
Abstract
Purpose
Skeletal muscle comprises different kinds of muscle fibres that can be classified as slow and fast fibres. The purpose of this study was to compare the yield, proliferation, and multi-potentiality of rat mesenchymal stem cells (MSCs) from the tibialis anterior (TA; fast muscle) and soleus (SO; slow muscle) in vitro.
Methods
The TA and SO muscles were harvested, and isolated cells were plated. After two hours, the cells were washed extensively to remove any cell that did not adhere to the cell culture plate. The adherent cells, namely MSCs, were then cultured. Both types of MSCs were differentiated toward the osteogenic, chondrogenic and adipogenic lineages using lineage specific induction factors.
Results
The colony-forming unit fibroblast (CFU-F) assay revealed that the SO contained significantly higher quantities of MSCs than the TA. The self-renewal capacity of MSCs derived from the TA was significantly higher at later passages (passage 9–11). Both types of MSCs exhibited similar cell surface antigens to bone marrow (BM)-derived MSCs and were positive for CD29, CD44, and CD90 and negative for CD11b, CD34, and CD45. TA-derived MSCs were superior in terms of osteogenic differentiation capacity, but there was no significant difference in chondrogenic and adipogenic differentiation capacity.
Conclusion
Our results demonstrated significant differences in the properties of muscle-derived MSCs from different muscle types (i.e. fast or slow muscles). The greater expandability and osteogenic differentiation ability of TA-derived MSCs suggests that fast muscle may be a better source for generating large numbers of MSCs for bone regeneration.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Regenerative medicine has created a new paradigm of future therapeutic options for musculoskeletal tissues, and has required cells, morphogenetic signals, scaffolds, and an appropriate mechanical environment [1]. Mesenchymal stem cells (MSCs) have been proven to be clinically promising for the repair and regeneration of tissue damaged by trauma or disease [2–4]. Bone marrow (BM) is currently the most commonly used and studied source of MSCs [4, 5]. Several reports have discussed isolating MSCs from adult skeletal muscle [6–10]. Skeletal muscle is one of the most abundant tissues in the body, comprising about 40 % of the body weight in a healthy individual. Given that these cells can be obtained via minimally invasive muscle biopsy, skeletal muscle may be an attractive and promising source of MSCs for use in regenerative medicine.
Skeletal muscle fibres can be classified as slow twitch type I and fast twitch type II fibres [11]. These fibres are distinguished by their production of ATP, number of mitochondria, and capillary density [11–13]. Muscles containing predominantly type I fibres (e.g. soleus [SO]) are called “slow” or “red”, whereas those containing predominantly type II fibres (tibialis anterior [TA]) are called “fast” or “white”. It has been reported that satellite cells, precursors to skeletal muscle cells, originating from slow and fast muscle are intrinsically different [14]. However, to date, whether differences exist in MSCs originating from slow and fast muscle has not been determined. The two commonly studied prototypic slow and fast muscles are TA and SO, respectively. In this study, we isolated rat MSCs from the TA and SO and performed an animal-matched quantitative comparison of their properties for yield, proliferation, and multi-potentiality.
Materials and methods
Isolation and culture of rat muscle-derived MSCs
Twelve-week-old male Sprague–Dawley rats (CLEA Japan, Tokyo, Japan) were used in this study. All animal procedures were performed under the approval and guidance of the Animal Care and Use Committee of Kobe University Graduate School of Medicine. The whole TA and SO muscles were harvested from one hind limb of the same rats (n = 21). Harvested TA and SO were weighed, and the muscle was minced and digested for two hours at 37 °C with 0.2 % collagenase type II (Worthington Biochemical Corp, Lakewood, NJ, USA). Isolated cells were plated in growth medium consisting of Dulbecco’s modified Eagle’s medium (DMEM)-high glucose (Sigma, St. Louis, MO, USA) supplemented with 10 % fetal bovine serum (FBS; Sigma) at a density of approximately 2 × 104 cells/cm² and incubated. After two hours, the cells were washed extensively with phosphate-buffered saline (PBS; Wako, Osaka, Japan) to remove any cell that did not adhere to the cell culture plastic [10]. The adherent cells were then cultured in growth medium. When subconfluent, the cells were trypsinized and passaged.
Colony-forming unit fibroblast (CFU-F) assays
To establish the frequency of the adherent cells in TA and SO muscles, the CFU-F assay was performed by plating cell suspensions in six-well plates (n = 5) [15]. Mononuclear cells (MNCs) (1 × 104) from the TA and SO of five rats were plated and cultured in a growth medium. After two hours, the cells were washed with PBS as described above. The adherent cells were then maintained in a growth medium. After seven days, the culture plates were stained with Giemsa solution (Muto Pure Chemicals, Tokyo, Japan). Colonies consisting of more than 50 cells were counted.
Growth kinetics
When cells at passage 0 reached subconfluence, cells were replated at 1 × 103 cells/cm². After 14 days, cells at passage 1 were passaged again. To examine long-term growth kinetics, the cells were passaged at 1 × 103 cells/cm² and counted with a hemocytometer every seven days until reaching passage 11 (n = 5).
Flow cytometry analysis
The surface antigen profiles of MSCs derived from the TA and SO at passage 2 or 3 were characterized by flow cytometry (n = 6). A total of 5 × 105 cells were incubated with the following phycoerythrin (PE)-conjugated anti-mouse antibodies: CD11b and CD29 (eBioscience, San Diego, CA, USA), CD44 and CD90 (BD Pharmingen, San Diego, CA, USA), and CD34 and CD45 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 30 minutes at 4 °C in the dark. Nonspecific PE-conjugated IgG was substituted as an isotype control. The fluorescence intensity of the cells was evaluated using a FACScalibur instrument (BD Biosciences, San Jose, CA, USA), and data were analysed using CellQuest Pro (BD Biosciences).
Multi-lineage differentiation
To induce osteogenic differentiation, cells were cultured in osteogenic medium composed of α-modified minimum essential medium (Sigma), 10 % FBS, 10 nM dexamethasone (Sigma), 50 μg/ml L-ascorbic acid-phosphate (Sigma), and 10 mM β-glycerophosphate (Sigma) [15]. After four weeks, osteogenic differentiation was evaluated by staining deposited calcium with 1 % Alizarin Red S (Hartman Leddon, Philadelphia, PA, USA). The expression of osteoblast-related genes—Runx2, alkaline phosphate (ALP), and osteopontin (OPN)—was also measured by real-time polymerase chain reaction (PCR).
To induce chondrogenic differentiation, three-dimensional pellet culture was performed. In a 15 ml polypropylene tube, 2.5 × 105 cells were pelleted by centrifugation. The cells were then cultured in chondrogenic medium consisting of DMEM-high glucose with 100 nM dexamethasone, 50 μg/ml L-ascorbic acid, 0.4 mM proline (Sigma), 1 % ITS + Premix (BD Biosciences), 10 ng/ml recombinant human transforming growth factor-β3 (TGF-β3; R & D Systems, Minneapolis, MN, USA), and 500 ng/ml recombinant human bone morphogenetic protein-6 (BMP-6; R & D Systems) [15]. For histological analysis, the pellets were embedded in paraffin and sectioned. After 21 d, chondrogenic differentiation was assessed by staining with Safranin-O (Chroma, Münster, Germany). The expression of chondrocyte-related genes—aggrecan, type II collagen (Col II), and Sox9—was also measured by real-time PCR.
To induce adipogenic differentiation, cells were cultured using a Cultrex adipogenic differentiation kit (Trevigen, Gaithersburg, MD). After 14 days, adipogenic differentiation was evaluated by the cellular accumulation of neutral lipid vacuoles that were stained with Oil-red O (Trevigen). The expression of adipocyte-related genes: lipoprotein lipase (LPL) and PPARγ2 was also measured by real-time PCR.
Real-time PCR analysis
Total RNA was extracted from each culture at days seven, 14, and 21 for osteogenic (n = 7), adipogenic (n = 4), and chondrogenic (n = 6) induction, respectively, using an RNeasy Mini Kit (Qiagen, Valencia, CA, USA). Undifferentiated TA-derived MSCs were used as a d-0 control. Total RNA was reverse transcribed into single-stranded cDNA using a high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA, USA). Real-time PCR was performed using an ABI PRISM 7700 Sequence Detection System and SYBR Green reagents (both from Applied Biosystems) following the recommended protocols. All primer sequences are shown in Table 1. Expression levels of all genes were normalized to GAPDH and expressed relative to the d-0 control (ΔΔCT methods; Applied Biosystems).
Analysis of capillary density in TA and SO
It was reported that MSC frequency in adipose associated blood vessels densities [16]. To elucidate the association between the CFU-F frequencies and blood vessel densities, we assessed cross-sectional capillary density in samples from the TA and SO. Samples were paraffin-embedded, sectioned, and immunohistochemically stained with fluorescein-labeled isolectin B4 (Vector Laboratories, Burlingame, CA, USA), an endothelial cell marker (n = 5). Nuclear staining was performed with 4′,6-diamidino-2-phenylindole solution (Nacalai Tesque, Kyoto, Japan). Capillaries were morphometrically examined under a fluorescent microscope, counted in five randomly selected fields, and averaged. The morphometric study was performed by two blinded examiners.
Statistical analysis
All the quantitative data were presented as the mean and standard error (SE). To assess differences, the Wilcoxon signed-rank test was used. The level of statistical significance was set at p <0.05.
Results
Cultured cell morphology
One gram of fresh TA and SO muscle yielded 1.2 ± 0.4 and 3.5 ± 1.8 million MNCs, respectively (Table 2). In the primary culture, the cells that were adherent within two hours gave rise to colonies which became visible between days three and five as shown by cells exhibiting a fibroblast-like spindle shape. These colonies resembled CFU-Fs observed in BM-derived MSCs (Fig. 1a and b). After two weeks, the cells merged and formed a subconfluent monolayer of fibroblastoid cells (Fig. 1c and d). No obvious differences in morphology were noted between adherent cells (hereafter called MSCs) derived from the TA and SO.

Phase contrast images of MSCs derived from the tibialis anterior (TA) (a) and soleus (SO) (b), developing CFU-F between days 3 and 5, and images of subconfluent fibroblastic cell monolayers of MSCs derived from the TA (c) and SO (d) at passage 0. Scale bar =400 μm
CFU-F assay
The CFU-F assay revealed that the colony number per 104 MNCs was 48.1 ± 9.8 and 94.3 ± 19.6 from the TA and SO, respectively (Table 2). The colony number per volume (mg) was 4.4 ± 0.6 and 24.6 ± 10.5 from the TA and SO, respectively (Table 2). Both colony number per 104 MNCs and colony number per volume of the SO were significantly higher than those of the TA (p <0.05).
Growth kinetics
The proliferation ability was retained even at passage 11 in both TA and SO MSCs (Fig. 2). The accumulated number of cells from the TA was significantly higher than the number of cells from the SO at days 70 (passage 9), 77 (passage 10), and 84 (passage 11) (p <0.05).

Growth kinetics of MSCs derived from the TA and SO. Starting from passage 1, each population of cells was replated at 1 × 103 cells/cm² every 7 days (n = 5)
Epitope profile
Flow cytometry analysis revealed that both TA and SO MSCs were strongly positive for MSC-related surface antigens such as CD29, CD44, and CD90 (Table 3). Both TA and SO MSCs were negative for hematopoietic-lineage markers such as CD11b, CD34, and CD45. The cell-surface antigen profiles of MSCs derived from the TA and SO were similar.
Multi-lineage differentiation potential
Osteogenic differentiation of both TA- and SO-derived MSCs was confirmed after a 28-day osteogenic induction by Alizarin Red S staining, which showed a mineralized matrix of rich calcium deposits (data not shown). After a 21-day chondrogenic induction, both types of MSC pellets had a similar spherical and glistening transparent appearance. Chondrogenic differentiation of both MSCs was confirmed by Safranin-O staining, which showed the presence of a proteoglycan-rich extracellular matrix in both types of pellets (data not shown). Finally, adipogenic differentiation of both types of MSCs was confirmed after a 14-day adipogenic induction by the accumulation of neutral lipid vacuoles indicated by Oil-red O staining (data not shown).
Osteogenic differentiation capacity
Osteogenic differentiation capacity of both TA- and SO-derived MSCs after a 21-day induction was further confirmed by real-time PCR (Fig. 3a). TA-derived MSCs showed significantly higher expression of Runx2 than SO-derived MSCs on days 14 and 21 and higher expression of ALP and OPN on day 14 (p <0.05).

Quantitative real-time PCR analysis of MSCs derived from the TA and SO after osteogenic (a) chondrogenic (b) and adipogenic (c) induction. The mRNA levels are normalized to GAPDH, expressed relative to those of the day 0 controls from TA-derived MSCs (value set at 1 for each marker), and presented as a fold change (*p <0.05 in the indicated group)
Chondrogenic differentiation capacity
The expression levels of aggrecan, Col II, and Sox9 in both types of MSCs were up-regulated by 21-day chondrogenic induction (Fig. 3b). There were no significant differences observed in the expression levels of aggrecan, Col II, and Sox9 on day 21.
Adipogenic differentiation capacity
Adipogenic differentiation capacity of both TA- and SO-derived MSCs after 14-day induction was further confirmed by the observed up-regulation of LPL and PPARγ2 expression (Fig. 3c). There were no significant differences observed in the expression levels of LPL and PPARγ2 on day 14.
Capillary density in SO and TA muscles
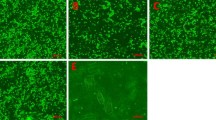
Vascular staining with isolectin B4 showed that the number of capillary vessels surrounding a muscle fiber was higher in the SO than in the TA (Fig. 4a). Capillary density (capillary number per muscle fibre) of the SO was significantly higher than that of the TA (p <0.05; Fig. 4b).

Vascular staining of the TA and SO muscle with isolectin B4. a Representative images of the TA and SO. Capillaries are indicated in green. Scale bar =100 μm. b Capillary density is expressed as the average number of capillaries per muscle fiber (n = 5; *p <0.05 within the indicated group)
Discussion
This is the first study investigating the differences in MSCs originating from fast (TA) and slow (SO) rat muscles from the same animal by analysing their potential for yield, proliferation, and multi-lineage differentiation in vitro. The CFU-F assay revealed that the SO contains a significantly greater number of MSCs than the TA. When cultured, the self-renewal capacity of MSCs derived from the TA was significantly higher at later passages (passage 9–11). Both types of MSCs exhibited similar cell surface antigens to BM-derived MSCs. We found that TA-derived MSCs were superior in terms of osteogenic differentiation potential, but there were no significant differences in chondrogenic and adipogenic differentiation potential. These results provide important information for selecting the optimal skeletal muscle type as a source of MSCs for regenerative medicine.
The physiological characteristics of skeletal muscle are determined by the proportion of fibre types. Slow muscle consists mostly of slow twitch fibres, whereas fast muscle mainly consists of fast twitch fibres [11, 12]. Slow twitch fibres express slow myosin ATPase activity and oxidative metabolism, present high fatigue resistance, and are implicated in postural maintenance and endurance. In contrast, fast twitch fibres express fast myosin ATPase activity and glycolytic metabolism, present low fatigue resistance, and participate in fast corporal movement. It has been shown that the number of capillary vessels surrounding a muscle fibre is higher for slow than for fast fibres [11, 13], which is consistent with our results (Fig. 4). This evidence supports our finding that MSCs originating from different muscle types are intrinsically different.
Cells isolated from the TA and SO exhibited typical MSC characteristics [17]: a fibroblastoid morphology, formation of CFU-F, high self-renewal capacity, expression of a typical set of surface proteins, and multi-lineage differentiation capacity. In skeletal muscle, several distinct mesenchymal stem/progenitor cell populations have been identified [18]. Satellite cells are quiescent myoblast precursors that lie adjacent to the myofibres beneath the basal lamina [19]. Although most satellite cells are committed only to the myogenic lineage, the small subset of satellite cells that co-express endothelial markers, myoendothelial cells, are capable of multi-lineage differentiation [20]. Other distinct populations of mesenchymal stem/progenitor cells have been identified using different isolation techniques [6–10]. For instance, Levy et al. isolated osteoprogenitor cells by separating cells from human skeletal muscle connective tissues and satellite cell fractions [6]. In the present study, we eliminated the contaminating population of satellite cells by selecting the most adherent cells two hours after initial plating on uncoated flasks.
Pericytes are cells that are intimately associated with capillaries and microvessels [21]. They are embedded within the basement membrane of microvessels and possess the potential to give rise to cells of multiple lineages [18]. It has been reported that there is a close relationship between MSCs and pericytes [6, 8, 22–24]. Crisan et al. isolated pericytes from skeletal muscle and found that MSCs and pericytes are close in proximity [23]. Hence, there is a possibility that the cells that we isolated from the TA and SO were indeed pericytes. This hypothesis would explain the higher frequency of MSCs in the SO than in the TA in the present study, as the capillary density of the SO was higher than that of the TA (Fig. 4b). da Silva Meirelles et al. demonstrated that adipose tissue-derived MSC frequency correlated directly with blood vessel densities [16]. However, further studies are required to clarify the direct relationship between muscle-derived MSCs and pericytes.
Although both types of MSCs were able to differentiate into osteo-, chondro- and adipo-lineage cells, the osteogenic differentiation ability of TA-derived MSCs was superior to that of SO-derived MSCs. In order to minimize the invasiveness of clinical interventions for regenerative medicine, it is necessary to be able to harvest a large quantity of MSCs with high differentiation potential from a minimal amount of tissue. The greater expandability and osteogenic differentiation ability of TA-derived MSCs suggests that fast muscle may be a better source for generating large numbers of MSCs for bone regeneration.
A potential limitation of our study was that all of the experiments were performed in vitro. In future studies, we will investigate whether both types of MSCs will exhibit the same regenerative functions in vivo. Another potential limitation is the use of cells from rat. Although we believe the transferability of our results from rat to the human, further studies are needed to determine whether differences exist in the properties between MSC-derived from fast and slow muscles in human.
References
Pećina M, Vukičević S (2014) Tissue engineering and regenerative orthopaedics (TERO). Int Orthop 38:1757–1760
Ivkovic A, Marijanovic I, Hudetz D, Porter RM, Pecina M, Evans CH (2011) Regenerative medicine and tissue engineering in orthopaedic surgery. Front Biosci 3:923–944
Wang X, Wang Y, Gou W, Lu Q, Peng J, Lu S (2013) Role of mesenchymal stem cells in bone regeneration and fracture repair: a review. Int Orthop 37:2491–2498
Marmotti A, de Girolamo L, Bonasia DE, Bruzzone M, Mattia S, Rossi R, Montaruli A, Dettoni F, Castoldi F, Peretti G (2014) Bone marrow derived stem cells in joint and bone diseases: a concise review. Int Orthop 38:1787–1801
Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S, Marshak DR (1999) Multilineage potential of adult human mesenchymal stem cells. Science 284:143–147
Levy MM, Joyner CJ, Virdi AS, Reed A, Triffitt JT, Simpson AH, Kenwright J, Stein H, Francis MJ (2001) Osteoprogenitor cells of mature human skeletal muscle tissue: an in vitro study. Bone 29:317–322
Qu-Petersen Z, Deasy B, Jankowski R, Ikezawa M, Cummins J, Pruchnic R, Mytinger J, Cao B, Gates C, Wernig A, Huard J (2002) Identification of a novel population of muscle stem cells in mice: potential for muscle regeneration. J Cell Biol 157:851–864
Mastrogiacomo M, Derubeis AR, Cancedda R (2005) Bone and cartilage formation by skeletal muscle derived cells. J Cell Physiol 204:594–603
Yoshimura H, Muneta T, Nimura A, Yokoyama A, Koga H, Sekiya I (2007) Comparison of rat mesenchymal stem cells derived from bone marrow, synovium, periosteum, adipose tissue, and muscle. Cell Tissue Res 327:449–462
Nesti LJ, Jackson WM, Shanti RM, Koehler SM, Aragon AB, Bailey JR, Sracic MK, Freedman BA, Giuliani JR, Tuan RS (2008) Differentiation potential of multipotent progenitor cells derived from war-traumatized muscle tissue. J Bone Joint Surg Am 90:2390–2398
Schiaffino S, Reggiani C (2011) Fiber types in mammalian skeletal muscles. Physiol Rev 91:1447–1531
Ennion S, Sant’ana Pereira J, Sargeant AJ, Young A, Goldspink G (1995) Characterization of human skeletal muscle fibres according to the myosin heavy chains they express. J Muscle Res Cell Motil 16:35–43
Hudlická O (1982) Growth of capillaries in skeletal and cardiac muscle. Circ Res 50:451–461
Manzano R, Toivonen JM, Calvo AC, Miana-Mena FJ, Zaragoza P, Muñoz MJ, Montarras D, Osta R (2011) Sex, fiber-type, and age dependent in vitro proliferation of mouse muscle satellite cells. J Cell Biochem 112:2825–2836
Lee SY, Miwa M, Sakai Y, Kuroda R, Matsumoto T, Iwakura T, Fujioka H, Doita M, Kurosaka M (2007) In vitro multipotentiality and characterization of human unfractured traumatic hemarthrosis-derived progenitor cells: a potential cell source for tissue repair. J Cell Physiol 210:561–566
da Silva ML, Sand TT, Harman RJ, Lennon DP, Caplan AI (2009) MSC frequency correlates with blood vessel density in equine adipose tissue. Tissue Eng Part A 15:221–229
Horwitz EM, Le Blanc K, Dominici M, Mueller I, Slaper-Cortenbach I, Marini FC, Deans RJ, Krause DS, Keating A (2005) Clarification of the nomenclature for MSC: The International Society for Cellular Therapy position statement. Cytotherapy 7:393–395
Jackson WM, Nesti LJ, Tuan RS (2010) Potential therapeutic applications of muscle-derived mesenchymal stem and progenitor cells. Expert Opin Biol Ther 10:505–517
Seale P, Sabourin LA, Girgis-Gabardo A, Mansouri A, Gruss P, Rudnicki MA (2000) Pax7 is required for the specification of myogenic satellite cells. Cell 102:777–786
Li G, Zheng B, Meszaros LB, Vella JB, Usas A, Matsumoto T, Huard J (2011) Identification and characterization of chondrogenic progenitor cells in the fascia of postnatal skeletal muscle. J Mol Cell Biol 3:369–377
Sims DE (1986) The pericyte—a review. Tissue Cell 18:153–174
da Silva ML, Caplan AI, Nardi NB (2008) In search of the in vivo identity of mesenchymal stem cells. Stem Cells 26:2287–2299
Crisan M, Yap S, Casteilla L, Chen CW, Corselli M, Park TS, Andriolo G, Sun B, Zheng B, Zhang L, Norotte C, Teng PN, Traas J, Schugar R, Deasy BM, Badylak S, Buhring HJ, Giacobino JP, Lazzari L, Huard J, Péault B (2008) A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell 3:301–313
Mravic M, Asatrian G, Soo C, Lugassy C, Barnhill RL, Dry SM, Peault B, James AW (2014) From pericytes to perivascular tumours: correlation between pathology, stem cell biology, and tissue engineering. Int Orthop 38:1819–1824
Acknowledgments
The authors wish to express sincere thanks to Ms. Kyoko Tanaka, Ms. Maya Yasuda, and Ms. Minako Nagata (Department of Orthopaedic Surgery, Kobe University Graduate School of Medicine) for their technical assistance.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Okumachi, E., Lee, S.Y., Niikura, T. et al. Comparative analysis of rat mesenchymal stem cells derived from slow and fast skeletal muscle in vitro . International Orthopaedics (SICOT) 39, 569–576 (2015). https://doi.org/10.1007/s00264-014-2569-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00264-014-2569-6