
Abstract
Cancer vaccines that utilize patient antigen-presenting cells to fight their own tumors have shown exciting promise in many preclinical studies, but have proven quite challenging to translate to clinical feasibility. Dendritic cells have typically been the cell of choice for such vaccine platforms, due to their ability to endocytose antigens nonspecifically, and their expression of multiple surface molecules that enhance antigen presentation. However, dendritic cells are present in low numbers in human peripheral blood and must be matured in culture before use in vaccines. Mature B lymphocytes, in contrast, are relatively abundant in peripheral blood, and can be quickly activated and expanded in overnight cultures. We devised an optimal stimulation cocktail that engages the B cell antigen receptor, CD40, TLR4 and TLR7, to activate B cells to present antigens from lysates of the recipient’s tumor cells, precluding the need for known tumor antigens. This B cell vaccine (Bvac) improved overall survival from B16F1 melanoma challenge, as well as reduced tumor size and increased time to tumor appearance. Bvac upregulated B cell antigen presentation molecules, stimulated activation of both CD4+ and CD8+ T cells, and induced T cell migration. Bvac provides an alternative cellular vaccine strategy that has considerable practical advantages for translation to clinical settings.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Immunotherapies for cancer treatment have shown exciting progress during the past decade, offering multiple approaches for harnessing the power of the immune system to combat numerous cancer types. Because each type of therapy has both strengths and weaknesses, particularly for distinct tumor types, cancer patients will be best served by the availability of a variety of therapeutic strategies for engaging immune cells to fight tumors. Thus, there remains a need for continuing development of diverse immunotherapies.
The therapeutic approach of cancer vaccines has a long history of exciting successes, but also disappointments in clinical translation. These vaccines are based upon various platforms [1], including nucleic acids [2], soluble molecules [3], and immune cells themselves [4]. The principle underlying the latter is to stimulate the cancer patient’s own immune cells to recognize and eliminate their tumor cells. This stimulation can occur within the patient, or by harvesting patient immune cells, stimulating them in vitro, and re-introducing them. Dendritic cells (DC) are well-appreciated for their ability to nonspecifically ingest and process antigen (Ag) and present it effectively in a cognate manner to activate effector T lymphocytes. DC have therefore been the natural focus of many studies of anti-tumor cellular vaccination and have shown great promise in preclinical models [4].
However, there remain significant challenges in adapting DC-based cellular vaccines from animal models to human clinical use. In the many models using inbred strains of laboratory mice, a virtually unlimited number of mice genetically identical to the vaccine recipient can be used as a source of DC, but in humans, DC must be autologous, are relatively rare in peripheral blood [5], and must be cultured in vitro to allow their differentiation into mature DC. Consequently, limitation in DC cell numbers is a major challenge for human DC vaccine delivery. Further, many preclinical mouse studies used well-established tumor models expressing highly immunogenic tumor Ags known to stimulate T cell responses, but this advantage is seldom present in the much more heterogeneous profile of individual human tumors. These considerable logistical challenges may explain why, after many years and a large number of published preclinical studies with positive outcomes, there is to date only a single FDA-approved cellular vaccination therapy for human cancer, the Provenge DC vaccine for prostate cancer. In clinical trials, Provenge demonstrated statistically improved survival of treated patients compared to controls, but by a modest four months, and tumor size was unaffected by treatment [https://www.fda.gov/media/78511/].
We thus considered whether B lymphocytes could effectively serve as a source of antigen-presenting cells (APC) in cellular anti-tumor vaccines. B cells offer several attractive features as APC vaccines from a logistical perspective [6]. B cells are relatively numerous in human peripheral blood [7], from which they are easily isolated. Although resting/naïve B cells do not have the high capacity for nonspecific Ag ingestion characteristic of DC, B cells serve quite effectively as T cell-stimulatory APC in a variety of immune responses when appropriately activated [8,9,10,11]. B cells can activate both CD4+ and CD8+ T cells and induce T cell memory. Importantly, B cells can stimulate anti-tumor T cells in preclinical models [12].
Our laboratory previously demonstrated that B cell vaccines stimulate both CD4+ and CD8+ T cell-mediated immune responses in mice by efficiently presenting foreign antigen, resulting in control of Listeria monocytogenes liver infections. Importantly, these proof-of-concept studies showed that B cell-based vaccines are comparable to DC-based vaccines in efficacy [13]. Our major goal is to exploit the advantages offered by B cells in cellular vaccine design. Consistent with our aim of improving the practical utility of cellular vaccines, we also wished to determine if B cell vaccines can be produced using recipient tumor cells as a source of Ag, rather than known, purified Ag, to further enhance their utility in a clinical setting.
To pursue these goals in the present study, we tested the prediction that appropriately stimulated B cells could provide an effective cellular vaccine (Bvac) against a well-characterized, aggressive mouse tumor (B16F1 melanoma), using tumor cell lysates as priming Ag. Because cellular vaccines are most likely to be efficacious in minimal residual disease, rather than eliminating a large tumor burden, we provided Bvac immediately prior to tumor injection. We found that mice treated with optimally activated Bvac and B16F1 tumor lysate had significantly improved survival, increased time to detectable tumor development, and smaller tumors, when subsequently challenged with B16F1. Bvac expressed a strong APC profile, stimulated both CD4+ and CD8+ T cell activation responses, and induced T cell migration in vitro. These results have promising implications for harnessing easily obtained and activated patient B cells to fight tumors.
Materials and methods
Preparation of B16F1 tumor cell lysate
B16F1 mouse melanoma cells [14] were provided by Dr. George Weiner, University of Iowa, Iowa City, IA. Adherent B16F1 cells were grown to confluency and disassociated using 2 mM EDTA in PBS. Cells were washed twice and suspended in 1 mL PBS, then transferred to cryo-vials. Cells were pelleted and resuspended in 100µL PBS. The cryo-vial was placed in a liquid nitrogen bath for 5 min, then thawed in a 37°C water bath; this cycle was repeated five times. After the final thaw, each lysate was sonicated to shear DNA and centrifuged at 4°C at 16,000 × g to pellet the insoluble fraction. The supernatant was harvested as tumor lysate, and its protein concentration determined via O.D. at 280 nm.
Mice
Six- to 8-week-old female C57BL/6 mice were purchased from Jackson Laboratories (Bar Harbor, ME) and housed in a pathogen-free barrier facility at The University of Iowa. Mice in this study were used in accordance with a protocol approved by the Iowa City VA Healthcare System’s Institutional Animal Care and Use Committee. Female mice were used because male mice had a tendency to chew subcutaneous tumors, leading to infections.
B cell isolation and Bvac preparation
Highly purified resting B cells were isolated from mouse splenocytes via EasySep magnetic negative selection kits (STEMCELL Technologies; Vancouver, BC, Canada). Purified B cells were suspended in RPMI 1640 medium with 10% heat-inactivated fetal calf serum, 2 mM fresh glutamine, 10−5 M 2-mercaptoethanol, and antibiotics (BCM-10) at 2 × 106 cells/mL and were stimulated with the following reagent combination: 0.5 µg/mL anti-mouse CD40 mAb (clone HM40, Ebioscience, Carlsbad, CA), 1 µg/mL R848 (TLR7 agonist; Enzo Scientific, Farmingdale, NY), 20 ng/mL E. coli LPS (TLR4 agonist; Sigma-Aldrich, St. Louis, MO). After 22 h of stimulation, either B16F1 tumor lysate (1 µg/mL; preparation described above) or a cocktail of two known melanoma peptides [15], Mage-AX (LGITYDGM) and Mage-A5 (HNTQYCNL) (3.33 µM, Selleckchem, Houston, TX) was added to the B cell cultures for an additional two hours. After 24 h of culture, B cells were washed two times with 1X PBS and prepared for injection. B cells prepared in this manner are hereafter referred to as Bvac.
Bvac administration and melanoma challenge
Bvac or control B cells (2 × 105) were washed and resuspended in PBS, then adoptively transferred via retro-orbital injection into naïve C57BL/6 mice. A vaccine boost of identical composition was performed 14 days after initial vaccination. Forty days after the second vaccination both B cell control and Bvac immunized mice were challenged with 25 × 104 B16F1 mouse melanoma cells in the side flank. Tumor growth was measured with digital calipers every other day, starting when tumor growth was established, until critical tumor burden (150 mm2) was reached, at which point the mice were euthanized.
Flow cytometry
Expression of various molecules described in the text was analyzed via flow cytometry as previously described [16]. T cells from activation assays were collected on an LSR II BD Biosciences flow cytometer, (Franklin Lakes, NJ). Cells from migration and expression assays were collected on an Accuri C6 benchtop flow cytometer (BD Biosciences, San Jose, CA). All Abs were specific for mouse molecules. Abs to CD80 (clone 1G10), CD86 (clone GL1), MHC class I (clone AF6-88.5.5.3), and MHC class II (clone M5/114) were purchased from Ebioscience (San Diego, CA). Abs specific for Integrin β2, l-Selectin, CXCR4, CCR7, and CXCR5 were obtained from R&D Systems (Minneapolis, MN). Abs specific for CD8a (clone 53-6.7), CD4 (clone GK1.5), CD49d (clone 9F10), CD11a (M17/4), CD90.2 (clone 30-H12), CD25 APC (clone PC61.5), and CD44 (clone BJ18) were purchased from Biolegend (San Diego, CA). Anti-CD45 (clone 30-F11), CD19 (clone 1D3), and anti-CD69 (clone FN50) mAbs were purchased from Ebioscience. Data were analyzed with FlowJo software (Tree Star, Ashland, OR).
Chemotaxis
To assess Bvac migration [17], 1 × 106 Bvac were placed into the upper chamber of 3-μm pore 12 well Transwell plates (Costar, Cambridge, MA). The chemokines CCL19 and CCL21 (R&D Systems), at a 10 ng/ml final concentration of each of diluted in 1 ml BCM10, or undiluted Bvac-conditioned medium (for T cell migration), were added to the lower chamber. Migration of Bvac was assessed after 3 h by counting Bvac in the lower chamber, on full speed for 1 min (Accuri A6) to tally detected events. T cell migration was assessed as above, in response to CCL19 and CCL21 or to Bvac-conditioned medium placed in lower chambers of transwell plates.
T cell activation
Bvac-tumor lysate were prepared as above. Highly purified CD4+ and CD8+ T cells were isolated from mouse splenocytes via EasySep magnetic negative selection (STEMCELL Technologies). 1 × 106 Bvac and 1 × 106 naïve CD4+ or CD8+ T cells were placed into a 48 well tissue culture plate in a 1:1 ratio. Alternatively, 1 × 106 Bvac were combined with both 5 × 105 CD4+ and 5 × 105 CD8+ T cells to better model more diverse cell populations present in a secondary lymphoid structure, maintaining the 1:1 B and T cell ratio. T cells were tested for activation after either 24 h or 5 days of co-culture. T cell populations were removed from cell culture, stained for surface antigens of interest with immunofluorescent mAbs, and analyzed by flow cytometry as previously described [16]. All co-cultured populations were stained with anti-CD19 mAb to enable exclusion of B cells from T cell analysis. Unstimulated B cells were used as a control for Bvac.
Expansion of B and T cell compartments after Bvac vaccination
Bvac-tumor lysate were prepared as noted in Bvac preparation above. Each cohort of nine mice was divided into the following groups for analysis: (1) three naive, (2) three Bvac-tumor lysate vaccinated and analyzed on Day 7 of treatment and (3) three Bvac-tumor lysate vaccinated and analyzed on Day 14 post vaccination. Spleen and the following lymph nodes were harvested from each mouse: superficial cervical, brachial, axillary, and inguinal, (lymph nodes were pooled to ensure adequate numbers for analysis). Cells extracted from tissues were stained with CD19, CD4, and CD8 specific-immunofluorescent mAbs, and cell proportions were determined via flow cytometry, as described above.
Results
Impact of Bvac immunization upon survival and tumor growth
We chose the aggressive B16F1 mouse melanoma tumor model to provide a rigorous test to assess the ability of Bvac to provide anti-tumor immunity. Because we predict that tumor vaccination holds the greatest promise as a treatment to eliminate minimal residual disease, rather than removing bulky or metastatic primary tumors, we challenged with tumor shortly after provision of Bvac. Two tumor Ag delivery platforms were tested. Bvac were exposed to either B16F1 tumor lysate produced as described in Methods, or to two purified B16F1 melanoma peptides, MAGE AX and MAGE A5 [15]. We tested both sources of Ag, because purified known tumor Ags (as used more commonly in preclinical experiments) are available for only a small proportion of human cancers, whereas tumor lysates can be derived from biopsy samples for a much larger group of tumors. Two separate cohorts were assessed, for a total of 10 naïve, 15 Bvac/tumor lysate-vaccinated and 10 Bvac/peptide-vaccinated mice. Mice were vaccinated twice, 14 days apart, and 40 days later challenged with subcutaneous injection of B16F1 cells as described in Methods. Unstimulated, naïve B cells were used as controls for Bvac. While many previous reports have performed vaccination with antigen preparations alone, our focus here was the efficacy of B cells as cellular vaccines, so mice injected only with B16F1 lysate or melanoma peptides were not assessed. Previous studies demonstrated that vaccination with B cells that received the activating stimuli without tumor antigens did not result in effective protection from B16F1 challenge [18].
Analysis of survival data revealed that mice vaccinated with Bvac-tumor lysate had improved survival (32 days), compared to naïve mice (23 days); this difference is statistically significant (Gehan–Breslow–Wilcoxon test) (Fig. 1a). Enhanced survival of mice receiving Bvac-peptide as a source of Ag (Fig. 1b) was not as striking as that of recipients of Bvac-tumor lysate, but was statistically significant, with an improved survival of 28 days. These results suggest that tumor cell lysate as a source of Ag offers a better stimulus for Bvac than does a more limited mixture of highly purified tumor-derived peptides (Fig. 1c). Analysis revealed that the mean time of first detectable tumor was 21.43 days for Bvac-lysate whereas in naïve mice the tumors first appeared on mean day 15.6. These results were statistically significant. Bvac-peptide-treated mice had a mean tumor appearance of 18.25 days. Finally, overall tumor burden was lower in both Bvac-immunized groups compared to naïve mice on day 20 post tumor challenge (Fig. 1d), the day the first animal was removed from the study as it exceeded the critical tumor burden criterion, and this parameter was statistically significant for both Bvac Ag groups. Because Bvac-lysate was superior to Bvac-peptide in inducing tumor protection, as well as the practical translational advantage of tumor lysate as an Ag source, Bvac-lysate was used as a source of Ag in all subsequent experiments; the term “Bvac” in these experiments refers to Bvac prepared with tumor cell lysate. Additionally, examination of the relationships between survival time versus tumor onset and survival time versus tumor burden revealed statistically significant correlations. This supports the conclusion that mice survive longer the later the tumor is established, and the smaller tumor burden they develop (Supplementary Table 1).

source of antigen had statistically higher survival (**p = 0.0030) than naïve mice. b Bvac that were stimulated with purified melanoma peptides MAGE AX and MAGE A5 as an antigen source had statistically higher survival (*p = 0.0401) than naïve mice. c Day of tumor onset, defined as the first day that tumor could be palpably measured, is shown for indicated groups of mice (each symbol represents one mouse). Delayed tumor onset was observed in Bvac-lysate (*p ≤ 0.0061) and highly trending in Bvac-peptide groups (ns = p ≤ 0.0600), as analyzed by (two-tailed unpaired t test). d Reduced tumor size was observed in Bvac-lysate (**p ≤ 0.0037) and Bvac-peptide groups (*p ≤ .0124) at day 20 post-tumor challenge, analyzed by (Two-tailed Unpaired t test)
Impact of Bvac upon host survival and tumor burden. a, b Comparison of survival between untreated (gray lines) and Bvac-treated (black lines) mice following challenge with B16F1 melanoma. Differences in survival curves between naïve and Bvac-treated mice were analyzed by the Gehan–Breslow–Wilcoxon test. a Bvac that were stimulated with tumor lysate as a
Expression of lymph node homing and migration molecules by Bvac
Bvac were examined for expression of cell surface Ags indicative of APC homing to secondary lymphoid organs or to sites of inflammation, which can include tumors [19]. Figure 2 demonstrates that Bvac had increased expression of cell surface molecules important in extravasation. CD62L, CD18, and CD11a were all upregulated on Bvac at both day 1 and day 5 post-stimulation, compared to naïve B cells. Bvac express the secondary lymphoid homing molecules CXCR4 and CCR7 [3, 19, 20] on day 5 after stimulation. Additionally, expression of CXCR5, an important molecule that promotes B and T cell interactions in secondary lymphoid structures [21] was noted on Bvac at day 5 of activation. We note that upregulation of these molecules was indistinguishable whether or not tumor lysate was included in the stimulation cocktail (not shown). A previous study of Bvac employed against infection with an intracellular pathogen shows that Bvac successfully migrate to secondary lymphoid organs in vivo as well [13, 18]. Two-tailed Unpaired t test statistical analysis of surface receptor upregulation MFI also revealed an increase in per cell expression of these relevant molecules on Bvac. Even those for which differences did not quite reach statistical significance due to mouse–mouse variability displayed a strong trend of increased expression.

Bvac expression of cell migration molecules. Unstimulated B cells were compared to Bvac for expression of the indicated surface molecules involved in extravasation to, and migration within, secondary lymphoid structures. (D0) = Freshly purified, naive B cells (D1) = 24 h post anti-Bvac stimulation, and (D5) = 120 h post stimulation. MFI indicates mean fluorescence intensity; flow cytometry results are representative of 2 similar experiments. Statistically significant upregulation (Two-tailed Unpaired t test) was observed for CD62L, CD11a, CXCR4, and CCR7 molecules. Trending significance was seen for upregulation of CD18 and CXCR5. Error bars represent mean ± SEM of replicate experiments
Ag presentation phenotype of Bvac
Compared to naive mouse B cells, Bvac stimulated for 24 h with anti-mCD40 mAb and TLR4/7 ligands displayed upregulation of surface molecules characteristic of effective APC (Fig. 3a). While resting mouse B cells express MHC class I molecules, expression is increased in response to Bvac stimulation. MHC class II molecules were expressed on ~ 25% of unstimulated B cells, and this proportion increased to ~ 75% of Bvac. Additionally, the expression of the T cell costimulatory molecules CD80 and CD86 was greatly enhanced on Bvac, from < 1% of cells expressing each of these molecules to ~ 40% positive for both following stimulation. In a separate experiment to access MFI of upregulated surface markers, (Fig. 3b), obvious upregulation with all molecules in Bvac is observed in histogram plots compared to untreated naïve B cells, and Bvac had high MFI significance compared to naïve B cells in expression of MHC I, CD80, and CD86 molecules. Increases in MHC II expression were not statistically significant, although expression was consistently detectably upregulated. Taken together, these data show that Bvac have the potential to present tumor Ags to T cells via MHC molecules, as well as providing costimulation through expression of CD28 ligands.

Bvac expression of surface molecules involved in antigen presentation. Naïve B cells cultured for 24 h in vitro with medium alone (left panels) or the Bvac stimuli described in Methods (right panels) were examined by immunofluorescent staining and flow cytometry for relative expression of the indicated Y-axis molecules; the X-axis represents staining for CD19 (a). MHCI expression was basally measurable on greater than 90% of resting B cells. (b) Statistically significant upregulation (Two-tailed Unpaired t test) was observed for MHCI, CD80 and CD86, as well as trending significance for upregulation of MHC II when MFI was analyzed. Error bars represent mean ± SEM of replicate experiments. Results are representative of 2 similar experiments
Bvac-induced T cell migration
Surface molecule expression data demonstrated that Bvac have the potential to reach environments in which they can interact with other immune cells, particularly T lymphocytes, which are important to many anti-tumor responses [22]. To assess ability of Bvac to induce the migration of naïve CD4+ and/or CD8+ T cells, we plated Bvac-derived culture supernatant, a CCL19/21 chemokine cocktail (positive control) or BCM10 medium (negative control) in the lower chamber of a Transwell plate. Naïve CD4+ and CD8+ T cells were placed in the upper chamber, and after 3 h, T cells that had migrated to the lower chamber were counted. Statistical analysis revealed that CD8+ T cells (Fig. 4a) migrated significantly compared to the negative control, with greater migration induced by Bvac supernatant than by the CCL19/21 cocktail. CD4+ T cell migration (Fig. 4b) was strongly trending toward significance. Additionally, activated Bvac themselves migrated robustly to the CCL19/21 cocktail gradient compared to BCM-10 alone (Fig. 4c). These results indicate the potential of Bvac to migrate toward and induce the migration of naïve T cell populations, enhancing their ability to interact with and present Ag effectively to T cells.

Impact of Bvac on T cell migration in vitro. a, b Freshly isolated naïve CD8+ and CD4+ T cells were exposed to Bvac supernatant, or CCL19/CCL21 chemokine gradients as a positive control. a Analysis (Two-tailed Unpaired t test) showed statistically significant CD8+ T cell migration induced by Bvac, (p ≤ 0.0072) and (p ≤ 0.0091) respectively. b CD4+ T cells also had increased migration, although this was not statistically significant. c Activated Bvac themselves migrated to the CCL19/21-positive control cocktail and (two-tailed unpaired t test) showed a strong statistical difference of (p ≤ 0.0055) compared to their spontaneous migration. Error bars represent mean ± SEM of replicate experiments. Results are representative of 2 similar experiments
Activation of naïve CD4+and CD8+ T cells by Bvac
To determine the potential of Bvac to activate T cells, we co-cultured Bvac with CD4+ and CD8+ T cells in vitro for 24 h. We identified activated CD4+ T cells by the upregulation of surface molecules CD25 and CD49d, and activated CD8+ T cells by the upregulation of CD25 and CD44 [23]. There was a substantial increase in activated CD4+ T cells following co-culture with Bvac for 24 h, compared to CD4+ T cells cultured with control B cells. Co-culture of Bvac with CD8+ T cells resulted in the appearance of a distinct CD25+CD44+ T cell population not detectable in B cell control-T cell co-cultures. These results demonstrate that Bvac has the potential to activate T cell populations that are both instrumental to orchestrating anti-tumor immune responses (CD4+ T cells) and cytotoxic activity against the tumor (CD8+ T cells) (Fig. 5).

Activation phenotype of T cells co-cultured with Bvac. Freshly isolated naïve CD4+ and CD8+ T cells were co-cultured in the presence of naïve B cells or Bvac. T cells and Bvac or naïve B cells were co-cultured in a 1:1 ratio for 24 h. (Upper Panels) CD4+ T cell activation, and (Lower Panels) CD8+ T cell action was defined as the upregulation of CD49d and CD25 for CD4+ T cells and upregulation of CD44 and CD25 by CD8+ T cells. Results are representative of 2 experiments with similar fold-increases in activation
Bvac-dependent expansion of B and T lymphocytes
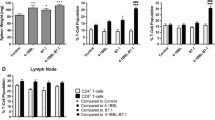
To understand the effect Bvac have on immune cell activation prior to tumor encounter, as could be the case when giving Bvac following elimination of primary tumors by more conventional treatment, we vaccinated mice with Bvac-tumor lysate as described in Methods. Lymph nodes harvested from each mouse were pooled (Fig. 6a). Analysis of these cells revealed significant CD4+ T cell expansion on day 7 following Bvac-tumor lysate treatment and trending CD8+ T cell expansion on day 14 post vaccination in lymph nodes (Fig. 6b). Bvac-tumor lysate treatment also resulted in highly significant CD19+ B cell expansion in the spleen and pooled lymph nodes on day 14 post vaccination. Taken together, these data demonstrate that Bvac expand B cell and T cell compartments after vaccination.

B and T cell expansion in secondary lymphoid structures post-Bvac immunization. a ANOVA analysis of T cells pooled from lymph nodes vaccinated with Bvac-lysate showed statically significant expansion of CD4+ T cells (*p ≤ 0.0185), with trending significance in expansion of CD8+ T cells. b ANOVA analysis of CD19+ B cell expansion in Bvac-lysate immunized mice showed statistically significant expansion in both spleen and pooled lymph node tissue; (**p ≤ 0.0070) and (**p ≤ 0.0087), respectively
Discussion
Various new and refined cancer immunotherapies currently provide treatment options to supplement or replace traditional surgical, chemotherapeutic, and radiological approaches [24,25,26]. Because tumors are derived from self tissue, it has been challenging to identify tumor Ags that the immune system will see as ‘non-self’, when presented in tumor vaccination. Some tumor types are inherently more antigenic, expressing targets not expressed by their normal cell counterparts, but this is unfortunately uncommon [27]. In tumors with high mutation rates, new Ags may arise during tumor growth, but outgrowth of subclones expressing such Ags may be selected against by the oncogenic process. Many preclinical mouse models of cancer use established tumors expressing well-defined and highly immunogenic Ags that can be targeted by cancer vaccines [28], but human tumors rarely have such Ags. For this reason, we chose not to use purified known tumor Ags in our Bvac stimulus; rather, we utilized tumor cell lysates as a polyclonal antigenic stimulus [1]. Lysates were generated by a freeze–thaw method followed by sonication. A similar protocol was used to generate cell lysate that was successfully loaded onto DCs to activate both CD4+ and CD8+ T cells [29]. The amount of lysate required could easily be obtained by a surgical resection of a primary tumor, or even by biopsy procedures, increasing the range of tumor types amenable to this approach. Using patient derived tumor lysate to activate Bvac may be most practical in the circumstance in which Bvac are used to fight minimal residual disease after primary treatment, as mentioned above. Bvac may serve as an immune system activator that breaks tolerance to self-antigens. It is quite encouraging that Bvac activated by tumor cell lysate performed as well or better than Bvac stimulated by purified antigenic peptides of the tumor. This result suggests that the potential breadth of clonal activation from Bvac-lysate is greater than for Bvac-peptide, as tumor lysate offers many potential neo-antigens for immune recognition and activation compared to just two melanoma peptide antigens.
Another key component of successful tumor vaccination is effective antigen presentation. To date, the cell of choice for this role has typically been the DC. DC play many key functions as APC in normal immune responses, due to their tremendous capacity for nonspecific Ag uptake, expression upon activation of numerous surface molecules that stimulate T lymphocytes in Ag presentation, and their secretion of cytokines and chemokines. It is thus not surprising that so many preclinical tumor vaccine studies have focused upon DC as the source of APC [30, 31]. However, B cells are highly efficient at receptor-mediated endocytosis. Activation signals delivered via TLRs, the BCR, and CD40 synergize to induce B cells to become highly effective APC, expressing abundant MHC molecules, adhesion molecules, chemokines and their receptors, and T cell costimulatory molecules such as members of the B7 and TNF receptor families [32, 33]. Activated B cells also secrete lymphokines and chemokines that attract activated T cells [28, 33]. Like activated T cells, B cells and Bvac are capable of secreting interferon-γ, (sometimes not considered in data interpretation), and this cytokinesis positively correlated with extended survival times in multiple cancers [2, 34]. Practical considerations that recommend B cells as APC for clinical applications include their relative abundance in human peripheral blood [7, 27] and the ease with which they can be substantially expanded with just overnight culture as we show here.
Bvac are durable and long lasting in secondary lymphoid organs up to 40 days post vaccination in an infection model [13, 18]. We thus postulated that Bvac may serve as APCs for cancer vaccine immunotherapy. In this study, we observed that mice vaccinated prophylactically with Bvac+B16F1 tumor lysate had significantly better survival following challenge with this highly aggressive model tumor. Both survival and inhibition of tumor growth were observed. This efficacy was associated with multiple characteristics of effective APC including rapid upregulation of APC molecules, as well as phenotypic characteristics promoting successful migration and extravasation to tissues, and receptors promoting attraction and activation of T lymphocytes.
Activation and expansion of the adaptive arms of the immune system are hallmarks of successful vaccination strategies [35], and this remains true for cancer vaccines [22, 36, 37]. Here we show expansion of both T and B cell compartments in secondary lymphoid tissue 7 and 14 days post Bvac immunization. T lymphocyte-driven control of tumor cells is a third desirable component of many successful tumor vaccines. For most tumors, both CD4+ and CD8+ T cell activation is needed to successfully orchestrate an immune response that eliminates the tumor [22, 37]. Many preclinical models use T cells expressing high-affinity transgenic TCRs to recognize tumor antigens. In contrast, in the present study, we deliberately relied upon the more physiologically relevant polyclonal T cell response to a mixture of tumor Ags provided by tumor cell lysate. It is very promising for potential clinical application that we could demonstrate clear activation of both CD4+ and CD8+ T cells with Bvac. Our study shows that Bvac can recruit and activate T cells, as well as upregulating surface molecules involved in effective antigen presentation. Further details of mechanism will be the subject of future investigation, as well as probing the causal relationship between features of Bvac and their tumor-protective ability.
This study documented the protective effects that Bvac provided in mice challenged with the aggressive B16F1 mouse melanoma. Future Bvac studies would benefit from the inclusion of additional cancer types, as mice challenged with slower growing tumors might demonstrate additional benefit from Bvac. The most promising use of cancer vaccines may be in eliminating minimal residual disease, which can lead to relapse and recurrence—currently, an often more challenging therapeutic problem than eliminating a primary tumor. As a secondary therapy, the Bvac platform could be used as an individualized prophylactic vaccine that is patient specific, activating a tailored and durable immune response unique to the specific tumor and syngeneic to the patient.
Availability of data and material
No new biological materials/reagents were generated as part of the reported work. No large data sets were generated. All primary laboratory notebooks are stored securely in our laboratory at The University of Iowa.
Abbreviations
- Ag:
-
Antigen
- APC:
-
Antigen presenting cell
- Bvac:
-
B cell-based cellular vaccine
- DC:
-
Dendritic cell
- mAb:
-
Monoclonal antibody
- MHC:
-
Major histocompatibility complex
- MFI:
-
Mean fluorescence intensity
References
Hollingsworth RE, Jansen K (2019) Turning the corner on therapeutic cancer vaccines. NPJ Vaccines 4:7. https://doi.org/10.1038/s41541-019-0103-y
Lopes A, Vandermeulen G, Preat V (2019) Cancer DNA vaccines: current preclinical and clinical developments and future perspectives. J Exp Clin Cancer Res 38(1):146. https://doi.org/10.1186/s13046-019-1154-7
Fan C, Joshi J, Li F, Xu B, Khan M, Yang J, Zhu W (2020) Nanoparticle-mediated drug delivery for treatment of ischemic heart disease. Front Bioeng Biotechnol 8:687. https://doi.org/10.3389/fbioe.2020.00687
Palucka K, Banchereau J (2013) Dendritic-cell-based therapeutic cancer vaccines. Immunity 39(1):38–48. https://doi.org/10.1016/j.immuni.2013.07.004
Wu C, Liu Y, Zhao Q, Chen G, Chen J, Yan X, Zhou YH, Huang Z (2010) Soluble CD40 ligand-activated human peripheral B cells as surrogated antigen presenting cells: A preliminary approach for anti-HBV immunotherapy. Virol J 7:370. https://doi.org/10.1186/1743-422X-7-370
Rossetti RAM, Lorenzi NPC, Yokochi K, Rosa M, Benevides L, Margarido PFR, Baracat EC, Carvalho JP, Villa LL, Lepique AP (2018) B lymphocytes can be activated to act as antigen presenting cells to promote anti-tumor responses. PLoS ONE 13(7):e0199034. https://doi.org/10.1371/journal.pone.0199034
von Bergwelt-Baildon MS, Vonderheide RH, Maecker B, Hirano N, Anderson KS, Butler MO, Xia Z, Zeng WY, Wucherpfennig KW, Nadler LM, Schultze JL (2002) Human primary and memory cytotoxic T lymphocyte responses are efficiently induced by means of CD40-activated B cells as antigen-presenting cells: potential for clinical application. Blood 99(9):3319–3325. https://doi.org/10.1182/blood.v99.9.3319
Colluru VT, McNeel DG (2016) B lymphocytes as direct antigen-presenting cells for anti-tumor DNA vaccines. Oncotarget 7(42):67901–67918. https://doi.org/10.18632/oncotarget.12178
Chen X, Jensen PE (2008) The role of B lymphocytes as antigen-presenting cells. Arch Immunol Ther Exp (Warsz) 56(2):77–83. https://doi.org/10.1007/s00005-008-0014-5
Kondo E, Gryschok L, Klein-Gonzalez N, Rademàcher S, Weihrauch MR, Liebig T, Shimabukuro-Vornhagen A, Kochanek M, Draube A, von Bergwelt-Baildon MS (2009) CD40-activated B cells can be generated in high number and purity in cancer patients: analysis of immunogenicity and homing potential. Clin Exp Immunol 155(2):249–256. https://doi.org/10.1111/j.1365-2249.2008.03820.x
Kruger S, Ilmer M, Kobold S, Cadilha BL, Endres S, Ormanns S, Schuebbe G, Renz BW, D’Haese JG, Schloesser H, Heinemann V, Subklewe M, Boeck S, Werner J, von Bergwelt-Baildon M (2019) Advances in cancer immunotherapy 2019—latest trends. J Exp Clin Cancer Res 38(1):268. https://doi.org/10.1186/s13046-019-1266-0
Candolfi M, Curtin JF, Yagiz K, Assi H, Wibowo MK, Alzadeh GE, Foulad D, Muhammad AK, Salehi S, Keech N, Puntel M, Liu C, Sanderson NR, Kroeger KM, Dunn R, Martins G, Lowenstein PR, Castro MG (2011) B cells are critical to T-cell-mediated antitumor immunity induced by a combined immune-stimulatory/conditionally cytotoxic therapy for glioblastoma. Neoplasia 13(10):947–960. https://doi.org/10.1593/neo.11024
Vanden Bush TJ, Buchta CM, Claudio J, Bishop GA (2009) Cutting Edge: Importance of IL-6 and cooperation between innate and adaptive immune receptors in cellular vaccination with B lymphocytes. J Immunol 183(8):4833–4837. https://doi.org/10.4049/jimmunol.0900968
Overwijk WW, Restifo NP (2001) B16 as a mouse model for human melanoma. Curr Protoc Immunol. doi:https://doi.org/10.1002/0471142735.im2001s39 (Chapter 20: Unit 20.1)
Eggert AO, Andersen MH, Voigt H, Schrama D, Kampgen E, Straten PT, Becker JC (2004) Characterization of mouse MAGE-derived H-2Kb-restricted CTL epitopes. Eur J Immunol 34(11):3285–3290. https://doi.org/10.1002/eji.200324618
Lin WW, Yi Z, Stunz LL, Maine CJ, Sherman LA, Bishop GA (2015) The adaptor protein TRAF3 inhibits IL-6 receptor signaling in B cells to limit plasma cell development. Sci Signal 8(392):ra88. https://doi.org/10.1126/scisignal.aaa5157
Moratz C, Kehrl JH (2004) In vitro and in vivo assays of B-lymphocyte migration. Methods Mol Biol 271:161–171. https://doi.org/10.1385/1-59259-796-3:161
Buchta CM (2014) Mechanisms of TLR signaling and cooperation in B lymphocytes. Doctoral Dissertation, The University of Iowa, Iowa City, IA, USA
von Bergwelt-Baildon M, Shimabukuro-Vornhagen A, Popov A, Klein-Gonzalez N, Fiore F, Debey S, Draube A, Maecker B, Menezes I, Nadler LM, Schultze JL (2006) CD40-activated B cells express full lymph node homing triad and induce T-cell chemotaxis: potential as cellular adjuvants. Blood 107(7):2786–2789. https://doi.org/10.1182/blood-2004-01-0113
Reif K, Ekland EH, Ohl L, Nakano H, Lipp M, Forster R, Cyster JG (2002) Balanced responsiveness to chemoattractants from adjacent zones determines B-cell position. Nature 416(6876):94–99. https://doi.org/10.1038/416094a
Cyster JG, Allen CDC (2019) B cell responses: cell interaction dynamics and decisions. Cell 177(3):524–540. https://doi.org/10.1016/j.cell.2019.03.016
Bird L (2019) Joint effort needed. Nat Rev Immunol 19(12):717. https://doi.org/10.1038/s41577-019-0242-4
Christiaansen AF, Dixit UG, Coler RN, Marie Beckmann A, Reed SG, Winokur PL, Zimmerman MB, Varga SM, Wilson ME (2017) CD11a and CD49d enhance the detection of antigen-specific T cells following human vaccination. Vaccine 35(33):4255–4261. https://doi.org/10.1016/j.vaccine.2017.06.013
Finn OJ (2018) A believer’s overview of cancer immunosurveillance and immunotherapy. J Immunol 200(2):385–391. https://doi.org/10.4049/jimmunol.1701302
Demaria O, Cornen S, Daeron M, Morel Y, Medzhitov R, Vivier E (2019) Harnessing innate immunity in cancer therapy. Nature 574(7776):45–56. https://doi.org/10.1038/s41586-019-1593-5
Kline J, Godfrey J, Ansell SM (2020) The immune landscape and response to immune checkpoint blockade therapy in lymphoma. Blood 135(8):523–533. https://doi.org/10.1182/blood.2019000847
Valilou SF, Rezaei N (2019) Tumor antigens. Vaccines for cancer immunotherapy: an evidence-based review on current status and future perspectives. Ind J Med Res 61–74. doi:https://doi.org/10.1016/B978-0-12-814039-0.00004-7
Day CP, Merlino G, Van Dyke T (2015) Preclinical mouse cancer models: a maze of opportunities and challenges. Cell 163(1):39–53. https://doi.org/10.1016/j.cell.2015.08.068
Herr W, Ranieri E, Olson W, Zarour H, Gesualdo L, Storkus WJ (2000) Mature dendritic cells pulsed with freeze-thaw cell lysates define an effective in vitro vaccine designed to elicit EBV-specific CD4+ and CD8+ T lymphocyte responses. Blood 96(5):1857–1864. https://doi.org/10.1182/blood.V96.5.1857.h8001857_1857_1864
Mosca PJ, Lyerly HK, Clay TM, Morse MA, Lyerly HK (2007) Dendritic cell vaccines. Front Biosci 12:4050–4060. https://doi.org/10.2741/2371
Nierkens S, Janssen EM (2011) Harnessing dendritic cells for tumor antigen presentation. Cancers (Basel) 3(2):2195–2213. https://doi.org/10.3390/cancers3022195
Barr TA, Gray M, Gray D (2012) B cells: programmers of CD4 T cell responses. Infect Disord Drug Targets 12(3):222–231. https://doi.org/10.2174/187152612800564446
Adler LN, Jiang W, Bhamidipati K, Millican M, Macaubas C, Hung SC, Mellins ED (2017) The other function: class II-restricted antigen presentation by B cells. Front Immunol 8:319. https://doi.org/10.3389/fimmu.2017.00319
Castro F, Cardoso AP, Goncalves RM, Serre K, Oliveira MJ (2018) Interferon-g at the crossroads of tumor immune surveillance or evasion. Front Immunol 9:847. https://doi.org/10.3389/fimmu.2018.00847
Grodeland G, Fossum E, Bogen B (2015) Polarizing T and B cell responses by APC-targeted subunit vaccines. Front Immunol 6:367. https://doi.org/10.3389/fimmu.2015.00367
Tsou P, Katayama H, Ostrin EJ, Hanash SM (2016) The emerging role of B cells in tumor immunity. Cancer Res 76(19):5597–5601. https://doi.org/10.1158/0008-5472.CAN-16-0431
Ostroumov D, Fekete-Drimusz N, Saborowski M, Kuhnel F, Woller N (2018) CD4 and CD8 T lymphocyte interplay in controlling tumor growth. Cell Mol Life Sci 75(4):689–713. https://doi.org/10.1007/s00018-017-2686-7
Acknowledgements
The authors are grateful to Drs. Emma Hornick, Laura Stunz and Claire Rosean for valuable discussion and Drs. Bruce Hostager and Emma Hornick for critical review of the manuscript.
Funding
This work was supported by Merit Review Award I01BX001702 from the Dept. of Veteran’s Affairs to GAB. GAB is a Senior Research Career Scientist of the VA. Ashley Zani received support from NSF REU grant 1559927. The study made use of the Flow Cytometry Facility, which is a Carver College of Medicine/Holden Comprehensive Cancer Center core research facility at the University of Iowa. The facility is funded through user fees and financial support from the Carver College of Medicine, Holden Comprehensive Cancer Center (HCCC), and Iowa City Veteran's Administration Medical Center. The HCCC is supported by NIH award P30CA086862.
Author information
Authors and Affiliations
Contributions
KLO contributed to conception and design, acquisition of data, analysis of data, interpretation of results, preparation of the manuscript, and approval of the submitted version of the manuscript. BMH and ANZ contributed to acquisition and analysis of data and approval of the submitted version of the manuscript. GAB contributed to conception and design, analysis of data, interpretation of results, preparation of the manuscript, approval of the final version of the manuscript, and provided oversight for conduct of this study.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Consent for publication
All listed authors have read and approved this manuscript for submission for publication.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Oxley, K.L., Hanson, B.M., Zani, A.N. et al. Activated B lymphocytes and tumor cell lysate as an effective cellular cancer vaccine. Cancer Immunol Immunother 70, 3093–3103 (2021). https://doi.org/10.1007/s00262-021-02914-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-021-02914-7