
Abstract
The CD57 antigen (alternatively HNK-1, LEU-7, or L2) is routinely used to identify terminally differentiated ‘senescent’ cells with reduced proliferative capacity and altered functional properties. In this article, we review current understanding of the attributes of CD57-expressing T-cells and NK cells in both health and disease and discuss how this marker can inform researchers about their likely functions in human blood and tissues in vivo. While CD57 expression on human lymphocytes indicates an inability to proliferate, these cells also display high cytotoxic potential, and CD57pos NK cells exhibit both memory-like features and potent effector functions. Accordingly, frequencies of CD57-expressing cells in blood and tissues have been correlated with clinical prognosis in chronic infections or various cancers and with human aging. Functional modulation of senescent CD57pos T-cells and mature CD57pos NK cells may therefore represent innovative strategies for protection against human immunological aging and/or various chronic diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Biology of CD57/HNK-1
The CD57 antigen, also called HNK-1, LEU-7 or L2, is a 100-115 kD terminally sulfated carbohydrate epitope that was originally reported as a marker of human natural killer cells [1]. HNK-1 is widely conserved in animals from insects to mammals, and expression is regulated both developmentally and spatially in specific areas of the nervous system, including migrating neural crest cells, rhombomeres, the cerebellum, and myelinated Schwann cells in motor neurons. Yamamoto et al. [2] have shown that the HNK-1 epitope is also widely expressed in the mouse brain, and mice that lack the enzyme responsible for HNK-1 biosynthesis (GlcAT-P gene deletion) exhibit alterations in several key higher brain functions, including spatial memory formation and synaptic plasticity. Cell surface carbohydrates function as binding sites in a variety of crucial cellular activities including recognition and adhesion to other cells, bacteria, viruses, and toxins. HNK-1 was originally detected by immunoblot/immunohistochemical analysis of cell adhesion glycoproteins in the human nervous system, including the binding partners neural cell adhesion molecule, myelin-associated glycoprotein, 5′-nucleotidase, ICAM5, NCAM, integrins, cytotactin, TAG-1, MAG, PMP-22, contactin, L1, P0 and telencephalin, as well as on chondroitin sulfate proteoglycans and glycolipids such as sulfoglucuronylglycolipid-1 and sulfoglucuronylglycolipid-2 [3]. HNK-1 is now also known to act as a ligand for laminin, L-selectin, and P-selectin, as well as for the cerebellar adhesion protein amphoterin. These observations suggest that HNK-1/CD57 is associated with cell–cell and/or cell–matrix interactions.
CD57 in T-cell ‘senescence’ and ‘exhaustion’
The majority of previous investigations into CD57 biology have assessed the role of this antigen in different populations of T-cells. CD57 has been shown to be present on both CD4 and CD8 T-cells in the late stages of differentiation [4]. Late-differentiated T cells are generally thought to exhibit a phenotype characterized by lack of a CD45RO isoform, reduced expression of costimulatory molecules CD27/CD28, loss of chemokine receptor CCR7, and acquisition of CD57 expression together with re-expression of CD45RA. Both CD57posCD4pos and CD57posCD8pos T-cells are presumed to lack proliferative capacity [4, 5] and are known to increase in frequency with chronic immune activation [4] as well as during normal aging [6]. However, the oligoclonal CD57posCD4pos T-cell subset accumulates at a significantly lower rate than does the CD57posCD8pos subset [7]. For this reason, researchers have focused their efforts on investigating the expression and functional role of CD57 on the CD8 T-cell population. In CD8 T-lymphocytes, CD57 identifies terminally differentiated cells that display reduced proliferative responses to TCR triggering in the presence of IL-2, IL-7 or IL-15, as well as increased susceptibility to antigen-induced apoptosis [8], but not to either spontaneous or Fas-induced apoptosis. CD57posCD8 T-cells have thus same characteristics as senescent fibroblasts [9]. CD57posCD8pos T-cells have short telomeres, low telomerase activity, and low expression of cell cycle-associated genes compared with CD57negCD8pos T-cells [3]. Although CD57posCD8pos T-cells can be defined as replicatively senescent, these cells are not functionally exhausted, thus emphasizing the fact that mechanisms of senescence and exhaustion are independently regulated. Exhaustion is characterized by the progressive loss of T-cell function due to a chronically high antigen load [10]. Exhausted T-cells maintain expression of CD45RO and low levels of CCR7 and CD27/CD28, but also up-regulate inhibitory receptors on the cell surface, including programmed cell death 1 (PD-1), cytotoxic T-lymphocyte antigen 4 (CTLA-4), T-cell immunoglobulin domain and mucin domain protein 3 (TIM-3), and lymphocyte activation gene 3 (LAG-3) [11]. IL-2 production and proliferative capacity are the first functions to be lost by exhausted T-cells, followed later by loss of TNF-α production and cytotoxic activity. Severe T-cell exhaustion can eventually result in impaired IFN-γ production and ultimately leads to cell deletion if the high antigen load persists for sufficient time [12]. Unlike exhausted lymphocytes, senescent CD57posCD8pos T-cells can still secrete cytokines such as TNF-α and IFN-γ upon encounter with cognate antigen [4, 8] and have been shown to possess high cytotoxic potential with strong expression of granzymes, granulysin, and perforin [13, 14]. It is important to note, however, that some previous studies may have over-estimated the cytotoxic potential of CD57posCD8pos T-cells by failing to simultaneously assess intracellular expression of cytotoxic molecules and surface expression of the degranulation marker CD107a, which allows direct quantitation of cytotoxic activity. T-cell localization and migration are determined by expression of chemokine receptors and adhesion molecules. When compared with their CD57neg counterparts, CD57posCD8pos T-lymphocytes express high levels of multiple adhesion molecules (integrin β7, αL and β2, CD11a, ICAM-1), but reduced expression of CD62L (L-selectin), suggesting preferential localization in the peripheral tissues and impaired ability to migrate to lymph nodes. Indeed, while CD57posCD8pos T-lymphocytes express only low levels of most chemokine receptors (CCR7 and CXCR4), they also display up-regulation of CX3CR1, which induces altered migration of viral Ag-specific CD8 T-cells to peripheral tissues and allows viral replication to continue unchecked in the lymphoid tissues [8]. These data are consistent with reports that CD57posCD8pos T-cells infiltrate the lungs of HIV-infected individuals [15] and support the hypothesis that CD57posCD8pos cells can interact with the local tissue microenvironment, but display reduced ability to migrate to lymph nodes.
Unlike αβ T-cells, it is unclear whether CD57 represents a senescence marker in γδ T-cells, which exhibit different patterns of expression in the two major subsets: Vδ2 cells are CD57neg, while Vδ1 cells are typically CD57pos [16]. Indeed, while the frequency of CD57pos αβ T-cells is known to increase with natural human aging, this is not the case for CD57pos γδ T-cells [17]. Further investigations are therefore required to identify alternative markers that can be used to determine whether γδ T-cells become senescent in persistent infections, cancer, or aging.
Defining T-cell ‘senescence’ through CD57 and KLRG1
Terminally differentiated CD57pos T-lymphocytes display key features of cellular senescence including proliferation defects, alteration of mitochondrial functions, and elevated expression of ROS, MAPK, and the DNA damage response-related protein γH2AX [18]. However, CD57 is not the only marker that can inform investigators about the functional properties of ‘senescent’ cells. For instance, the inhibitory receptor KLRG1 (killer cell lectin-like receptor G1) is also thought to identify human CD4 and CD8 T-cells that are capable of secreting cytokines but unable to proliferate or clonally expand upon stimulation [19, 20]. In the blood of young individuals, KLRG1 is expressed on a significant proportion of CD8 T-cells and to a lesser extent on CD4 T-cells, but also by ~50 % of NK cells [20]. KLRG1 expression on CD8 T-cells increases dramatically with age, with greater than 90 % of CD8 T-cells displaying this marker in individuals over 65 years of age [19]. Indeed, KLRG1 expression increases not only with age, but also with factors known to induce T-cell activation and differentiation, as observed during persistent infections and cancer.
How CD57 and KLRG1 interrelate is not completely understood. Ibegbu and colleagues [21] investigated how expression of KLRG1 and CD57 correlated with the dysfunctional properties of virus-specific human T-cells. They showed that while a CD57negKLRG1pos subset retained the ability to proliferate and secrete IFN-γ in response to cognate antigen, the CD57posKLRG1pos subset failed to proliferate and displayed only trace IFN-γ secretion, suggesting that these were terminally differentiated senescent T-cells. Further study of how KLRG1 and CD57 expression are associated with altered CD8 T-cell function could therefore lead to the identification of effective methods for therapeutic modulation of these cells in a variety of disease settings.
KLRG1 is expressed on 30–60 % of murine NK cells and by a fraction of murine T-cells [22]. As in humans, this marker is also commonly used to identify senescent T-cells in rodent models [23]. Unlike acquisition of KLRG1, up-regulation of CD57 on chronically activated T-cells is an immunological characteristic of humans and other primates, but not of mice [24]. While CD28negCD8pos T-cells have been identified in mice, they are not the result of persistent antigenic stimulation and do not express CD57. Indeed, age-associated clonal expansion of IFNγ-secreting CD8 T-lymphocytes can be readily observed in mice, but these cells retain proliferative capacity and express high levels of CD28 [25]. This could perhaps be explained by the short lifespan of rodents compared with the longevity of humans. It is also important to note here that thymic activity has evolved differently in mice and humans; rapid thymic involution in humans limits production of naïve T-cells after birth, whereas mice maintain substantial production of naïve T-cells postnatally and may therefore be less affected by T-cell senescence. Moreover, pathogen exposure is far higher for humans than for laboratory mice, especially those housed in specific pathogen-free facilities. For these reasons, most flow cytometry protocols for the identification of senescent T-cells use CD57 and KLRG1 in humans, but only KLRG1 in mice.
Physiological and pathological role of CD57pos T-cells
CD57 in chronic infection and aging
CD8pos T-cells may up-regulate CD57 and become senescent during chronic infection with pathogens including HIV, HCMV, measles, HBV, HCV, B19 parvovirus, and tuberculosis [26, 27]. CD57posCD8pos T-cells have been reported to exert both beneficial and detrimental effects on host immunity during persistent viral infections. Expanded CD57posCD8pos T-cells may mediate effective protection against viruses by expressing high levels of cytotoxic molecules and cytokines [28], but these oligoclonally expanded cells have also been proposed to dominate the immune response to HIV and may therefore lead to higher incidence of opportunistic infections and cancers [29]. Exhaustion of HIV-specific T-cells that lack proliferative activity is a hallmark of chronic infection. Brenchley et al. [4] have shown that HIV-specific CD8 T-cells expressing CD57 are unable to proliferate upon antigen-specific stimulation in vitro and instead undergo activation-induced cell death, suggesting that CD57 denotes senescence in this setting. However, the functional differences between senescent and exhausted T-cells are likely to be context-dependent and thus remain poorly understood. Indeed, Petrovas et al. [9] have reported that HIV-specific CD8 T-cells are predominantly PD1high but express variable levels of CD57, with elevated expression being associated with increased resistance to both spontaneous and Fas-mediated apoptosis.
Progressive oligoclonal accumulation of CD57posCD8pos T-lymphocytes occurs not only during persistent infection but also during natural human aging [6]. We confirmed the expansion of terminally differentiated CD27negCD57pos T-cells in our cohort of elderly Asian individuals (the Singapore Longitudinal Aging Study, SLAS; Fig. 1a). At birth, nearly all T-cells are CD57neg, but CD57posCD8pos T-cells accumulate over time to comprise approximately 20 % of the total population in healthy adults and peaking at 50–60 % after 80 years of age [30]. This age-related increase in the CD57posCD8pos T-cell subset is a key component of the ‘immune risk’ profile that predicts a higher all-cause 2-year mortality in octogenarians and nonagenarians [31]. A high proportion of these late-differentiated CD8 T-cells are specific for HCMV antigens, which could lead to reduced antigenic diversity and could represent a major component of age-related immunosenescence [32]. The relationship between CD57 expression, HCMV infection, TCR repertoire and human physiological aging remains incompletely understood, particularly regarding the cellular changes induced by the infection and/or by age itself. This is in part due to conclusions being drawn from studies that used different antibody panels to define T-cell subsets of unknown homogeneity in statistically underpowered cohorts. We could wonder whether the loss of immunity in elderly could be explained by cumulative consequences of memory inflation due to persistent CMV infection and of attrition of naïve T-cells. More recently, Wertheimer et al. [33] compared CMVneg and CMVpos donors of similar ages and ethnicities to dissect the consequences of aging and CMV infection on T-cell development. They showed that physiological aging is accompanied by a reduction in naïve CD8 cells, while CMV infection is associated with an increase in the effector memory CD8 subset. The impact of aging and CMV on CD4 cell subset distribution was limited in this cohort. These data demonstrated that aging and CMV exert distinct yet synergistic effects on the CD8 T-cell pool.

Functional significance of CD57 in the differentiation of T-cells and NK cells. a Correlation between the late-differentiated stage (CD27negCD57pos) of T-cells and the late-differentiated stage (CD62LnegCD57pos) of NK cells in elderly (r = 0.3255; p = 0.06) and young individuals (r = 0.5307; p = 0.0037). b Correlation between the frequencies of CD27negCD28negCD45RAnegCD57posCD4pos T-cells and CD27negCD28negCD45RAposCD57posCD8pos T-cells (r = 0.4921; p < 0.0001;) in young individuals. c, d Pearson correlation between CMV IgG titers in young individuals with frequencies of CD57pos senescent T-cells in the CD4 subset (r = 0.4864; p < 0.0001; c) and CD8 subset (r = 0.3547; p = 0.001; d). p values <0.05 were considered significant
In our elderly Asian cohort, more than 90 % of individuals were seropositive for CMV; hence, this group was not suitable for delineating the influences of viral infection from those of aging. We therefore recruited a young Asian cohort to discern the impact of CMV infection on the terminal differentiation of effector T-cells using a range of markers including CD27, CD28, and CD45RA. In CMVneg donors, we observed that CD57 expression was enriched in different subsets: CD27negCD28negCD45RAneg for CD4 T-cells and CD27negCD28negCD45RApos for CD8 T-cells, with the frequencies of these populations being strongly correlated with each other as well as with CMV IgG titer (Fig. 1b–d). These data confirmed that CMV infection is a major driving factor in accelerating T-cell differentiation even in young individuals.
Flu vaccination offers limited protection in elderly donors, perhaps due to CD8 T-cell senescence limiting the vaccine response and increasing susceptibility to influenza virus. In the cohort of elderly individuals, proliferation and cytokine secretion by flu-specific CD8 T-cells was significantly decreased compared with young individuals, whereas CMV-specific CD8 T-cell function was preserved. This impaired function of flu-specific CD8 T-cells was associated with increased expression of the senescence markers CD57 and KLRG1, as well as with elevated expression of the transcription factor T-bet which regulates T-cell terminal differentiation [34]. A deeper understanding of the role played by CD57 in shaping T-cell responses to infectious challenges might therefore provide new insights into the rational design of vaccination strategies. This could be particularly beneficial in elderly individuals, who display increased mortality and morbidity associated with common viral infections such as influenza.
CD57 in cancer biology
Cancer risk increases with aging and is thought to be associated with a deficiency of anti-tumor immunity. Accordingly, CD8 T-cell phenotype and function in the blood and tumor mass have been the subject of intensive studies aiming to better predict cancer patient survival and responsiveness to chemotherapy. However, it remains unclear how CD57 expression influences the anti-tumor responses of CD4 helper T-cells and CD8 cytotoxic T-cells.
A CD28negCD8pos T-cell population, enriched in CD57pos cells and putative CD8 Treg, has been identified in both the peripheral blood and tumor microenvironment of patients with solid cancers or hematological malignancies. Indeed, expansion of CD28negCD8pos T-cells in the peripheral blood is a negative prognosticator in patients with advanced renal cell carcinoma [35], melanoma [36], advanced gastric cancer [37], and bladder carcinoma [38]. Filaci et al. [39] analyzed the phenotype and functions of CD28negCD8pos T-cells infiltrating primary tumors and/or satellite lymph nodes in a series of 42 human cancers. Despite their finding that expansion and tumor infiltration by CD8 T-lymphocytes were generally associated with prolonged survival [40], the presence of numerous CD28negCD8pos T-cells was instead associated with poor survival. Similarly, in head and neck cancer, a direct link has been established between cancer development and the expansion of late-differentiated T cells, which decline to normal levels after tumor removal irrespective of patient CMV status [41].
Van den Hove et al. [42] reported that compared with healthy donors, 42 % of patients with untreated hematological malignancies display increased expression of CD57 on CD4 and CD8 T-lymphocytes. Selective expansion of CD57pos T-cells among circulating CD4pos T-lymphocytes has also been repeatedly detected in patients with Hodgkin’s lymphoma [43], chronic lymphocytic leukemia [44], and chronic leukemia of B-cell origin [42]. Functional analyses have revealed that these CD57posCD4pos T-cells display a typical Th1 cytokine profile upon TCR stimulation, but also retain the ability to secrete IL-2, which is not the case for terminal-differentiated CD57pos T-cells in healthy donors [42]. This characteristic could perhaps explain the CD57pos T-cell expansion observed in the tumor microenvironment. Moreover, the cytotoxic activity of circulating CD8pos and CD4pos T-lymphocytes was almost exclusively exerted by the CD57pos subset. Despite the apparent preservation of potent effector functions, these CD57pos T-cells seem unable to inhibit the growth of malignant cells and may even dampen immune responses against tumor-associated antigens, perhaps by competing for resources such as cytokines or nutrients [42].
Where researchers have reported a positive correlation between CD57posCD28negCD8pos T-cells and cancer survival, the investigations focused on untreated patients with myeloma [45] or patients treated with thalidomide for relapsed/refractory multiple myeloma [46]. The location or the nature of the transformed cells in the bone marrow, the therapies administered, and/or ethnic/genetic determinants may explain this apparent discrepancy compared with other types of cancer. Nonetheless, in light of the above studies, it is possible to conclude that solid and hematological cancer progression are strongly influenced by the presence of CD57posCD8pos T-cells in the tumor or circulation, irrespective of patient CMV status [47]. Simple screening for CD57 expression could therefore be used to predict and identify patients likely to respond to chemoradiation therapy [48] or those with high probability of developing an aggressive clinical course in chronic lymphocytic leukemia [47] or squamous cell carcinoma following renal transplant [49].
Several questions remain unresolved concerning the role of CD57-expressing T-cells in cancer. A more detailed analysis of gene expression, phenotype, metabolism, and functions of CD57pos T-cells should allow us to determine how cancer-associated ‘senescent-like’ T-cells differ from those induced by CMV infection or aging. Further study of the interactions between CD57pos T-cells and tumor-associated antigen-specific T-cells will also inform the development of novel cell depletion strategies for cancer immunotherapy. Finally, while the mechanisms that induce the expansion of CD57pos T-cells in cancer are still unknown, identification of these mechanisms may present opportunities to disrupt this process to prevent immune evasion by the tumor.
CD57 in transplantation and autoimmunity
Allogeneic solid organ and bone marrow transplantations are routinely accompanied by the oligoclonal expansion of an immunosuppressive CD28negCD8pos population (likely to be CD57posCD8pos T-cells) [50, 51]. Accumulation of these T-cells is associated with better graft acceptance and stable function in liver transplant recipients, as well as with a reduced need for immunosuppressive drugs [50]. In addition, Sabnani et al. [52] showed that in patients without evidence of allograft rejection or viral syndrome, 71 % of cardiac transplant patients and 44 % of renal transplant patients exhibited monoclonal expansion of CD57posCD8pos T-lymphocytes. CD57posCD8pos T-cells frequency may therefore represent a useful biomarker for adjusting immunosuppressive therapy following transplantation.
Several autoimmune conditions are characterized by an increase in highly cytotoxic CD28negCD8pos (putative CD57pos) T-cells in the peripheral blood, including Graves’ disease, ankylosing spondylitis, and rheumatoid arthritis, as well as their accumulation in diseased tissues in dermatomyositis and polymyositis. These cells may play an active role in the autoimmune response and appear to be associated with more severe disease manifestations [7, 53]. Alternatively, Mikulkova et al. [54] reported that type 1 diabetes mellitus and multiple sclerosis were instead associated with a decrease in the CD28negCD8pos (putative CD57pos) peripheral T-cell population. Lower frequencies of this cell subset have been also observed in systemic lupus erythematosus [55]. These data suggest that the CD28negCD8pos (putative CD57pos) T-cell population is heterogeneous and includes functionally opposing subsets (cytotoxic vs. immunosuppressive) whose relative frequencies may determine patient outcomes [7]. Further phenotypic analysis will be required before we can reliably detect and manipulate these opposing populations in different disease settings and potentially harness their positive functions (and/or disrupt deleterious effects) for therapeutic benefit in the clinic.
CD57 as a marker of mature NK cells
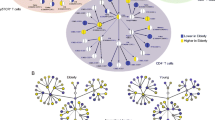
While it is now well established that CD57 identifies terminally differentiated T-cells, this marker was originally thought to denote cells with natural killer activity and exhibits variable expression among different subsets of NK cells. CD57 is expressed by CD16posCD56dim cytotoxic NK cells and CD16posCD56neg inflammatory NK cells, whereas CD16negCD56bright regulatory NK cells do not express this marker even during chronic infections [56, 57]. The acquisition of CD57 thus follows the natural differentiation of NK cells (from regulatory to cytotoxic to inflammatory NK cells) [53]. Like T-cells, NK cell expression of CD57 could be considered a marker of terminal differentiation, albeit not clearly associated with senescence in this population (Fig. 2). Indeed, antiretroviral therapy in chronic HIV infection was able to restore the normal phenotype and functions of NK cells in treated patients [58], and the balance between regulatory, cytotoxic, and inflammatory NK cells was similar to that observed in peripheral blood from healthy donors. The restoration of NK cell subset balance by interventional therapies against chronic infections (HCV) or autoimmune disorders (myasthenia gravis or dermatomyositis) therefore warrants further investigation.

CD57 expression during T and NK cells differentiation. Proposed model of CD57 acquisition during T-lymphocyte and NK cell differentiation. Phenotypic and functional characteristics of T-cells and NK cells expressing CD57 are summarized. The question mark in the figure corresponds to evidence reported for specific infections that has yet to be validated in other pathologies
The strong association of CD57 with cell adhesion and the acquisition of homing molecules restricted to inflamed peripheral tissues (CXCR1, CX3CR1) suggest a local role for CD57pos NK cells during inflammation [59]. Since CD57 is a marker of NK cell maturation, its expression is coupled with the loss of antigens typical of CD56bright NK cells (NKp46, Nkp30, NKG2D, CD62L) and up-regulation of markers including CD16, KIR, and LIR-1. The recent use of mass cytometry has confirmed the diversity of NKR surface expression by CD57pos NK cells [60]. As expected, CD57 was detected in mature NK cell subsets and was closely associated with NKG2C and CD8 expression.
It has been reported that infection with viruses including HIV and CMV could drive the expansion of CD57posNKG2Chigh NK cells [61–64]. It has therefore been proposed that CD57posNKG2Chigh NK cells might represent human CMV-specific ‘memory’ NK cells, analogous to the CMV-specific Ly49Hpos NK cell population identified in mice. While it is interesting to note that LIR-1 can mediate recognition of the MHC class I homolog UL18 in CMV-infected cells [65], the role of this interaction in the development of memory NK cells is still unclear and requires further study. Deletion of NKG2C is associated with a decrease of CD57pos NK cells, and NKG2C-deficient children exhibit elevated anti-HCMV IgG titers likely due to unchecked viral replication [66]. The induction of CD57posNKG2Chigh NK cells during acute HCMV infection and the persistence of this population even during viral control have prompted researchers to investigate the role of this population during acute infection with EBV [67] or following vaccination [68]. In these cases, the main innate population involved was CD56dimCD57neg NK cells in CMV seronegative donors. Expression of CD8 [69] and Siglec-7 [70] has been described as being associated with CD57 expression on mature NK cells in HIV patients, but the role of these molecules during other viral infections requires further investigation. The mechanisms that drive the expansion of human memory NK cells are also poorly documented, although a recent study identified that the frequency of tumor-infiltrating CD57pos NK cells correlates with expression levels of the NK cell chemoattractant cytokine IL-37 in primary hepatocellular carcinoma as well as a mouse cancer model [71].
Characteristics of CD57-expressing NK cells
Differentiation of CD57negCD56dim into CD57posCD56dim NK cells is thought to be associated with a loss of proliferative response to inflammatory cytokines in vitro, perhaps due to the down-regulation of the corresponding receptors including IL-2Rβ, IL-12Rβ, and IL-18Rα [56, 57]. However, measurement of the proliferation marker Ki-67 in cytotoxic NK cells did not reveal any differences in the division of CD57negCD56dimCD16pos and CD57posCD56dimCD16pos subsets [72]. These data suggested that mature NK cells were able to undergo normal progression through the cell cycle in vivo following exposure to inflammatory cytokines or specific stimuli such as CMV. It is still unclear whether the proliferative potential of NK cells and the acquisition of CD57 are related in vivo, either during acute infection or after control of viremia.
Elevated expression of CD16 by CD57posCD56dimCD16pos NK cells renders them hyper-responsive to CD16 ligation, which mimics antibody-dependent cell-mediated cytotoxicity (ADCC). Mature NK cells exhibit considerable cytotoxic potential, as indicated by increased expression of the degranulation marker CD107a, granzyme B, and perforin. It has been reported that following exposure to inflammatory cytokines, CD57negCD56dimCD16pos NK cells secrete more IFN-γ than do CD57posCD56dimCD16pos NK cells, but this difference is not observed after cross-linking with CD16 [56]. Licensed NK cells have even been reported to aid dendritic cell maturation and prime antigen T-cells via a mechanism that may involve the TNF ligand family member TNFSF14 [73, 74], although this interaction requires further investigation and seems more likely to occur in peripheral tissues than in lymph nodes due to the profile of NK cell chemokine receptor expression.
Clinical relevance of CD57 NK cells
There is accumulating evidence that CD57pos NK cell frequency and function are clinically relevant in a range of different human diseases. Le Garff-Tavernier et al. reported the progressive accumulation of CD57posCD56dimCD16pos NK cells during aging, with the mature subset comprising up to 60 % of total NK cell pool in European adults (18–60 years old). Mature CD57pos NK cell expansion and accumulation may be a consequence of cumulative lifetime exposure to infections including HIV, HBV, HCV, chikungunya virus, and hantavirus, although the influence of co-infection with HCMV is difficult to distinguish [75]. The ratio of CD57neg: CD57pos NK cells (or NKG2CposCD57neg/NKG2CposCD57pos) is significantly modulated during acute and chronic HCMV infection, as well as after virus reactivation following hematopoietic stem cell transplantation (HSCT). It is still not clear whether this disturbed population ratio is caused by the virus itself or by the associated inflammatory response driving NK cell differentiation. It is also possible that disruption of the normal balance of NK cell populations represents a virus-induced ‘immune escape’ mechanism that limits the killing of HCMV-infected target cells by the CD57pos subset [76].
In CMV-infected patients receiving solid organ transplantation, increased peripheral frequency of mature NK cells is associated with a better prognosis. A slower disease progression is also observed in chronic HIV patients presenting with elevated frequencies of poly-functional CD57pos NK cells [69]. In order to better understand infection resistance in highly HIV-exposed seronegative (HESN) subjects, Lima et al. [77] sought to determine how innate immune responses in these individuals differed from those of chronic HIV-infected patients. Unlike patients with chronic infection, HESN patients that exhibited CD56bright regulatory NK cells with a CD127posNKG2Dpos phenotype also displayed peripheral expansion of CD57posCD56dimCD16pos cytotoxic NK cells, consistent with a role for these cells in mediating resistance to HIV.
Initial functional characterization of NK cells focused on their ability to kill malignant cells displaying reduced MHC class I surface expression and up-regulation of ‘danger signals’ including stress-related molecules [78]. It seems likely therefore that NK cell differentiation and modulation of their cytotoxic potential will critically alter their role in tumor surveillance. Accordingly, increased peripheral frequency of mature CD57pos NK cells has been correlated with improved patient survival in leukemia, lymphoma, and carcinoma [53, 79]. Indeed, a recent study also reported that local expansion of CD57posCD56dimCD16pos NK cells in tumor-infiltrated lymph nodes as mediated by CCL22 and IL-6 was correlated with improved survival in melanoma patients [80].
Contrasting with data from studies of tumor immunology, analyses of NK cell biology in autoimmunity, allergy, and inflammatory disorders have shown that CD57posCD56dimCD16pos NK cells are depleted from the circulation in pathologies including atopic dermatitis [81] and psoriasis [82]. In parallel, the affected tissues are characterized by localized expansion of CD57negCD56dimCD16pos NK cells, as identified in the inflamed skin in psoriasis [83], the synovial fluid in rheumatoid arthritis patients [84], and the pancreas in type 1 diabetes [85]. These data demonstrate that it is possible to restrain NK cell maturation in both the circulation and inflamed peripheral tissues, although it is currently unknown how pro-inflammatory and/or auto-reactive lymphocytes can mediate these effects.
Together, the data discussed here urge further study of how ‘memory’ CD57pos NK cells can protect against viral infections, tumor progression, and autoimmune disorders. This may in turn lead to the development of novel immunotherapies that can modulate NK cell function for patient benefit in a wide range of clinical settings. For example, one approach could aim to convert/expand mature and long-lasting NK cells for adoptive transfer into human patients. In this context, it might prove possible to drive in vitro differentiation of cytotoxic NK cells using CMV derivatives and/or various cytokines and chemokines including IL-37, TNFSF14, IL-6, and CCL22. Vaccine strategies that exploit the potent functions of innate leukocytes and specifically boost the expansion of mature cytotoxic NK cells will be exciting avenues to explore in future.
CD57 and therapy: promises and caveats
In pathologies that are characterized by an accumulation of CD57pos T-cells, it will first be necessary to determine the clinical relevance of this phenomenon and then to identify methods of decreasing the frequency of these cells for therapeutic benefit. With this approach, it may prove possible to identify the molecular and signaling pathways that drive the generation CD57pos T-cells and perhaps disrupt these in disease settings, while simultaneously sparing other CD57pos cell types with likely beneficial functions in the innate and non-immune compartments. Recent studies have described the role of Zeb2 in the terminal maturation of NK and CD8 T-cells for melanoma rejection or during chronic infection [86, 87]. The modulation of the transcriptional network including Zeb2 and T-bet may favor formation and preservation of memory T-cells. An alternative strategy might be to focus on the causes of T-cell terminal differentiation or senescence. This might involve patient treatment with drugs that promote the clearance of chronic viral infections (HIV, HCV) or that prevent the reactivation of latent infections (CMV) in order to decrease senescent cell numbers. However, this strategy would need to be carefully optimized to avoid a drug-induced increase in terminal-differentiated CD57pos T-cells, such as that induced by sub-optimal antiretroviral therapy in HIV patients [88, 89]. It might also prove possible to directly reverse senescence, perhaps by inhibiting p38 MAPK signaling, which has been reported to increase telomerase activity and decrease TCR-induced apoptosis in human CD27negCD45RAposCD4pos memory T-cells [90]. Again, this approach would need to consider that different subsets of CD57pos T-cells exhibit differential sensitivities to p38 inhibition, as well as distinct profiles of cell signaling, metabolic activity, gene expression and antigen specificities; hence, the effects of therapies that target the p38 pathway will need to be carefully evaluated.
Previous reports have indicated that the combined action of p38 inhibition and checkpoint blockade can restore the proliferation of senescent CD8 T-cells [91]. Effects on exhausted CTLs and senescent T-cells may also have contributed to the promising results obtained during recent clinical trials in cancer patients treated with antibodies against inhibitory molecules such as PD-1. Further investigations will be needed to validate this hypothesis. Finally, it may be possible to ‘purge’ specific tissues of senescent T-cells by inducing their migration or sequestration to alternative sites such as the bone marrow [92], although efforts to neutralize the signals that drive the generation of TEMRA including α-interferons [93] or IL-7 [92] seem more likely to yield results in the near term.
Conclusion
The CD57 antigen is commonly used to identify populations of late-differentiated ‘senescent’ cells with defined cell phenotypes and effector functions, but patterns of expression differ significantly between NK cells and T-cells (Fig. 2). CD57 expression is most prominent in terminally differentiated effector T-cells and mature cytotoxic NK cells. Aging and/or repetitive stimulation of these populations due to persist infection might trigger expansion of these subsets. Accordingly, restoring the function of senescent CD57pos T-cells or expanding memory CD57pos NK cells may offer therapeutic benefit in elderly patients or individuals with chronic viral infections.
Abbreviations
- CTLA-4:
-
Cytotoxic T-lymphocyte antigen 4
- FLU:
-
Influenza virus
- HBV:
-
Hepatitis B virus
- HCMV:
-
Human cytomegalovirus
- HCV:
-
Hepatitis C virus
- HESN:
-
Highly HIV-exposed seronegative subjects
- HIV:
-
Human immunodeficiency virus
- KLRG1:
-
Killer cell lectin-like receptor G1
- LAG-3:
-
Lymphocyte activation gene 3
- PD-1:
-
Programmed cell death 1
- SLAS:
-
Singapore Longitudinal Aging Studies
- SPF:
-
Specific pathogen-free
- TIM-3:
-
T-cell immunoglobulin domain and mucin domain protein 3
References
Abo T, Balch CM (1981) A differentiation antigen of human NK and K cells identified by a monoclonal antibody (HNK-1). J Immunol 127(3):1024–1029
Yamamoto S, Oka S, Inoue M, Shimuta M, Manabe T, Takahashi H, Miyamoto M, Asano M, Sakagami J, Sudo K, Iwakura Y, Ono K, Kawasaki T (2002) Mice deficient in nervous system-specific carbohydrate epitope HNK-1 exhibit impaired synaptic plasticity and spatial learning. J Biol Chem 277(30):27227–27231
Focosi D, Bestagno M, Burrone O, Petrini M (2010) CD57+ T lymphocytes and functional immune deficiency. J Leukoc Biol 87(1):107–116
Brenchley JM, Karandikar NJ, Betts MR, Ambrozak DR, Hill BJ, Crotty LE, Casazza JP, Kuruppu J, Migueles SA, Connors M, Roederer M, Douek DC, Koup RA (2003) Expression of CD57 defines replicative senescence and antigen-induced apoptotic death of CD8+ T cells. Blood 101(7):2711–2720
Palmer BE, Blyveis N, Fontenot AP, Wilson CC (2005) Functional and phenotypic characterization of CD57+ CD4+ T cells and their association with HIV-1-induced T cell dysfunction. J Immunol 175(12):8415–8423
Bandres E, Merino J, Vazquez B, Inoges S, Moreno C, Subira ML, Sanchez-Ibarrola A (2000) The increase of IFN-gamma production through aging correlates with the expanded CD8(+high) CD28(−) CD57(+) subpopulation. Clin Immunol 96(3):230–235
Strioga M, Pasukoniene V, Characiejus D (2011) CD8+ CD28− and CD8+ CD57+ T cells and their role in health and disease. Immunology 134(1):17–32
Le Priol Y, Puthier D, Lecureuil C, Combadiere C, Debre P, Nguyen C, Combadiere B (2006) High cytotoxic and specific migratory potencies of senescent CD8+ CD57+ cells in HIV-infected and uninfected individuals. J Immunol 177(8):5145–5154
Petrovas C, Chaon B, Ambrozak DR, Price DA, Melenhorst JJ, Hill BJ, Geldmacher C, Casazza JP, Chattopadhyay PK, Roederer M, Douek DC, Mueller YM, Jacobson JM, Kulkarni V, Felber BK, Pavlakis GN, Katsikis PD, Koup RA (2009) Differential association of programmed death-1 and CD57 with ex vivo survival of CD8+ T cells in HIV infection. J Immunol 183(2):1120–1132
Akbar AN, Henson SM (2011) Are senescence and exhaustion intertwined or unrelated processes that compromise immunity? Nat Rev Immunol 11(4):289–295
Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, Betts MR, Freeman GJ, Vignali DA, Wherry EJ (2009) Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol 10(1):29–37
Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R (2003) Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol 77(8):4911–4927
Chattopadhyay PK, Betts MR, Price DA, Gostick E, Horton H, Roederer M, De Rosa SC (2009) The cytolytic enzymes granyzme A, granzyme B, and perforin: expression patterns, cell distribution, and their relationship to cell maturity and bright CD57 expression. J Leukoc Biol 85(1):88–97
Chiang SC, Theorell J, Entesarian M, Meeths M, Mastafa M, Al-Herz W, Frisk P, Gilmour KC, Ifversen M, Langenskiold C, Machaczka M, Naqvi A, Payne J, Perez-Martinez A, Sabel M, Unal E, Unal S, Winiarski J, Nordenskjold M, Ljunggren HG, Henter JI, Bryceson YT (2013) Comparison of primary human cytotoxic T-cell and natural killer cell responses reveal similar molecular requirements for lytic granule exocytosis but differences in cytokine production. Blood 121(8):1345–1356
Sadat-Sowti B, Parrot A, Quint L, Mayaud C, Debre P, Autran B (1994) Alveolar CD8+ CD57+ lymphocytes in human immunodeficiency virus infection produce an inhibitor of cytotoxic functions. Am J Respir Crit Care Med 149(4 Pt 1):972–980
De Rosa SC, Mitra DK, Watanabe N, Herzenberg LA, Herzenberg LA, Roederer M (2001) Vdelta1 and Vdelta2 gammadelta T cells express distinct surface markers and might be developmentally distinct lineages. J Leukoc Biol 70(4):518–526
Vasudev A, Ying CT, Ayyadhury S, Puan KJ, Andiappan AK, Nyunt MS, Shadan NB, Mustafa S, Low I, Rotzschke O, Fulop T, Ng TP, Larbi A (2014) gamma/delta T cell subsets in human aging using the classical alpha/beta T cell model. J Leukoc Biol 96(4):647–655
Henson SM, Lanna A, Riddell NE, Franzese O, Macaulay R, Griffiths SJ, Puleston DJ, Watson AS, Simon AK, Tooze SA, Akbar AN (2014) p38 signaling inhibits mTORC1-independent autophagy in senescent human CD8(+) T cells. J Clin Invest 124(9):4004–4016
Ouyang Q, Wagner WM, Voehringer D, Wikby A, Klatt T, Walter S, Muller CA, Pircher H, Pawelec G (2003) Age-associated accumulation of CMV-specific CD8+ T cells expressing the inhibitory killer cell lectin-like receptor G1 (KLRG1). Exp Gerontol 38(8):911–920
Voehringer D, Koschella M, Pircher H (2002) Lack of proliferative capacity of human effector and memory T cells expressing killer cell lectinlike receptor G1 (KLRG1). Blood 100(10):3698–3702
Ibegbu CC, Xu YX, Harris W, Maggio D, Miller JD, Kourtis AP (2005) Expression of killer cell lectin-like receptor G1 on antigen-specific human CD8+ T lymphocytes during active, latent, and resolved infection and its relation with CD57. J Immunol 174(10):6088–6094
McMahon CW, Zajac AJ, Jamieson AM, Corral L, Hammer GE, Ahmed R, Raulet DH (2002) Viral and bacterial infections induce expression of multiple NK cell receptors in responding CD8(+) T cells. J Immunol 169(3):1444–1452
Voehringer D, Blaser C, Brawand P, Raulet DH, Hanke T, Pircher H (2001) Viral infections induce abundant numbers of senescent CD8 T cells. J Immunol 167(9):4838–4843
Vallejo AN (2005) CD28 extinction in human T cells: altered functions and the program of T-cell senescence. Immunol Rev 205:158–169
Ortiz-Suarez A, Miller RA (2002) A subset of CD8 memory T cells from old mice have high levels of CD28 and produce IFN-gamma. Clin Immunol 104(3):282–292
Kared H, Camous X, Larbi A (2014) T cells and their cytokines in persistent stimulation of the immune system. Curr Opin Immunol 29:79–85
Papagno L, Spina CA, Marchant A, Salio M, Rufer N, Little S, Dong T, Chesney G, Waters A, Easterbrook P, Dunbar PR, Shepherd D, Cerundolo V, Emery V, Griffiths P, Conlon C, McMichael AJ, Richman DD, Rowland-Jones SL, Appay V (2004) Immune activation and CD8+ T-cell differentiation towards senescence in HIV-1 infection. PLoS Biol 2(2):E20
Scheinberg P, Melenhorst JJ, Brenchley JM, Hill BJ, Hensel NF, Chattopadhyay PK, Roederer M, Picker LJ, Price DA, Barrett AJ, Douek DC (2009) The transfer of adaptive immunity to CMV during hematopoietic stem cell transplantation is dependent on the specificity and phenotype of CMV-specific T cells in the donor. Blood 114(24):5071–5080
Maurer T, Ponte M, Leslie K (2007) HIV-associated Kaposi’s sarcoma with a high CD4 count and a low viral load. N Engl J Med 357(13):1352–1353
Weekes MP, Wills MR, Mynard K, Hicks R, Sissons JGP, Carmichael AJ (1999) Large clonal expansions of human virus-specific memory cytotoxic T lymphocytes within the CD57(+) CD28(−) CD8(+) T-cell population. Immunology 98(3):443–449
Wikby A, Ferguson F, Forsey R, Thompson J, Strindhall J, Lofgren S, Nilsson BO, Ernerudh J, Pawelec G, Johansson B (2005) An immune risk phenotype, cognitive impairment, and survival in very late life: impact of allostatic load in Swedish octogenarian and nonagenarian humans. J Gerontol A Biol Sci Med Sci 60(5):556–565
Franceschi C, Bonafe M, Valensin S (2000) Human immunosenescence: the prevailing of innate immunity, the failing of clonotypic immunity, and the filling of immunological space. Vaccine 18(16):1717–1720
Wertheimer AM, Bennett MS, Park B, Uhrlaub JL, Martinez C, Pulko V, Currier NL, Nikolich-Zugich D, Kaye J, Nikolich-Zugich J (2014) Aging and cytomegalovirus infection differentially and jointly affect distinct circulating T cell subsets in humans. J Immunol 192(5):2143–2155
Dolfi DV, Mansfield KD, Polley AM, Doyle SA, Freeman GJ, Pircher H, Schmader KE, Wherry EJ (2013) Increased T-bet is associated with senescence of influenza virus-specific CD8 T cells in aged humans. J Leukoc Biol 93(6):825–836
Characiejus D, Pasukoniene V, Kazlauskaite N, Valuckas KP, Petraitis T, Mauricas M, Den Otter W (2002) Predictive value of CD8highCD57+ lymphocyte subset in interferon therapy of patients with renal cell carcinoma. Anticancer Res 22(6B):3679–3683
Characiejus D, Ukoniene VP, Auskaite RJ, Azlauskaite N, Aleknavicius E, Mauricas M, Den Otter W (2008) Peripheral blood CD8highCD57+ lymphocyte levels may predict outcome in melanoma patients treated with adjuvant interferon-alpha. Anticancer Res 28(2B):1139–1142
Akagi J, Baba H (2008) Prognostic value of CD57(+) T lymphocytes in the peripheral blood of patients with advanced gastric cancer. Int J Clin Oncol 13(6):528–535
Characiejus D, Pasukoniene V, Jacobs JJ, Eidukevicius R, Jankevicius F, Dobrovolskiene N, Mauricas M, Van Moorselaar RJ, Den Otter W (2011) Prognostic significance of peripheral blood CD8highCD57+ lymphocytes in bladder carcinoma patients after intravesical IL-2. Anticancer Res 31(2):699–703
Filaci G, Fenoglio D, Fravega M, Ansaldo G, Borgonovo G, Traverso P, Villaggio B, Ferrera A, Kunkl A, Rizzi M, Ferrera F, Balestra P, Ghio M, Contini P, Setti M, Olive D, Azzarone B, Carmignani G, Ravetti JL, Torre G, Indiveri F (2007) CD8+ CD28− T regulatory lymphocytes inhibiting T cell proliferative and cytotoxic functions infiltrate human cancers. J Immunol 179(7):4323–4334
Gutkin DW, Shurin MR (2014) Clinical evaluation of systemic and local immune responses in cancer: time for integration. Cancer Immunol Immunother 63(1):45–57
Tsukishiro T, Donnenberg AD, Whiteside TL (2003) Rapid turnover of the CD8(+) CD28(−) T-cell subset of effector cells in the circulation of patients with head and neck cancer. Cancer Immunol Immunother 52(10):599–607
Van den Hove LE, Vandenberghe P, Van Gool SW, Ceuppens JL, Demuynck H, Verhoef GE, Boogaerts MA (1998) Peripheral blood lymphocyte subset shifts in patients with untreated hematological tumors: evidence for systemic activation of the T cell compartment. Leuk Res 22(2):175–184
Atayar C, Poppema S, Visser L, van den Berg A (2006) Cytokine gene expression profile distinguishes CD4+/CD57+ T cells of the nodular lymphocyte predominance type of Hodgkin’s lymphoma from their tonsillar counterparts. J Pathol 208(3):423–430
Serrano D, Monteiro J, Allen SL, Kolitz J, Schulman P, Lichtman SM, Buchbinder A, Vinciguerra VP, Chiorazzi N, Gregersen PK (1997) Clonal expansion within the CD4+ CD57+ and CD8+ CD57+ T cell subsets in chronic lymphocytic leukemia. J Immunol 158(3):1482–1489
Sze DM, Brown RD, Yuen E, Gibson J, Ho J, Raitakari M, Basten A, Joshua DE, de St Fazekas, Groth B (2003) Clonal cytotoxic T cells in myeloma. Leuk Lymphoma 44(10):1667–1674
Mileshkin L, Honemann D, Gambell P, Trivett M, Hayakawa Y, Smyth M, Beshay V, Ritchie D, Simmons P, Milner AD, Zeldis JB, Prince HM (2007) Patients with multiple myeloma treated with thalidomide: evaluation of clinical parameters, cytokines, angiogenic markers, mast cells and marrow CD57+ cytotoxic T cells as predictors of outcome. Haematologica 92(8):1075–1082
Nunes C, Wong R, Mason M, Fegan C, Man S, Pepper C (2012) Expansion of a CD8(+) PD-1(+) replicative senescence phenotype in early stage CLL patients is associated with inverted CD4:CD8 ratios and disease progression. Clin Cancer Res 18(3):678–687
Dobrovolskiene NT, Cicenas S, Kazlauskaite N, Miseikyte-Kaubriene E, Krasko JA, Ostapenko V, Pasukoniene V, Strioga MM (2015) CD8CD57 T-cell population as an independent predictor of response to chemoradiation therapy in extensive-stage small cell lung cancer. Lung Cancer 90(2):326–333
Bottomley M, Harden P, Wood K (2015) CD57 expression in CD8 T cells and development of cutaneous squamous cell carcinoma in renal transplant recipients: a prospective cohort study. Lancet 385(Suppl 1):S23
Lin YX, Yan LN, Li B, Wang LL, Wen TF, Zeng Y, Wang WT, Zhao JC, Yang JY, Xu MQ, Ma YK, Chen ZY, Bai YJ (2009) A significant expansion of CD8+ CD28− T-suppressor cells in adult-to-adult living donor liver transplant recipients. Transplant Proc 41(10):4229–4231
Vlad G, Cortesini R, Suciu-Foca N (2008) CD8+ T suppressor cells and the ILT3 master switch. Hum Immunol 69(11):681–686
Sabnani I, Zucker MJ, Tsang P, Palekar S (2006) Clonal T-large granular lymphocyte proliferation in solid organ transplant recipients. Transplant Proc 38(10):3437–3440
Nielsen CM, White MJ, Goodier MR, Riley EM (2013) Functional Significance of CD57 Expression on human NK Cells and relevance to disease. Front Immunol 4:422
Mikulkova Z, Praksova P, Stourac P, Bednarik J, Strajtova L, Pacasova R, Belobradkova J, Dite P, Michalek J (2010) Numerical defects in CD8+ CD28− T-suppressor lymphocyte population in patients with type 1 diabetes mellitus and multiple sclerosis. Cell Immunol 262(2):75–79
Tulunay A, Yavuz S, Direskeneli H, Eksioglu-Demiralp E (2008) CD8+ CD28−, suppressive T cells in systemic lupus erythematosus. Lupus 17(7):630–637
Lopez-Verges S, Milush JM, Pandey S, York VA, Arakawa-Hoyt J, Pircher H, Norris PJ, Nixon DF, Lanier LL (2010) CD57 defines a functionally distinct population of mature NK cells in the human CD56dimCD16+ NK-cell subset. Blood 116(19):3865–3874
Bjorkstrom NK, Riese P, Heuts F, Andersson S, Fauriat C, Ivarsson MA, Bjorklund AT, Flodstrom-Tullberg M, Michaelsson J, Rottenberg ME, Guzman CA, Ljunggren HG, Malmberg KJ (2010) Expression patterns of NKG2A, KIR, and CD57 define a process of CD56dim NK-cell differentiation uncoupled from NK-cell education. Blood 116(19):3853–3864
Brunetta E, Hudspeth KL, Mavilio D (2010) Pathologic natural killer cell subset redistribution in HIV-1 infection: new insights in pathophysiology and clinical outcomes. J Leukoc Biol 88(6):1119–1130
Carrega P, Ferlazzo G (2012) Natural killer cell distribution and trafficking in human tissues. Front Immunol 3:347
Strauss-Albee DM, Horowitz A, Parham P, Blish CA (2014) Coordinated regulation of NK receptor expression in the maturing human immune system. J Immunol 193(10):4871–4879
Lugli E, Marcenaro E, Mavilio D (2014) NK Cell Subset Redistribution during the Course of Viral Infections. Front Immunol 5:390
Mela CM, Goodier MR (2007) The contribution of cytomegalovirus to changes in NK cell receptor expression in HIV-1-infected individuals. J Infect Dis 195(1):158–159; author reply 159-160
Guma M, Cabrera C, Erkizia I, Bofill M, Clotet B, Ruiz L, Lopez-Botet M (2006) Human cytomegalovirus infection is associated with increased proportions of NK cells that express the CD94/NKG2C receptor in aviremic HIV-1-positive patients. J Infect Dis 194(1):38–41
Sun JC, Beilke JN, Lanier LL (2009) Adaptive immune features of natural killer cells. Nature 457(7229):557–561
Prod’homme V, Griffin C, Aicheler RJ, Wang EC, McSharry BP, Rickards CR, Stanton RJ, Borysiewicz LK, Lopez-Botet M, Wilkinson GW, Tomasec P (2007) The human cytomegalovirus MHC class I homolog UL18 inhibits LIR-1+ but activates LIR-1− NK cells. J Immunol 178(7):4473–4481
Goodier MR, White MJ, Darboe A, Nielsen CM, Goncalves A, Bottomley C, Moore SE, Riley EM (2014) Rapid NK cell differentiation in a population with near-universal human cytomegalovirus infection is attenuated by NKG2C deletions. Blood 124(14):2213–2222
Hendricks DW, Balfour HH Jr, Dunmire SK, Schmeling DO, Hogquist KA, Lanier LL (2014) Cutting edge: NKG2C(hi)CD57+ NK cells respond specifically to acute infection with cytomegalovirus and not Epstein–Barr virus. J Immunol 192(10):4492–4496
White MJ, Nielsen CM, McGregor RH, Riley EH, Goodier MR (2014) Differential activation of CD57-defined natural killer cell subsets during recall responses to vaccine antigens. Immunology 142(1):140–150
Ahmad F, Hong HS, Jackel M, Jablonka A, Lu IN, Bhatnagar N, Eberhard JM, Bollmann BA, Ballmaier M, Zielinska-Skowronek M, Schmidt RE, Meyer-Olson D (2014) High frequencies of polyfunctional CD8+ NK cells in chronic HIV-1 infection are associated with slower disease progression. J Virol 88(21):12397–12408
Brunetta E, Fogli M, Varchetta S, Bozzo L, Hudspeth KL, Marcenaro E, Moretta A, Mavilio D (2009) The decreased expression of Siglec-7 represents an early marker of dysfunctional natural killer-cell subsets associated with high levels of HIV-1 viremia. Blood 114(18):3822–3830
Zhao JJ, Pan QZ, Pan K, Weng DS, Wang QJ, Li JJ, Lv L, Wang DD, Zheng HX, Jiang SS, Zhang XF, Xia JC (2014) Interleukin-37 mediates the antitumor activity in hepatocellular carcinoma: role for CD57+ NK cells. Sci Rep 4:5177
Lopez-Verges S, Milush JM, Schwartz BS, Pando MJ, Jarjoura J, York VA, Houchins JP, Miller S, Kang SM, Norris PJ, Nixon DF, Lanier LL (2011) Expansion of a unique CD57(+)NKG2Chi natural killer cell subset during acute human cytomegalovirus infection. Proc Natl Acad Sci USA 108(36):14725–14732
Bhatnagar N, Ahmad F, Hong HS, Eberhard J, Lu IN, Ballmaier M, Schmidt RE, Jacobs R, Meyer-Olson D (2014) FcgammaRIII (CD16)-mediated ADCC by NK cells is regulated by monocytes and FcgammaRII (CD32). Eur J Immunol 44(11):3368–3379
Holmes TD, Wilson EB, Black EV, Benest AV, Vaz C, Tan B, Tanavde VM, Cook GP (2014) Licensed human natural killer cells aid dendritic cell maturation via TNFSF14/LIGHT. Proc Natl Acad Sci USA 111(52):E5688–E5696
Le Garff-Tavernier M, Beziat V, Decocq J, Siguret V, Gandjbakhch F, Pautas E, Debre P, Merle-Beral H, Vieillard V (2010) Human NK cells display major phenotypic and functional changes over the life span. Aging Cell 9(4):527–535
Borysiewicz LK, Rodgers B, Morris S, Graham S, Sissons JG (1985) Lysis of human cytomegalovirus infected fibroblasts by natural killer cells: demonstration of an interferon-independent component requiring expression of early viral proteins and characterization of effector cells. J Immunol 134(4):2695–2701
Lima JF, Oliveira LM, Pereira NZ, Mitsunari GE, Duarte AJ, Sato MN (2014) Distinct natural killer cells in HIV-exposed seronegative subjects with effector cytotoxic CD56(dim) and CD56(bright) cells and memory-like CD57(+) NKG2C(+) CD56(dim) cells. J Acquir Immune Defic Syndr 67(5):463–471
Vivier E, Ugolini S, Blaise D, Chabannon C, Brossay L (2012) Targeting natural killer cells and natural killer T cells in cancer. Nat Rev Immunol 12(4):239–252
Balch CM, Tilden AB, Dougherty PA, Cloud GA (1983) Depressed levels of granular lymphocytes with natural killer (NK) cell function in 247 cancer patients. Ann Surg 198(2):192–199
Ali TH, Pisanti S, Ciaglia E, Mortarini R, Anichini A, Garofalo C, Tallerico R, Santinami M, Gulletta E, Ietto C, Galgani M, Matarese G, Bifulco M, Ferrone S, Colucci F, Moretta A, Karre K, Carbone E (2014) Enrichment of CD56(dim)KIR+ CD57+ highly cytotoxic NK cells in tumour-infiltrated lymph nodes of melanoma patients. Nat Commun 5:5639
Matsumura G (1990) Leu7 (HNK-1)-positive cells in peripheral blood and natural killer cell activity in patients with atopic dermatitis. Nihon Hifuka Gakkai Zasshi 100(1):57–62
Batista MD, Ho EL, Kuebler PJ, Milush JM, Lanier LL, Kallas EG, York VA, Chang D, Liao W, Unemori P, Leslie KS, Maurer T, Nixon DF (2013) Skewed distribution of natural killer cells in psoriasis skin lesions. Exp Dermatol 22(1):64–66
Ottaviani C, Nasorri F, Bedini C, de Pita O, Girolomoni G, Cavani A (2006) CD56brightCD16(−) NK cells accumulate in psoriatic skin in response to CXCL10 and CCL5 and exacerbate skin inflammation. Eur J Immunol 36(1):118–128
Dalbeth N, Callan MF (2002) A subset of natural killer cells is greatly expanded within inflamed joints. Arthritis Rheum 46(7):1763–1772
Dotta F, Censini S, van Halteren AG, Marselli L, Masini M, Dionisi S, Mosca F, Boggi U, Muda AO, Del Prato S, Elliott JF, Covacci A, Rappuoli R, Roep BO, Marchetti P (2007) Coxsackie B4 virus infection of beta cells and natural killer cell insulitis in recent-onset type 1 diabetic patients. Proc Natl Acad Sci USA 104(12):5115–5120
van Helden MJ, Goossens S, Daussy C, Mathieu AL, Faure F, Marcais A, Vandamme N, Farla N, Mayol K, Viel S, Degouve S, Debien E, Seuntjens E, Conidi A, Chaix J, Mangeot P, de Bernard S, Buffat L, Haigh JJ, Huylebroeck D, Lambrecht BN, Berx G, Walzer T (2015) Terminal NK cell maturation is controlled by concerted actions of T-bet and Zeb2 and is essential for melanoma rejection. J Exp Med 212(12):2015–2025. doi:10.1084/jem.20150809
Omilusik KD, Best JA, Yu B, Goossens S, Weidemann A, Nguyen JV, Seuntjens E, Stryjewska A, Zweier C, Roychoudhuri R, Gattinoni L, Bird LM, Higashi Y, Kondoh H, Huylebroeck D, Haigh J, Goldrath AW (2015) Transcriptional repressor ZEB2 promotes terminal differentiation of CD8+ effector and memory T cell populations during infection. J Exp Med 212(12):2027–2039. doi:10.1084/jem.20150194
Lee SA, Sinclair E, Jain V, Huang Y, Epling L, Van Natta M, Meinert CL, Martin JN, McCune JM, Deeks SG, Lederman MM, Hecht FM, Hunt PW (2014) Low proportions of CD28− CD8+ T cells expressing CD57 can be reversed by early ART initiation and predict mortality in treated HIV infection. J Infect Dis 210(3):374–382
Serrano-Villar S, Sainz T, Lee SA, Hunt PW, Sinclair E, Shacklett BL, Ferre AL, Hayes TL, Somsouk M, Hsue PY, Van Natta ML, Meinert CL, Lederman MM, Hatano H, Jain V, Huang Y, Hecht FM, Martin JN, McCune JM, Moreno S, Deeks SG (2014) HIV-infected individuals with low CD4/CD8 ratio despite effective antiretroviral therapy exhibit altered T cell subsets, heightened CD8+ T cell activation, and increased risk of non-AIDS morbidity and mortality. PLoS Pathog 10(5):e1004078
Di Mitri D, Azevedo RI, Henson SM, Libri V, Riddell NE, Macaulay R, Kipling D, Soares MV, Battistini L, Akbar AN (2011) Reversible senescence in human CD4+ CD45RA+ CD27− memory T cells. J Immunol 187(5):2093–2100
Henson SM, Macaulay R, Riddell NE, Nunn CJ, Akbar AN (2015) Blockade of PD-1 or p38 MAP kinase signaling enhances senescent human CD8(+) T-cell proliferation by distinct pathways. Eur J Immunol 45(5):1441–1451
Libri V, Azevedo RI, Jackson SE, Di Mitri D, Lachmann R, Fuhrmann S, Vukmanovic-Stejic M, Yong K, Battistini L, Kern F, Soares MV, Akbar AN (2011) Cytomegalovirus infection induces the accumulation of short-lived, multifunctional CD4+ CD45RA+ CD27+ T cells: the potential involvement of interleukin-7 in this process. Immunology 132(3):326–339
Lanna A, Coutavas E, Levati L, Seidel J, Rustin MH, Henson SM, Akbar AN, Franzese O (2013) IFN-alpha inhibits telomerase in human CD8(+) T cells by both hTERT downregulation and induction of p38 MAPK signaling. J Immunol 191(7):3744–3752
Acknowledgments
The study was supported by a research grant from the Agency for Science, Technology and Research (SIgN collaborative Grant No. 10–036) and by the Singapore Immunology Network. Anis Larbi is a scholar of the International Society for Advancement of Cytometry (ISAC). Serena Martelli is funded by the A*STAR Research Attachment Program (ARAP) and the Vice Chancellor Scholarship, University of Southampton.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Additional information
This article is part of the Symposium-in-Writing “Natural killer cells, ageing and cancer”, a series of papers published in Cancer Immunology, Immunotherapy.
Rights and permissions
About this article

Cite this article
Kared, H., Martelli, S., Ng, T.P. et al. CD57 in human natural killer cells and T-lymphocytes. Cancer Immunol Immunother 65, 441–452 (2016). https://doi.org/10.1007/s00262-016-1803-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-016-1803-z