
Abstract
Background
Chronic obstructive pulmonary disease (COPD) is a risk factor for lung cancer (LC). Myeloid-derived suppressor cells (MDSCs) down-regulate the T cell receptor ζ chain (TCR ζ) through l-arginine deprivation and lead to T cell dysfunction and deficient antitumor immunity. We hypothesized that abnormally high levels of MDSCs in COPD patients may alter tumor immunosurveillance.
Methods
We compared the proportion of circulating MDSCs (Lin-HLA-DR-/CD33+/CD11b+) (by flow cytometry), arginase I (ARG I) serum levels (by ELISA), and expression levels of TCR ζ on circulating lymphocytes (by flow cytometry) in 28 patients with LC, 62 subjects with COPD, 41 patients with both LC and COPD, 40 smokers with normal spirometry and 33 non-smoking controls. T cell proliferation assays were performed in a subgroup of participants (CFSE dilution protocol).
Results
We found that: (1) circulating MDSCs were up-regulated in COPD and LC patients (with and without COPD); (2) MDSCs expansion was associated with TCR ζ down-regulation in the three groups; (3) in LC patients, these findings were independent of COPD and tobacco smoking exposure; (4) TCR ζ down-regulation correlates with T cell hyporesponsiveness in COPD and LC patients.
Conclusions
These results suggest that tumor immunosurveillance might be impaired in COPD and may contribute to the increased risk of LC reported in these patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Tobacco smoking is the main risk factor for both chronic obstructive pulmonary disease (COPD) [1] and lung cancer (LC) [2]. Yet, not all smokers develop COPD or LC, and importantly, the risk of LC is increased in patients with COPD [2–4]. Shared genetic determinants and/or chronic inflammation can link COPD and LC pathogenically [2, 5, 6].
It is well established that chronic inflammation favors the initiation and progression of many types of tumors through a variety of mechanisms [7–10]. On the one hand, inflammatory cells produce a number of molecules, including reactive oxygen and nitrogen species, cytokines, chemokines, growth factors and prostaglandins that can cause DNA mutations, genomic instability, proliferation of premalignant and malignant cells, tumor neovascularization, invasion and metastasis [7]. On the other hand, chronic inflammation can also favor cancer development by impairing tumor immunosurveillance and antitumor immunity. In this context, recent studies have shown that cancer-related inflammation is associated with a defective T cell effector function which is mediated, in part, by myeloid-derived suppressor cells (MDSCs) [11–13].
MDSCs are a heterogeneous mixture of mature and immature granulocytic and monocytic cells produced by the bone marrow characterized by their capacity to suppress T cell responses [14]. In humans, when activated by mediators such as interferon-γ (IFN-γ), interleukin 4 (IL-4), interleukin 13 (IL-13) and transforming growth factor β (TGF β), MDSCs release arginase I (ARG I) to the circulation and the microenvironment, inducing T cell dysfunction by l-arginine deprivation, which in turn reduces the expression of the T cell receptor ζ chain (TCR ζ), inhibit T cell proliferation and cytokine production [15, 16], hence helping a variety of malignancies to escape antitumor immunity [17].
MDSCs are also important immune regulators in chronic inflammatory (non-malignant) conditions [14]. We previously showed that COPD patients present up-regulation of circulating MDSCs, increased serum levels of ARG I and down-regulation of TCR ζ in circulating lymphocytes [18].
The TCR is a multisubunit complex comprising the clonotypic α and β antigen recognition chains, associated with the invariant chain subunits that form heterodimers CD3 γ-ε and CD3 δ-ε, and the invariant ζ-ζ homodimers (TCR ζ). Following TCR engagement by the αβ chains, the TCR ζ is phosphorylated and functions as a key transmembrane structure that triggers intracellular signal transduction pathways leading to nuclear factor kappa B (NF-kB) activation downstream and its translocation to the nucleus determining full T cell activation, secretion of cytokines and cell proliferation. Loss of TCR ζ expression is associated with T cell hyporesponsiveness and has been reported in T lymphocytes of hosts with tumors, autoimmune and infectious diseases, all of which have chronic inflammation as a common denominator. In cancer patients, this defect is present in tumor microenvironment, tumor-involved lymph nodes and peripheral blood [12].
Based on these previous observations, we hypothesized that this pattern of expansion and activation of circulating MDSCs associated to TCR ζ chain expression reduction can contribute to the increased incidence of LC reported in COPD [2–4]. To test this hypothesis, we compared the circulating levels of MDSCs, the serum concentration of ARG I and the expression of TCR ζ in circulating T cells in five groups of individuals: (1) LC patients without COPD; (2) COPD patients without LC; (3) patients with both LC and COPD; (4) smokers with normal spirometry without LC; and (5) never smokers with normal spirometry and without LC. Results of groups 2, 4 and 5 have been published elsewhere [18] and are included here because they represent an important reference for the analysis of the new LC-COPD-related observations.
Methods
Study design and ethics
This is an observational, cross-sectional study that complied with the Declaration of Helsinki and Good Clinical Practice Guidelines. The project was approved by the Ethics Committee of Balearic Islands, Spain, and all participants signed their informed consent.
Study population
Participants were consecutively recruited from hospital out-patient clinics, pulmonary function laboratory attendees and primary care clinics. Subjects aged 40–80 years having a minimum smoking cumulative exposure of >10 pack-years. (except non-smoking controls) were invited to participate. COPD diagnosis and severity were defined using post-bronchodilator spirometry according to the GOLD recommendations [1]. COPD patients were stable and free of exacerbations for ≥3 months. We defined former smokers as those participants who had quit smoking completely for at least 1 year before the study. LC diagnosis was confirmed through histological specimens and classified according to the WHO classification [19]. All LC patients were current or former smokers. Tumor clinical stage and type were established before the patient had received any anticancer treatment. TNM (tumor, node, metastases) staging was determined according to the 7th edition of the TNM Classification of Lung Cancer [20]. Subjects with a history of asthma, atopy, allergic rhinitis, autoimmune diseases, renal disorders, other malignancies, infectious diseases and/or using immunomodulatory drugs were excluded from the study.
Lung function
Spirometry was performed (Micro 6000 Spirometer, Medisoft, France) according to international guidelines [21]. In LC patients, reported spirometric values here correspond to preoperative values. Reference values were those of a Mediterranean population [22].
Peripheral blood sampling and processing
Peripheral blood was obtained by peripheral venipuncture and collected into sterile tubes. Differential and absolute white blood cells count (WBC) was taken using a CELL-DYN Sapphire automatic analyzer (Abbot, USA) following the manufacturer’s instructions. Serum was obtained by centrifugation of non-heparinized whole blood during 15 min at 3000 rpm, and samples were stored at −70 °C until further analysis. Heparinized blood was used to isolate peripheral blood mononuclear cells (PBMCs) by gradient centrifugation (lymphocyte separation medium, PAA Laboratory, Pasching, Austria) as previously described [18].
Flow cytometry
Circulating T cell subpopulations, MDSCs, expression of TCR ζ and CD3 ε chains, and T cell proliferation assay were analyzed with an Epics XL-MCL (Beckman Coulter, Miami, USA) flow cytometer using the Expo32 ADC software, as detailed below.
Circulating T cell subpopulations
CD4 and CD8 lymphocytes were quantified in whole blood samples incubated with combinations of anti-CD3-PE-, anti-CD4-PE-Cy5- and anti-CD8-FITC-conjugated monoclonal antibodies (BD Pharmingen, NJ, USA) and expressed as a proportion of the total amount of lymphocytes [18].
Characterization of circulating MDSCs
PBMCs were washed, re-suspended in 1× phosphate-buffered saline (PBS) (Dulbecco’s PBS, PAA Laboratory, Pasching, Austria) and incubated with a combination of anti-lineage cocktail 2 (Lin) (a mixture of monoclonal antibodies against CD3, CD14, CD19, CD20 and CD56)—FITC (BD Biosciences, San Jose, CA), anti-MHC class II (HLA-DR)-ECD (Beckman Coulter, France), anti-CD33-PECy5 and anti-CD11b-PE (BD Pharmingen, NJ, USA). After 30 min of incubation at room temperature in the dark, PBMCs were fixed (BD FACS™ BD Biosciences), washed and re-suspended in PBS. MDSCs were characterized as Lin-/HLA-DR-/CD33+/CD11b+ [14, 18, 23] (Fig. 1) and reported as percentage of the total amount of PBMCs.

Representative flow cytometry dot plots of MDSCs in peripheral blood in a non-smoker control (a), COPD patient (b) and LC patient without COPD (c). A1 represents PBMCs negative for HLA-DR and the lineage cocktail 2 (Lin), and A2 identifies Lin-/HLA-DR-/CD33+/CD11b+ MDSCs. For further explanations, see text
TCR ζ and CD3 ε chain expression
PBMCs were first incubated with an anti-CD3 ε-FITC (BD Pharmingen, NJ, USA) monoclonal antibody. Cells were then fixed, permeabilized and labeled with an anti-TCR ζ-PE (Abcam, Cambridge, UK) monoclonal antibody using the fixation/permeabilization kit (e-Biosciences, CA, USA), according to the manufacturer’s instructions. The level of expression of the CD3 ε and TCR ζ was determined on total CD3ε+ cells. The intensity of surface TCR ζ and CD3 ε expression was expressed as mean fluorescence intensity (MFI).
T cell proliferation assay
A carboxyfluorescein succinimidyl ester (CFSE) dilution protocol was used to evaluate the T cell proliferation on cultures of CFSE-labeled PBMCs. Briefly, PBMCs were labeled during 5 min at room temperature (RT) (25 °C) with 1 μg of CFSE (Invitrogen), following the manufacturer’s instructions. 2 × 105 CFSE-labeled PBMCs per well were cultured in 96-well plates and stimulated with phytohemagglutinin 1 % in DMEM supplemented with 10 % heat inactivated fetal calf serum (FCS), 50 IU/ml penicillin, 50 µg/ml streptomycin and 292 µg/ml l-glutamine (Gibco®, UK). After 96 h of culture at 37 °C in a 5 % CO2 atmosphere, PBMCs were harvested, washed and stained with anti-CD3 PE-Cy7 (Beckman Coulter). T cell proliferation index was calculated on total CFSE+CD3ε+ cells using ModFit LT™ software (Verity Software House).
Serum concentration of ARG I
Serum ARG I levels were quantified by ELISA using a specific human ARG I (liver type) antibody (BioVendor, Brno, Czech Republic), as previously described [18].
Statistical analysis
Results are presented as mean ± standard error of the mean (SEM), median values [range] or proportion, as appropriate. Categorical data were analyzed using Chi-square (χ 2) test, whereas for continuous variables ANOVA (or Kruskal–Wallis test) was used for parametrically (or nonparametrically) distributed variables, respectively. A p value <0.05 was considered statistically significant.
Results
Characterization of participants
As shown in Table 1, we included in the study 28 patients with LC (but not COPD), 62 patients with COPD (but not LC), 41 patients with both LC and COPD, 40 smokers with normal spirometry and 33 non-smoking controls. Age was similar across groups albeit smokers with normal spirometry were slightly younger. This latter group as well as that of non-smokers included more females. Body mass index (BMI) was similar in all groups. Cumulative smoking exposure (pack-years) was similar in LC and COPD patients (alone or in combination), smaller in smokers and, by definition, absent in non-smokers. The proportion of current smokers was lowest in LC patients without COPD and highest in LC patients with COPD (Table 1). Airflow limitation was moderate to severe in COPD patients, with or without LC (Table 1). Total leukocytes were increased in smokers, COPD and LC patients, with a predominance of neutrophils in COPD and LC patients. No differences were found in the proportion of CD4 and CD8 T cells between groups. Finally, Table 2 shows the histological LC type distribution and TNM staging was similar in LC patients irrespective of the presence or absence of COPD.
Circulating levels of MDSCs
Figure 2 (Panel a) shows that the level of circulating MDSCs (as % of PBMCs) was similar in patients with LC, COPD or both, all of them higher than those of never smokers. Also, MDSCs levels were significantly higher in patients with LC and COPD than in smokers with normal spirometry. We did not find significant differences in the proportion of circulating MDSCs in LC patients without COPD according to their smoking status (current (0.56 [2.74] %) vs. former (0.28 [0.84] %, p = 0.36)), indicating that this finding was independent of tobacco smoking exposure. No difference was found in LC patients (with and without COPD) according to the tumor type or cancer staging (data not shown).

Circulating levels of MDSCs (a), serum levels of arginase I (b), expression of TCR ζ chain (c) and CD3 ε (d) in circulating T lymphocytes in the five groups studied. Only significant differences (p < 0.05) are shown. For further explanations, see text
Serum levels of arginase I
Similar to our observations on MDSCs, serum ARG I levels were not significantly different between LC, COPD or LC + COPD patients but they were significantly higher than in non-smokers (Fig. 2, panel b). Smokers with normal spirometry also showed higher levels of serum ARG I than non-smokers. In LC subjects without COPD, serum ARG I levels were similar in current and former smokers. We did not find any association between serum ARG I and the histological type or stages of LC (with and without COPD).
Expression levels of TCR ζ and CD3 ε chains on circulating T cells
The expression of both TCR ζ and CD3 ε was only determined in 28 LC patients (16 with and 12 without COPD, respectively), 17 COPD patients without LC, 16 smokers with normal spirometry and 12 non-smokers. Their clinical and demographic characteristics were similar to those of the entire population (Table 1). The surface expression levels of TCR ζ chain were again similar in patients with LC, COPD or both, but significantly lower than those of smokers with normal spirometry and non-smokers (Fig. 2, Panel c) [18]. By contrast, the expression of CD3 ε was similar in the five groups studied (Fig. 2, Panel d).
T cell proliferation
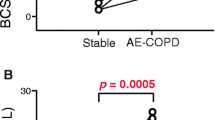
To determine whether the lower expression of TCR ζ correlates with impaired T cell function, T cell proliferation assay using a CFSE dilution protocol was performed in three cases (two COPD patients and one lung cancer patient without COPD) and three matched non-smokers. First, the level of expression of TCR ζ was measured by using the method described above. As shown in Fig. 3, peripheral T cells from COPD and LC patients showed lower expression of TCR ζ than non-smokers (Panel a), and this finding was associated with a profound alteration of T cell responsiveness (Panel b).

Expression of TCR ζ chain (a) and T cell proliferation assay using CFSE dilution protocol (b) in three cases (two COPD patients and one LC patient) and three healthy controls. Histograms and flow cytometry dot plots of T cell proliferation representative of LC, COPD and non-smoker control subjects (c). For further explanation, see text
Discussion
The main novel findings of this study are that: (1) The circulating levels of MDSCs and the ARG I serum concentration are increased, and the surface expression of TCR ζ in circulating lymphocytes is reduced, to a similar extent in patients with LC, COPD and both LC and COPD (Fig. 2), and (2) TCR ζ down-regulation is associated with T cell hyporesponsiveness in COPD and LC patients (Fig. 3). These observations support our working hypothesis and suggest that there is an impaired host tumor immunosurveillance in COPD that, we speculate, can contribute to explain the increased incidence of LC described in these patients [3, 4].
Previous studies
Our results confirm previous studies in patients with different types of cancer, including LC, showing increased levels of circulating MDSCs in these patients [14, 23–25]. In fact, several studies have identified circulating MDSCs as a prognostic factor in a variety of malignancies, including LC [23, 27].
MDSCs down-regulate antitumor immunity and facilitate tumor development [14, 17, 26] because they suppress T cell function through the reduction in TCR ζ expression via ARG I [14–16, 23, 24, 26]. TCR is a complex T lymphocyte molecular structure with different subunits critical for antigen recognition and T cell effector function [12]. The ζ chain is indispensable for coupling antigen recognition by the TCR to a number of signal transduction pathways [12]. In vitro and in vivo experiments have demonstrated that TCR ζ down-regulation is mediated by MDSCs and that among all TCR subunits, the ζ chain is the only one modulated by MDSCs [12, 30, 31]. It has been suggested that sustained antigen exposure and the resulting chronic inflammatory state that ensues are the main underlying causes for TCR ζ down-regulation, and that this represents a natural homeostatic mechanism to attenuate T cell function [12, 31]. In fact, in keeping with recent evidence that MDSCs also modulate the immune response in inflammatory non-malignant conditions [14], our group has recently described the up-regulation of circulating MDSCs associated with TCR ζ down-regulation in COPD [18].
Interpretation of current findings
We observed (Fig. 2) that the circulating levels of MDSCs and the serum concentration of ARG I (a surrogate marker of MDSCs activation) were increased, and the surface expression of TCR ζ in circulating lymphocytes was reduced, to a similar extent in patients with LC, COPD or both. Reduced expression of TCR ζ was associated with impaired T cell proliferation in COPD and LC patients. Based on the molecular studies discussed above, we propose that this can functionally cause a reduced cancer immunosurveillance state in COPD and that this can contribute to explain pathogenically the increased incidence of LC repeatedly reported in COPD [2–4].
Chronic obstructive pulmonary disease is often associated with low-grade chronic systemic inflammation [29]. A recent population-based study showed that COPD patients with systemic inflammation have an increased risk of developing a number of comorbidities, including LC [34]. Many inflammatory factors, such as granulocyte–macrophage colony-stimulating factor (GM-CSF), granulocyte colony-stimulating factor (G-CSF), macrophage colony-stimulating factor (M-CSF), IFN-γ, interleukin 1β (IL-1β), vascular endothelial growth factor (VEGF) and interleukin 3 (IL-3), are increased in both COPD and LC [28, 29] and have the capacity to condition myelopoiesis in the bone marrow, block myeloid cell differentiation and cause MDSCs accumulation [14, 17, 26]. Similarly, a variety of potential tumor antigens and auto-antigens [32, 33] may cause sustained antigen exposure, TCR ζ down-regulation and impaired T cell function [12, 31].
Arginase I is constitutively expressed in human polymorphonuclear leukocytes (PMNs) and stored in gelatinase granules [35, 36]. Activated PMNs, but not resting PMNs, induce ARG I-dependent T cell suppression by concomitantly releasing both gelatinase and azurophil granules to the circulation and the microenvironment. The latter granules contain key serine proteases required to activate ARG I probably by proteolytic cleavage [37, 38]. Different studies demonstrated that human MDSCs are a subpopulation of low-density activated PMNs that copurify with PBMC on a density gradient due to degranulation [38–40]. Based on these observations, we speculate that MDSCs, but no PMNs, are the source of raised serum ARG I in the present study.
We observed that, in smokers subjects, the TCR ζ expression was similar to non-smokers despite high levels of ARG I. In our previous study, we hypothesized that this may be related to a transitory and nonspecific response, as it was related to current exposition to cigarette smoke [18]. Alternatively, the serum ARG I in smokers may be released as inactive proenzyme from gelatinase granules in the absence of activating proteinases from azurophil granules [37]. This observation is compatible with previous studies reporting differential PMNs degranulation in COPD patients with increased serum and sputum levels of azurophilic proteinases such as neutrophil elastase (NE) and myeloperoxidase (MPO) [41].
Considering all these arguments, we propose that chronic systemic inflammation in COPD contributes to alter tumor immunosurveillance by enhancing the circulating levels of MDSCs.
Strengths and limitations
The present study has some strengths and limitations that deserve comments. The relative large number of well-characterized patients included allowed the analysis of the relationship between MDSCs, LC and COPD. Likewise, the inclusion of the smoking and non-smoking controls was important for the correct interpretation of findings, given the fact that current smoking up-regulates MDSCs in smokers [18], but it did not influence MDSCs proportion in LC without COPD, indicating an independent association between LC and MDSCs. Among the potential limitations of our study, we acknowledge that smoking controls were slightly younger and were less exposed to smoking than COPD and LC patients. However, we do not think that these differences influence our results since there was no correlation between age and the proportion of circulating MDSCs (data not shown), and cumulative smoking exposure was significant (albeit smaller) in smoker controls. Secondly, we postulate that the expansion of MDSCs and the reduction in TCR ζ expression may be causally related. However, we recognize that further studies are needed to fully elucidate the precise molecular mechanisms of the MDSCs suppressive effect on T cells.
Conclusion
The circulating levels of MDSCs and serum concentration of ARG I are increased, and TCR ζ expression in circulating lymphocytes decreased, to a similar extent in patients with LC, COPD or both. These observations are compatible with impaired tumor immunosurveillance in COPD. Hence, we propose that this can potentially facilitate tumor initiation and growth in COPD, contributing to explain the increased incidence of LC reported in COPD patients.
Abbreviations
- ARG I:
-
Arginase I
- CFSE:
-
Carboxyfluorescein succinimidyl ester
- COPD:
-
Chronic obstructive pulmonary disease
- G-CSF:
-
Granulocyte colony-stimulating factor
- GM-CSF:
-
Granulocyte–macrophage colony-stimulating factor
- IFN-γ:
-
Interferon-γ
- IL-13:
-
Interleukin 13
- IL-1β:
-
Interleukin 1β
- IL-3:
-
Interleukin 3
- IL-4:
-
Interleukin 4
- LC:
-
Lung cancer
- M-CSF:
-
Macrophage colony-stimulating factor
- MDSCs:
-
Myeloid-derived suppressor cells
- MFI:
-
Mean fluorescence intensity
- MPO:
-
Myeloperoxidase
- NE:
-
Neutrophil elastase
- NF-kB:
-
Nuclear factor kappa B
- PBMCs:
-
Peripheral blood mononuclear cells
- PMNs:
-
Polymorphonuclear leukocytes
- TCR ζ:
-
T cell receptor zeta chain
- TGF β:
-
Transforming growth factor β
- TNM:
-
Tumor, node, metastases
- VEGF:
-
Vascular endothelial growth factor
- WBC:
-
White blood cells
References
Vestbo J, Hurd SS, Agustí AG, Jones PW, Vogelmeier C, Anzueto A et al (2013) Global strategy for the diagnosis, management and prevention of chronic obstructive pulmonary disease, gold executive summary. Am J Respir Crit Care Med 187:347–365. doi:10.1164/rccm.201204-0596PP
Brody JS, Spira A (2006) Chronic obstructive pulmonary disease, inflammation and lung cancer. Proc Am Thorac Soc 3:535–537. doi:10.1513/pats.200603-089MS
Mannino DM, Aguayo SM, Petty TL, Redd SC (2003) Low lung function and incident lung cancer in the United States: from the First National Health and Nutrition Examination Survey follow-up. Arch Intern Med 163:1475–1480. doi:10.1001/archinte.163.12.1475
Young RP, Hopkins RJ, Christmas T, Black PN, Metcalf P, Gamble GD (2009) COPD prevalence is increased in lung cancer, independent of age, sex and smoking history. Eur Respir J 34:380–386. doi:10.1183/09031936.00144208
Rodríguez-Roisin R, Soriano JB (2008) Chronic obstructive pulmonary disease with lung cancer and/or cardiovascular disease. Proc Am Thorac Soc 5:842–847. doi:10.1513/pats.200807-075TH
Agustí A, Faner R (2012) Systemic inflammation and comorbidities in chronic obstructive pulmonary disease. Proc Am Thorac Soc 9:43–46. doi:10.1513/pats.201108-050MS
Grivennikov SI, Greten FR, Karin M (2010) Immunity, inflammation, and cancer. Cell 140:883–899. doi:10.1016/j.cell.2010.01.025
O’Byrne KJ, Dalgleish AD (2001) Chronic immune activation and inflammation as the cause of malignancy. Br J Cancer 85:473–483. doi:10.1054/bjoc.2001.1943
Baron JA, Sandler RS (2000) Nonsteroidal anti-inflammatory drugs and cancer prevention. Annu Rev Med 51:511–523. doi:10.1146/annurev.med.51.1.511
Mantovani A, Allavena P, Sica A, Balkwill F (2008) Cancer-related inflammation. Nature 454:436–444. doi:10.1038/nature07205
Ostrand-Rosenberg S, Sinha P (2009) Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol 182:4499–4506. doi:10.4049/jimmunol.0802740
Baniyash M (2004) TCR ζ-chain downregulation: curtailing an excesive inflammatory immune response. Nat Rev Immunol 4:675–687. doi:10.1038/nri1434
Swann JB, Smyth MJ (2007) Immune surveillance of tumors. J Clin Invest 117:1137–1146. doi:10.1172/JCI31405
Gabrilovich DI, Nagaraj S (2009) Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 9:162–174. doi:10.1038/nri2506
Bronte V, Zanovello P (2005) Regulation of immune responses by l-arginine metabolism. Nat Rev Immunol 5:641–654. doi:10.1038/nri1668
Rodriguez PC, Ochoa AC (2008) Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol Rev 222:180–191. doi:10.1111/j.1600-065X.2008.00608.x
Rabinovich GA, Gabrilovich D, Sotomayor EM (2007) Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol 25:267–296. doi:10.1146/annurev.immunol.25.022106.141609
Scrimini S, Pons J, Agustí A, Soriano JB, Cosio BG, Torrecilla JA et al (2013) Differential effects of smoking and COPD upon circulating myeloid derived suppressor cells. Respir Med 107:1895–1903. doi:10.1016/j.rmed.2013.08.002
Beasley MB, Brambilla E, Travis WD (2005) The 2004 World Health Organization classification of lung tumors. Semin Roentgenol 40:90–97. doi:10.1053/j.ro.2005.01.001
Lababede O, Meziane M, Rice T (2011) Seventh edition of the cancer staging manual and stage grouping of lung cancer: quick reference chart and diagrams. Chest 139:183–189. doi:10.1378/chest.10-1099
Miller MR, Hankinson J, Brusasco V, Burgos F, Casaburi R, Coates A et al (2005) Standardisation of spirometry. Eur Respir J 26:319–338. doi:10.1183/09031936.05.00034805
Roca J, Sanchis J, Agusti-Vidal A, Segarra F, Navajas D, Rodriguez-Roisin R, Casan P, Sans S (1986) Spirometric reference values for a Mediterranean population. Bull Eur Physiopathol Respir 22:217–224
Poschke I, Kiessling R (2012) On the armament and appearances of human myeloid derived suppressor cells. Clin Immunol 144:250–268. doi:10.1016/j.clim.2012.06.003
Liu CY, Wang YM, Wang CL, Feng PH, Ko HW, Liu YH et al (2010) Population alterations of L-arginase- and inducible nitric oxide synthase-expressed CD11b+/CD14−/CD15+/CD33+ myeloid-derived suppressor cells and CD8+ T lymphocytes in patients with advanced-stage non-small cell lung cancer. J Cancer Res Clin Oncol 136:35–45. doi:10.1007/s00432-009-0634-0
Almand B, Clark JI, Nikitina E, van Beynen J, English NR, Knight SC et al (2001) Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol 166:678–689. doi:10.4049/jimmunol.166.1.678
Gabrilovich DI, Ostrand-Rosenberg S, Bronte V (2012) Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 12:253–268. doi:10.1038/nri3175
Feng PH, Lee KY, Chang YL, Chan YF, Kuo LW, Lin TY et al (2012) CD14(+)S100A9(+) monocytic myeloid-derived suppressor cells and their clinical relevance in non-small cell lung cancer. Am J Respir Crit Care Med 186:1025–1036. doi:10.1164/rccm.201204-0636OC
Gan WQ, Man SF, Senthilselvan A, Sin DD (2004) Association between chronic obstructive pulmonary disease and systemic inflammation: a systematic review and ameta-analysis. Thorax 59:574–580. doi:10.1136/thx.2003.019588
Agustí A, Edwards LD, Rennard SI, MacNee W, Tal-Singer R, Miller BE et al (2012) Persistent systemic inflammation is associated with poor clinical outcomes in COPD: a novel phenotype. PLoS One 7:e37483. doi:10.1371/journal.pone.0037483
Ezernitchi AV, Vaknin I, Cohen-Daniel L, Levy O, Manaster E, Halabi A et al (2006) TCR zeta down-regulation under chronic inflammation is mediated by myeloid suppressor cells differentially distributed between various lymphatic organs. J Immunol 177:4763–4772. doi:10.4049/jimmunol.177.7.4763
Bronstein-Sitton N, Cohen-Daniel L, Vaknin I, Ezernitchi AV, Leshem B, Halabi A et al (2003) Sustained exposure to bacterial antigen induces interferon-γ-dependent T cell receptor ζ down-regulation and impaired T cell function. Nat Immunol 4:957–964. doi:10.1038/ni975
Finn OJ (2008) Cancer immunology. N Engl J Med 358:2704–2715. doi:10.1056/NEJMra072739
Cosio MG, Saetta M, Agusti A (2009) Immunologic aspects of chronic obstructive pulmonary disease. N Engl J Med 360:2445–2454. doi:10.1056/NEJMra0804752
Thomsen M, Dahl M, Lange P, Vestbo J, Nordestgaard BG (2012) Inflammatory biomarkers and comorbidities in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 186:982–988. doi:10.1164/rccm.201206-1113OC
Munder M, Mollinedo F, Calafat J, Canchado J, Gil-Lamaignere C, Fuentes JM et al (2005) Arginase I is constitutively expressed in human granulocytes and participates in fungicidal activity. Blood 105:2549–2556. doi:10.1182/blood-2004-07-2521
Jacobsen LC, Theilgaard-Mönch K, Christensen EI, Borregaard N (2007) Arginase I is expressed in myelocytes/metamyelocytes and localized in gelatinase granules of human neutrophils. Blood 109:3084–3087. doi:10.1182/blood-2006-06-032599
Rotondo R, Bertolotto M, Barisione G, Astigiano S, Mandruzzato S, Ottonello L et al (2011) Exocytosis of azurophil and arginase I-containing granules by activated polymorphonuclear neutrophils is required to inhibit T lymphocyte proliferation. J Leukoc Biol 89:721–727. doi:10.1189/jlb.1109737
Rodriguez PC, Ernstoff MS, Hernandez C, Atkins M, Zabaleta J, Sierra R et al (2009) Arginase I-producing myeloid-derived suppressor cells in renal cell carcinoma are a subpopulation of activated granulocytes. Cancer Res 69:1553–1560. doi:10.1158/0008-5472
Schmielau J, Finn OJ (2001) Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of t-cell function in advanced cancer patients. Cancer Res 61:4756–4760
Cloke T, Munder M, Taylor G, Muller I, Kropf P (2012) Characterization of a novel population of low-density granulocytes associated with disease severity in HIV-1 infection. PLoS One 7(11):e48939. doi:10.1371/journal.pone.0048939
Hoenderdos K, Condliffe A (2013) The neutrophil in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 48:531–539. doi:10.1165/rcmb.2012-0492TR
Acknowledgments
This project was supported, in part, by Acción Transversal en cancer, Instituto de Salud Carlos III (ISCIII); Fondo de Investigación Sanitaria (FIS) 08/673; Asociación Española Contra el Cáncer (AECC, Baleares) and Grups Competitius MAR (Govern Balear) Ref PRE-R-22528-2011. Authors thank all participants for their willingness to contribute to medical research, as well as Angel Rios, Rocío Córdova and Amanda Iglesias for their commitment and quality of their work. Finally, we truly appreciate the reviewers’ comments and observations, and we are certain to that they have contributed significantly to enrich our manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interests
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Scrimini, S., Pons, J., Agustí, A. et al. Expansion of myeloid-derived suppressor cells in chronic obstructive pulmonary disease and lung cancer: potential link between inflammation and cancer. Cancer Immunol Immunother 64, 1261–1270 (2015). https://doi.org/10.1007/s00262-015-1737-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-015-1737-x