites might also exert an effect on the OH bond at the V (O3) site by means of their coordination with the O5 site, which shares a weak hydrogen bond with the O3 site (Gatta et al. 2012; Fig. 1). In general, any variation in the short-range environment around an OH bond that results in the shortening (i.e., strengthening) of the OH bond is expected to result in an increase in its vibrational energy and thus in an increase in its Raman shift. Conversely, variations that result in the lengthening (i.e., weakening) of the OH bond are expected to result in a decrease in its Raman shift. As a result, the position of an OH bond’s Raman band is inversely proportional to the charge and electronegativity of the coordinated cations. Band assignments were thus made following the theory that OH bonds coordinated to cations of higher charge and electronegativity will produce bands of lower Raman shifts (Martínez-Alonso et al. 2002).
Bands in the 3400–3600 cm−1 wavenumber range are assigned to OH at the V (O3) sites and those above this range to OH at the W (O1) site, based on their relative intensities (Gonzalez-Carreño et al. 1988; Gatta et al. 2014). Bands in the range 3284–3301 and 3367–3370 cm−1 have been associated with tetrahedral B in olenite (Kutzschbach et al., in review). In the O–H stretching Raman spectrum of EB28 (Fig. 5), there is a broad, low-intensity feature around 3400 cm−1 that can be deconvoluted into two broad, low-intensity bands at 3381 and 3446 cm−1. The former band (3381 cm−1) can be indicative of the presence of tetrahedral B, as observed for olenite by Kutzschbach et al. (in review), and is consistent with the presence of tetrahedral B detected by EMPA (Table 4). A small feature at the same position in the spectrum of EB29 is also discernible. The broad band at 3446 cm−1 was associated with the presence of vacancies at the X-site and a coordination environment of X☐YZAl3 by Kutzschbach et al. (in review). This assignment is consistent with assignments made in the O–H stretching Raman spectrum of magnesio-foitite (Fig. 5; Table 8; MF2), whereby the band at 3459 cm−1 is assigned to YZAl3, reflecting the effect of the magnesio-foitite component of EB28 on the V (O3) site. As the bands at 3381 and 3446 cm−1 are of low intensity and only present in the EB28 dravite sample, it is ignored in the subsequent discussion.
W (O1) O–H stretching Raman bands
The five investigated tourmalines have between two and five bands assigned to the higher frequency, W (O1) site range (Fig. 5; Tables 7, 8, 9). Each of these five bands must reflect a unique bonding environment around the W (O1) site in terms of the occupancy of the X and Y sites. Given the restricted composition of the synthetic tourmalines, the Y sites can only be occupied by Al3+ or Mg2+ and the X site can only be vacant or occupied by K+ (EB13), Na+ (EB28 and EB29), or Ca2+ (CN11; Table 4). The W (O1) bands at 3769 and 3817 cm−1 (Fig. 5) are unique to K-dravite (EB13), unambiguously associating these bands with the presence of K at the X site. Similarly, the W (O1) bands at 3723, 3739–3740, and 3776–3778 cm−1 are unique to dravite (EB28 and EB29; Fig. 5) and are therefore assigned to Na at the X site. The W (O1) bands in the range 3618–3667 cm−1 are common to all synthetic tourmaline and are therefore assigned to the presence of vacancies at the X site. The increased intensity of these bands in the spectrum of X-vacant magnesio-foitite (MF2) supports this assignment. There are no bands assigned to Ca at the X site because the W (O1) site in oxy-uvite is deprotonated and therefore does not produce a corresponding band in the O–H stretching range of the Raman spectrum. However, W (O1) bands corresponding to a vacant X site (3618–3667 cm−1) are present (CN11; Fig. 5), indicating that the W (O1) site in oxy-uvite is locally protonated in the presence of a vacant X site.
The presence of multiple W (O1) bands associated with each X site composition reflects variations in the amount of Mg and Al at the Y sites coordinated to the W (O1) site. In the structural formulae for the ideal, ordered endmembers of dravite and K-dravite, the Y site is occupied entirely by Mg. However, based on the SREF of EB13, we expect significant Mg-Al disorder between the Y and Z sites resulting in the presence of significant Al at the Y site. We therefore assign the three bands associated with a K- or Na-occupied X site (Tables 7, 8; EB13, EB28, and EB29) to a coordination environment of YMgYMgYMg, YMgYMgYAl, and YMgYAlYAl, with decreasing wavenumbers, respectively.
Considering the Raman spectra of all investigated tourmalines (Fig. 5), there are a total of three W (O1) bands (>3600 cm−1) assigned to vacancies at the X site: 3618–3632, 3636–3650, and 3657–3667 cm−1. As in the case of the X-occupied bands, assignments were made with the proportion of Al (and therefore charge) increasing with decreasing Raman shift (Tables 7, 8, 9). In the Raman spectrum of magnesio-foitite (MF2), the band assigned to X☐YMgYMgYAl is absent (3636–3650 cm−1; Table 9; Fig. 5), and only the two Y-site configurations X☐YMgYMgYMg and X☐YMgYAlYAl are present. The absence of the 3636–3650 cm−1 band in the spectrum of magnesio-foitite has been observed in previous investigations of synthetic magnesio-foitite (Veličkov 2002) and therefore seems to be characteristic of the magnesio-foitite endmember. Our Raman band assignments suggest that the Y-site composition of the synthetic magnesio-foitite (~YMg1.4Al1.6; Table 3) as determined by SREF reflects a combination of YMg3 and YMgAl2 Y-site trimers, in an approximately 2:3 proportion (Table 9). This implies a long-range ordering of Mg and Al at the Y sites for magnesio-foitite. This is not the case for the X-vacant components in the synthesized K-dravite (EB13) and dravite (EB28, and EB29) samples, none of which has a band assigned to the configuration X☐YMgYMgYMg. From bond valence arguments, vacancies at the X site in these tourmalines are expected to be associated with Al at the Y site, with additional Al possibly resulting from Mg–Al disorder between the Y and Z sites. Therefore, it is reasonable that the band assigned to X☐YMgYMgYMg is absent from the synthetic K-dravite and dravite. In the case of oxy-uvite (CN11), all three X-vacant bands are observed in the O–H stretching Raman spectrum, with that at 3618–3632 cm−1 having the highest intensity. This high intensity band is assigned to X☐YMgYAlYAl. In the SREF of CN11, a Y-site occupancy of 1.5 Al and 1.5 Mg was refined, indicating a significant degree of Mg–Al disorder between the Y and Z sites. This disorder, which puts additional Al at the Y site, combined with the preferential occurrence of Al at the Y site in the presence of a vacant X site justifies the configuration X☐YMgYAlYAl as the highest intensity W (O1) band for oxy-uvite.
V (O3) O–H stretching Raman bands
The Raman spectrum of each tourmaline crystal has three bands fitted beneath the broad V (O3) band (3400–3600 cm−1; Fig. 5). The positions of the fitted bands in the spectra of dravite, K-dravite, and oxy-uvite (EB28, EB29, EB13, and CN11) are in good agreement with each other, and also with the Raman spectra of dravite and uvite from natural rocks (e.g., Fantini et al. 2014). In contrast, the V (O3) bands in the spectrum of magnesio-foitite (MF2) are all shifted to lower wavenumbers. For this reason, the assignments of the V (O3) bands in the latter are performed separately and subsequently compared to the other tourmalines. The positions of the V (O3) bands are principally influenced by their respective coordinating environment: one Y-site octahedron and two Z-site octahedra (Fig. 1). In the ideal, ordered endmembers of K-dravite, dravite, and oxy-uvite, the Z site is fully occupied by Al and the Y site by Mg (YMgZAlZAl). Al–Mg disorder between the Z and Y sites results in additional Mg occurring at the Z site (YMgZMgZAl) and additional Al occurring at the Y site (YAlZAlZAl). These three coordination environments were assigned to the three fitted V (O3) bands in the O–H stretching Raman spectra of K-dravite (EB13), dravite (EB28 and EB29), and oxy-uvite (CN11) with Mg content increasing with the band’s wavenumber (Table 7). The assignment of the 3549–3558 cm−1 band to YMgZAlZAl is consistent with the results of Bosi et al. (2015). The same coordination environments were assigned in the same order to the three V (O3) bands fitted in the spectrum of magnesio-foitite (MF2).
Controls on O–H stretching Raman band position
Band assignments for each hydroxyl site were consistently based on the principle that the Raman shift of a particular band should be inversely proportional to the net charge of the coordinated polyhedra. Comparison of the band position to the assigned coordination environment provides additional insight into this correlation. The Raman shift of the V (O3) O–H stretching vibration is more sensitive to the net charge (or electronegativity) of the coordinated polyhedra than those assigned to the W (O1) site, as reflected by the steeper slope of the V (O3) curve in Fig. 6. For both sites, increasing the charge/electronegativity of the coordination environment results in a consistent shift in the position of the Raman band regardless of the occupancy of the X site, as reflected in the curves of each endmember for the W (O1) and V (O3) bands in Fig. 6 being approximately parallel. It is therefore reasonable that substitution of elements of different charge/electronegativity at the Y and Z sites will produce bands at positions consistent with the observed trend for each endmember. This will in turn facilitate band assignments in tourmalines with complex chemistries, more typical of naturally occurring tourmalines.

Raman shift of O–H stretching bands plotted against the net charge of the coordinated polyhedra (YZAlMg2, YZAl2Mg, and YZAl3; or YMg3, YMg2Al, and YMgAl2). Only data points associated with the endmember X-site composition are shown for the W (O1) bands (i.e., XK for K-dravite, XNa for dravite, X☐ for magnesio-foitite). As no O1 bands in the Raman spectrum of oxy-uvite were assigned to XCa, data points for oxy-uvite in the W (O1) plot are not available
The overall shift of the V (O3) bands in the spectrum of magnesio-foitite (MF2) to lower wavenumbers reflects the lengthening (weakening) of the OH bond at this site (Fig. 6). This is likely a further indication of the deformation of the Si6O18 ring in the presence of a vacant X site. The V (O3) OH bond is affected by the Si6O18 ring via the hydrogen bond between H3 and the O5 site (Fig. 1). The shift of the V (O3) bands to lower wavenumbers was also observed for synthetic magnesio-foitite and foitite investigated by Veličkov (2002). In contrast, the overall lengthening of the V (O3) OH bonds was not observed in the Raman spectrum of the naturally occurring magnesio-foitite investigated by Fantini et al. (2014), which has an average X-site composition of X☐0.56Na0.31Ca0.13 (Fantini et al. 2014). The significant amount of Na at the X site of the natural magnesio-foitite is sufficient to make the Raman spectra in the lattice and O–H stretching range more comparable to those of synthetic dravite.
Calculating tourmaline compositions from their O–H stretching Raman spectra
For a group of bands assigned to a particular hydroxyl-bearing site (V or W), the integrated peak intensity under a given band is proportional to the occurrence of the local bonding environment assigned to it. For example, the integrated peak intensity under the band assigned to XKYMg2Al (Fig. 5, Tables 7, 8) is the highest of all bands assigned to the W (O1) site in EB13. We therefore expect this to be the most common coordination environment for hydroxyl-occupied W (O1) sites in EB13. Furthermore, the relative integrated peak intensity can be linked to the proportion of its assigned compositional environment within the structure. Since the Mg and Al are the only two elements present at the Y and Z sites, their relative amounts in each tourmaline can thus be calculated from the relative integrated peak intensities. In addition, dravite and K-dravite have vacancies and either Na or K at their X sites, respectively. The ratio of the two possible X-site compositions can be calculated from the ratio of the integrated peak intensities. Following this reasoning, the compositions of the X, Y, and Z sites were calculated from the O–H stretching region of the Raman spectra for each tourmaline (Tables 7, 8). Since oxy-uvite does not have W (O1) bands assigned to an X site occupied by Ca, the X-site composition cannot be calculated. However, the relative integrated peak intensities of the W (O1) bands assigned to vacancies at the X site are sufficient to calculate the Y site composition. This approach assumes that the X-site composition has no bearing on the local Y-site composition, an assumption that is unlikely true. Instead, because of local charge balance of the vacant X site, we expect the calculated Y-site composition to have increased Al relative to the true value. A comparison of the calculated values to those determined by SREF indicates this to be the case (Table 7). The V (O3) bands of oxy-uvite are unaffected by this bias and can be used to calculated the net Y and Z site composition following the same procedure as for the other tourmalines. The good agreement between the calculated Mg–Al ratios and those measured by EMP or refined by SREF (Tables 7, 9) supports the Raman band assignments. It should be noted that partially deprotonated V (O3) or W (O1) sites will not contribute a signal to the Raman spectrum and will therefore not contribute to the calculated compositions, introducing error into the calculation of all investigated tourmalines.
Influence of X-site composition on crystal structure
The composition of the X site affects tourmaline’s long-range crystal structure. This is reflected in the expansion of the unit cell volume from 1560 ± 3 Å3 for tourmaline with a vacant X site to 1570 ± 1 Å3 or 1573 ± 1 Å3 with the intermediate in size Na+ or Ca2+ ions, respectively, to 1590 ± 2 Å3 with the large K+ ion (averages of powder and single-crystal unit cell parameter determinations; Table 5). The gradual increase in unit cell volume is mainly accounted for by changes in the length of the c-axis (Table 5) and is expected to increase with increasing K content (Berryman et al. 2015). Tourmaline’s extension, or contraction, parallel to its c-axis in response to the size of the X-site-occupying ion reflects tourmaline’s increased elasticity in this direction (Dietrich 1985, and references therein).
Even though tourmaline’s crystal structure expands with increasing size of the X-site-occupying ion, the Raman spectra show minimal changes in the lattice vibrations of oxy-uvite, dravite, and K-dravite. In particular, the lattice vibration Raman spectrum of K-dravite is very similar to that of dravite (Fig. 4), suggesting that the overall structure is not strongly influenced by changing the size of the X-site-occupying ion. The differences observed in the spectra of magnesio-foitite (MF2) and oxy-uvite (CN11; Fig. 4) likely reflect the different charge of the X site, rather than the ion size. Nonetheless, comparison of the average bond lengths of tourmaline with the largest unit cell, K-dravite, and that with the smallest, magnesio-foitite, reveals that the Y and Z sites are only slightly expanded in the former, with the T site remaining unchanged (Table 6).
Changes in tourmaline’s structure with changing X-site composition appear to be localized around the X-site polyhedron. This is demonstrated by the large shifts in the band positions assigned to O–H stretching at the W (O1) site in the Raman spectra of K-dravite, dravite, and magnesio-foitite. The Raman shifts of the W (O1) bands assigned to the K-occupied X site in K-dravite are overall of higher value than those assigned to the Na-occupied X site in dravite (Fig. 6). The OH bond at the W (O1) site is oriented toward the X site (Fig. 1), and the value of the Raman shift is proportional to the energy of the OH bond. The overall increase in the Raman shift of the bands from dravite to K-dravite therefore reflects an increase in the energy of the O–H vibration at the W (O1) site. This strengthening likely results from the larger K+ ion repelling the H1 ion away from X site toward O1, thereby decreasing the O1–H1 bond length and increasing its bond strength. In oxy-uvite, the 2+ charge of Ca at the X site locally results in the deprotonation of the neighboring W (O1) site. In contrast, given a vacant X site, the neighboring W (O1) site is not deprotonated. This is reflected in the O–H stretching Raman spectrum of oxy-uvite (Fig. 5) by the presence of W (O1) bands assigned to vacancies at the X site but not those assigned to Ca (Table 7). In all the investigated tourmalines, the W (O1) bands assigned to vacancies at the X site occur at wavenumbers lower than those assigned to K and Na at the X site (Table 7, magnesio-foitite in Fig. 6). In addition, they are at a wavenumber lower than expected for an isolated OH group (3735 cm−1; Branscomb 1966). This indicates that in the presence of a vacant X site, the OH bond at the W (O1) site is lengthened relative to an isolated hydroxyl molecule, reducing its energy.
The presence of a mostly vacant X site, as in magnesio-foitite, affects more than the neighboring W (O1) site. The Si6O18 ring structure changes shape as reflected by changes in its assigned bands in the lattice vibration Raman spectra. In turn, the hydrogen bond between H at the O3 site and O5 in the Si6O18 ring (Fig. 1) is weakened, leading to its increased alignment with tourmaline’s c-axis. The O3–H3 bond is lengthened and its associated O–H stretching bands shifted to lower wavenumbers. The 2+ charge of Ca also leads to changes in the Si6O18 ring and, correspondingly, a reduction in the H bond between the H3 and O5 sites. However, in oxy-uvite, these changes were of lesser degree than in magnesio-foitite and did not affect the length of the OH bonds at the V (O3) site.
Conclusions
The combination of long- and short-range structural data collected by SREF and Raman spectroscopy, respectively, allows the effect of the X-site composition on tourmaline’s crystal structure to be investigated. In particular, the use of synthetic tourmalines with restricted compositions largely eliminates the ambiguity in refining the occupancy of different sites from their respective scattering power in the SREF and in assigning bands in the Raman spectra to particular coordination environments. As a result, band assignments for K-dravite, dravite, oxy-uvite, and magnesio-foitite were made and used to calculate the Mg–Al ratio of the Y and Z sites of each investigated tourmaline, giving values consistent with the net Mg–Al ratio determined by EMPA and the specific site occupancies determined by SREF. Moreover, a combination of the data collected by SREF and that by Raman spectroscopy demonstrated that the composition of tourmaline’s X site, which leads to the expansion or contraction of the crystal structure, is accommodated locally around the X site. With increasing size of the X-site-occupying ion, the X-site-coordination polyhedron expands, impinging on the hydroxyl at the O1 site leading to its shortening. The other sites in the crystal structure are not measurably affected. Increasing the charge of the X-site-occupying ion from a 1+ to a 2+ ion, as in the case of oxy-uvite, can result in the local deprotonation of the neighboring W (O1) site. The absence of an X-site-occupying ion, as is the case for the magnesio-foitite endmember or for magnesio-foitite components in the other tourmalines, leads to the lengthening of the H1–O1 bond. In addition, the magnesio-foitite endmember has demonstrable variations in the bonding environment around the other crystallographic sites, leading to distinct differences in its lattice vibration Raman spectrum. This structural difference also affects the O3–H3 bond, shifting the associated V (O3) Raman bands to lower wavenumbers. However, the band positions of vibrations assigned to the O1–H1 bond were consistent with bands assigned to vacant X sites in the other investigated tourmalines. Combined, SREF and polarized Raman spectroscopy of single crystals are powerful tools for investigating subtle changes in crystal structure and help to elucidate the feedback between structure and composition in tourmaline supergroup minerals.
References
Armstrong JT (1995) CITZAF: a package of correction programs for the quantitative electron microbeam X-ray analysis of thick polished materials, thin films, and particles. Microbeam Anal 4:177–200
Berryman E, Wunder B, Rhede D (2014) Synthesis of K-dominant tourmaline. Am Mineral 99:539–542
Berryman EJ, Wunder B, Wirth R, Rhede D, Schettler G, Franz G, Heinrich W (2015) An experimental study on K and Na incorporation in dravitic tourmaline and insight into the formation environment of diamoniferous tourmaline from the Kokchetav, Massif, Kazakhstan. Contrib Mineral Petrol 169:28. doi:10.1007/s00410-015-116-9
Bloodaxe ES, Hughes JM, Dyar MD, Grew ES, Guidotti CV (1999) Linking structure and chemistry in the Schorl-Dravite series. Am Mineral 84:922–928
Bosi F, Lucchesi S (2007) Crystal chemical relationships in the tourmaline group: structural constraints on chemical variability. Am Mineral 92:1054–1063
Bosi F, Skogby H, Lazor P, Reznitskii L (2015) Atomic arrangements around the O3 site in Al- and Cr-rich oxy-tourmalines: a combined EMP, SREF, FTIR and Raman study. Phys Chem Mineral 42:441–453. doi:10.1007/s00269-015-0735-z
Branscomb LM (1966) Photodetachment cross section, electron affinity, and structure of the negative hydroxyl ion. Phys Rev 148:11. doi:10.1103/PhysRev.148.11
Buerger M, Parrish W (1937) The unit cell and space group of tourmaline (an example of the inspective equi-inclination treatment of trigonal crystals). Am Mineral 22:1139–1150
Dietrich RV (1985) The tourmaline group. Van Nostrand Reinhold Company Inc., New York
Donnay G, Buerger MJ (1950) The determination of the crystal structure of tourmaline. Acta Crystallogr A 3:379–388
Ertl A, Tillmanns E (2012) The [9]-coordinated X site in the crystal structure of tourmaline-group minerals. Z Kristallogr 227:456–459
Ertl A, Tillmanns E, Ntaflos T, Francis C, Giester G, Körner W, Hughes JM, Lengauer C, Prem M (2008) Tetrahedrally coordinated boron in Al-rich tourmaline and its relationship to pressure-temperature conditions of formation. Eur J Mineral 20:881–888
Ertl A, Marschall HR, Giester G, Henry DJ, Schertl H-P, Ntaflos T, Luvizotto GL, Nasdala L, Tillmanns E (2010) Metamorphic ultrahigh-pressure tourmaline: structure, chemistry, and correlations to P–T conditions. Am Mineral 95:1–10
Ertl A, Giester G, Ludwig T, Meyer H-P, Rossman GR (2012) Synthetic B-rich olenite: correlations of single-crystal structural data. Am Mineral 97:1591–1597
Fantini C, Tavares MC, Krambrock K (2014) Raman and infrared study of hydroxyl sites in natural uvite, fluor-uvite, magnesio-foitite, dravite and elbaite tourmalines. Phys Chem Miner 41:247–254
Fischer RX, Tillmanns E (1988) The equivalent isotropic displacement factor. Acta Crystallogr A C44:775–776
Gasharova B, Mihailova B, Konstantinov L (1997) Raman spectra of various types of tourmaline. Eur J Miner 9:935–940
Gatta GD, Danisi RM, Adamo I, Meven M, Diella V (2012) A single-crystal neutron and X-ray diffraction study of elbaite. Phys Chem Miner 39:577–588
Gatta GD, Bosi F, McIntyre GJ, Skogby H (2014) First accurate location of two proton sites in tourmaline: a single-crystal neutron diffraction study of oxy-dravite. Mineral Mag 78:681–692
Geisinger KL, Spackman MA, Gibbs GV (1987) Exploration of structure, electron density distribution, and bonding in coesite with fourier and pseudoatom refinement methods using single-crystal X-ray diffraction. J Phys Chem 91:3237–3244
Genkina EA, Malinovskii YA (1983) Refinement of the structure of pinooite: location of hydrogen atoms. Kristallografiya (Sov Phys Cryst) 28:803–805
Gonzalez-Carreño T, Fernández M, Sanz J (1988) Infrared and electron microprobe analysis of tourmalines. Phys Chem Mineral 15:452–460
Grice JD, Ercit TS, Hawthorne FC (1993) Povondraite, a redefinition of the tourmaline ferridravite. Am Mineral 78:433–436
Hamburger GE, Buerger MJ (1948) The structure of tourmaline. Am Mineral 33:532–540
Hawthorne FC, Henry DJ (1999) Classification of the minerals of the tourmaline group. Eur J Mineral 11:201–215
Hawthorne FC, Selway JB, Kato A, Matsubara S, Shimizu M, Grice JD, Vajdak J (1999) Magnesiofoitite, ☐(Mg2Al)Al6(Si6O18)(BO3)3(OH)4, a new alkali-deficient tourmaline. Can Mineral 37:1439–1443
Henry DJ, Dutrow BL (1996) Metamorphic tourmaline and its petrologic applications. In: Grew ES, Anvitz LM (eds) Boron: mineralogy, petrology and geochemistry, reviews in mineralogy and geochemistry, vol 33., Mineral Soc AmChantilly, Virginia, pp 503–557
Henry DJ, Novák M, Hawthorne FC, Ertl A, Dutrow BL, Uher P, Pezzotta F (2011) Nomenclature of the tourmaline-supergroup minerals. Am Mineral 96:895–913
Kahlenberg V, Veličkov B (2000) Structural investigations on a synthetic alkali-free hydrogen-deficient Fe-tourmaline (foitite). Eur J Mineral 12:947–953
Larson AC, Von Dreele RB (1987) Generalized structure analysis system. Los Alamos National Laboratory Report LA-UR-86-748
Lussier A, Ball NA, Hawthorne FC, Henry DJ, Shimizu R, Ogasawara Y, Ota T (2014) Maruyamaite, IMA 2013-123. CNMNC Newsletter No. 20, June 2014. Mineral Mag 78:549–558
MacDonald DJ, Hawthorne FC (1995) The crystal chemistry of Si−Al substitution in tourmaline. Can Mineral 33:849–858
Martínez-Alonso S, Rustad JR, Goetz AFH (2002) Ab initio quantum mechanical modeling of infrared vibrational frequencies of the OH group in dioctahedral phyllosilicates. Part II: main physical factors governing the OH vibrations. Am Mineral 87:1224–1234
McKeown DA (2008) Raman spectroscopy, vibrational analysis, and heating of buergerite tourmaline. Phys Chem Miner 35:259–270
Mihailova B, Gasharova B, Konstantinov L (1996) Influence of non-tetrahedral cations on Si–O vibrations in complex silicates. J Raman Spectrosc 27:829–833
Mirwald PW, Massonne H-J (1980) Quartz-coesite transition and the comparative friction measurements in piston-cylinder apparatus using talc-alsimag-glass (TAG) and NaCl high pressure cells: a discussion. Neues Jahrbuch für Mineralogie 1980:469–477
Peacor DR, Rouse RC, Grew ES (1999) Crystal structure of boralsilite and its relation to a family of boroaluminosilicates, sillimanite, and andalusite. Am Mineral 84:1152–1161
Pertlik F, Ertl A, Körner W, Brandstätter F, Schuster R (2003) Na-rich dravite in the marbles from Friesach, Carintha, Austria: chemistry and crystal structure. Neues Jahrbuch Mineralogie Monatshefte 2003:277–288
Rosenberg PE, Foit FF Jr (1979) Synthesis and characterization of alkali-free tourmaline. Am Mineral 64:180–186
Shannon RD (1976) Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr A 32:751–767
Sheldrick GM (1997) SHELXL-97, a program for crystal structure refinement. University of Göttingen, Germany
Skogby H, Bosi F, Lazor P (2012) Short-range order in tourmaline: a vibrational spectroscopic approach to elbaite. Phys Chem Miner 39:811–816
Veličkov B (2002) Kristallchemie von Fe,Mg-Turmalinen: Synthese und spektroskopische Untersuchungen. Dissertation, Technische Universität Berlin
von Goerne G, Franz G (2000) Synthesis of Ca-tourmaline in the system CaO–MgO–Al2O3–SiO2–B2O3–H2O–HCl. Mineral Petrol 69:161–182
von Goerne G, Franz G, Wirth R (1999) Hydrothermal synthesis of large dravite crystals by the chamber method. Eur J Mineral 11:1061–1077
von Goerne G, Franz G, Heinrich W (2001) Synthesis of tourmaline solid solutions in the system Na2O–MgO–Al2O3–SiO2–B2O3–H2O–HCl and the distribution of Na between tourmaline and fluid at 300 to 700 C and 200 MPa. Contrib Miner Petrol 141:160–173
Žáček V, Frýda J, Petrov A, Hyršl J (2000) Tourmalines of the povondraite—(oxy)dravite series from the cap rock of meta-evaporite in Alto Chapare, Cochabamba, Bolivia. J Czech Geol Soc 45:3–12
Zhao C, Liao L, Xia Z, Sun X (2012) Temperature-dependent Raman and infrared spectroscopy study on iron-magnesium tourmalines with different Fe content. Vib Spectrosc 62:28–34
Zhukhlistov AP, Zvyagin BB, Soboleva SV, Fedotov AF (1973) The crystal structure of the dioctahedral mica 2M2 determined by high voltage electron diffraction. Clays Clay Miner 21:465–470
Acknowledgements
The authors are grateful to H.-P. Nabein for assistance in generating the powder XRD data and conducting the hydrothermal synthesis experiments; U. Dittmann for preparing the samples for EMPA; G. Franz for critical reading of the final manuscript; and D. Henry and B. Gasharova for their careful reviews. This study was supported by funding from the Deutsche Forschungsgemeinschaft granted to G. Franz and WH (FR 557/31-1; HE 2015/16-1) and by the Austrian Science Fund (FWF) project No. P-26903-N19 granted to AE. EJB is grateful for a postgraduate scholarship awarded by the Natural Sciences and Engineering Research Council of Canada.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Berryman, E.J., Wunder, B., Ertl, A. et al. Influence of the X-site composition on tourmaline’s crystal structure: investigation of synthetic K-dravite, dravite, oxy-uvite, and magnesio-foitite using SREF and Raman spectroscopy. Phys Chem Minerals 43, 83–102 (2016). https://doi.org/10.1007/s00269-015-0776-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00269-015-0776-3