
Abstract
Purpose
The objective of this phase IIa, open-label, single-centre, single-arm, two-stage clinical trial was to evaluate the safety and activity of 177-lutetium DOTATATE (LuDO) molecular radiotherapy in neuroblastoma.
Methods
Children with relapsed or refractory metastatic high-risk neuroblastoma were treated with up to four courses of LuDO. The administered activity was 75 to 100 MBq kg−1 per course, spaced at 8- to 12-week intervals. Outcomes were assessed by the International Neuroblastoma Response Criteria (primary outcome), progression-free survival (PFS), and overall survival (OS).
Results
The trial recruited 21 patients; eight received the planned four courses. There was dose-limiting haematologic toxicity in one case, but no other significant haematologic or renal toxicities. None of 14 evaluable patients had an objective response at 1 month after completion of treatment (Wilson 90% CI 0.0, 0.16; and 95% CI is 0.0, 0.22). The trial did not therefore proceed to the second stage. The median PFS was 2.96 months (95% CI 1.71, 7.66), and the median OS was 13.0 months (95% CI 2.99, 21.52).
Conclusion
In the absence of any objective responses, the use of LuDO as a single agent at the dose schedule used in this study is not recommended for the treatment of neuroblastoma. There are several reasons why this treatment schedule may not have resulted in objective responses, and as other studies do show benefit, the treatment should not be regarded as being of no value. Further trials designed to overcome this schedule’s limitations are required.
Trial registration
ISRCTN98918118; URL: https://www.isrctn.com/search?q=98918118
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neuroblastoma most commonly affects younger children [1]. Patients are stratified by age [2], stage [3], and molecular pathology [4] into low-, intermediate-, and high-risk groups [5]. High-risk neuroblastoma requires systemic therapies (induction [6] and high-dose chemotherapy [7,8] followed by immunotherapy [9,10]) and local approaches (surgery [11] and radiotherapy [12]). Around 25% respond poorly to induction chemotherapy, or may progress on initial treatment, and require alternative treatments before high-dose (myeloablative) chemotherapy [13]. These are referred to as refractory patients. Others may respond well initially, but relapse before or following high-dose chemotherapy, and are referred to as relapsed patients [14]. Patients in both groups have low survival rates, so new treatments are needed.
Neuroblastoma is radiosensitive [15], and radiotherapy is important [16]; however, full coverage of disseminated disease with external beam radiotherapy is not feasible. Molecular radiotherapy can potentially target all cells expressing the relevant receptor. 131-Iodine-labelled meta-iodobenzylguanidine (131I-mIBG) therapy [17], targeting the norepinephrine transporter molecule [18], is widely used [19].
Peptide receptor radionuclide therapy (PRRT) targets somatostatin receptors. Radiolabelled somatostatin analogues are used for adult neuroendocrine cancers [20,21]. 177-Lutetium DOTATATE (LuDO) prolongs survival in metastatic gastroenteropancreatic neuroendocrine tumours [22,23]. Toxicity is limited to myelotoxicity and nephrotoxicity [24].
Neuroblastoma cells express somatostatin receptors [25]. We used 68-gallium DOTATATE (GaDO) positron emission tomography (PET) computed tomography (CT) imaging to select patients with relapsed or refractory neuroblastoma with a high level of GaDO avidity, and treated six with LuDO molecular radiotherapy [26]. Two had partial metabolic responses. The role of PRRT in neuroblastoma has been reviewed by others [27].
Objectives
The objective of the study was to evaluate the safety and activity of LuDO molecular radiotherapy in metastatic high-risk relapsed or refractory neuroblastoma.
Patients and methods
Trial design
This is a phase IIa, open-label, single-centre, single-arm, two-stage clinical trial.
Patients
Patients (aged > 18 months and < 18 years) with histologically confirmed relapsed or refractory metastatic high-risk neuroblastoma were eligible if they had an estimated life expectancy > 3 months and good performance status (Lansky > 50% for patients ≤ 12 years of age; Karnofsky > 50% for those > 12 years of age) and had recovered from any major prior surgery. Tracer uptake in tumour deposits, measured by SUVmax on GaDO PET CT, had to be at least as high as in the liver. A 2-week washout from any prior treatment, with haematologic recovery, was required. Confirmation of adequate organ function was required prior to commencing trial treatment. Written informed consent from parents, or patients if aged 18 years or older, and agreement to abide by the radiation protection rules for comforters and carers [28] were required.
Exclusion criteria included as follows: patients not fit enough to undergo treatment; pregnant or lactating patients; concurrent treatment with any anti-tumour agents; and prior treatment with radiolabelled somatostatin analogues (any other prior treatment was permitted).
Disease and normal organ function assessments
Baseline disease assessments included 123I-mIBG scintigraphy, which was scored according to SIOPEN criteria [29]; GaDO PET CT; cross-sectional imaging with CT or magnetic resonance imaging (MRI) of the primary tumour or bulky metastatic sites if appropriate; urinary catecholamine metabolites; and bone marrow aspirates and trephine biopsies. Routine haematology and biochemistry profiles and glomerular filtration rate (GFR) measurement were also performed. A pregnancy test was required in females of reproductive potential.
All patients were imaged with GaDO PET CT according to local protocol, on a Discovery STE PET CT system (GE Healthcare, Chicago, IL, USA). Patients were injected intravenously with at least 100 MBq GaDO for adequate image quality. Imaging took place 45 to 60 min after injection. A total body protocol for transmission and emission imaging was acquired. CT was performed at 80–140 kVp and modulated mA. PET was performed in 3-dimensional mode with 4 min per bed position. Iterative reconstruction with 21 subsets was performed with attenuation correction. Patients who required general anaesthesia had images acquired with the same protocol.
Following each course of treatment, routine bloods were performed weekly. A GFR was calculated prior to subsequent courses [30]. The GaDO and 123I-mIBG imaging were performed prior to the third course of treatment, to ensure that no further LuDO was given if the disease had progressed. One month following completion of the planned four courses of LuDO, reassessments were performed as at baseline.
Treatment schedule
Patients were admitted to a radiation protection suite on the paediatric oncology ward, and remained there, as inpatients, until there were no longer any radiation restrictions. The investigational medicinal product LuDO ([177Lu-DOTA0,Tyr3]octreotate; United States Adopted Name (USAN) lutetium (177Lu) DOTATATE; also called lutetium oxodotreotide in Europe; brand name Lutathera®) was supplied by Advanced Accelerator Applications, Saint-Genis-Pouilly, France, a Novartis company. Hydration with 0.9% sodium chloride 3 l m−2 24 h−1 was commenced 6 h prior to the LuDO administration and continued for 24 h. Thirty minutes before treatment, anti-emetics were given, and an amino acid solution infusion (l-lysine hydrochloride 2.5% w/v, l-arginine hydrochloride 2.5% in water for injection) 1 l 1.73 m−1 was commenced and administered over 4 h (that is 250 ml 1.73 m−1 h−1) to protect the kidneys. The LuDO was administered intravenously over about 20 min.
The aim was to give a total of four courses. The inter-course interval was a minimum of 8 weeks, and this could be extended to a maximum of 12 weeks if necessary to allow for haematologic recovery.
Dose prescription
The dosing schedule was designed to mitigate the main potential toxicities of LuDO: myelosuppression and late nephrotoxicity. Dosimetry was performed in respect of both whole-body radiation absorbed dose, which may correlate with haematological toxicity, and kidney radiation absorbed dose, which may correlate with nephrotoxicity. Blood dose was not measured. We recognize that the relationship between absorbed dose and toxicity is controversial. With 131I-mIBG therapy for neuroblastoma, some authors have demonstrated a relationship [31], while others have not [32]. The aim was to err on the side of safety and tolerability and to administer an activity of LuDO per course which would result in a whole-body dose of about 0.5 Gy, with a cumulative whole-body dose of about 2 Gy over four courses, so that peripheral blood stem cell support was not required; and not to exceed a cumulative renal dose of 23 Gy, to avoid nephrotoxicity. No account was taken of any prior external beam radiotherapy which may have affected renal function, except for ensuring that all patients had an adequate GFR before trial entry. Toxicity and dose-limiting toxicity criteria are shown in Table 1.
For the first administration, a weight-based activity of 75 MBq kg−1 was used. Following administration, a series of radiation measurements were taken using a ceiling-mounted monitor according to a standard protocol to enable the whole-body radiation dose to be calculated. Subsequently, over the next 4 days, a series of at least three whole-body scans were acquired to enable the whole-body dose to be verified by a second method, as well as to confirm uptake in known areas of disease. In addition, single-photon emission computed tomography (SPECT) CT scans were performed to allow tumour and normal organs (principally the kidney) absorbed radiation doses to be estimated.
The administered activity for the second and subsequent courses was calculated on the basis of the whole-body dose measured following the previous administration, and the haematological toxicity (Table 1).
Dosimetry
Whole-body dosimetry was performed using in-room ceiling scintillation monitors (Southern Scientific Ltd., Henfield, UK). The first measurement was performed immediately after administration of LuDO, prior to the patient emptying his or her bladder. Subsequent measurements were taken by comforters and carers, and activity at these time points normalized to the first measurement. For each fraction of treatment, a time-activity graph was generated and the whole-body dose calculated from the product of cumulated activity and relevant s-factor.
Whole-body planar scans and SPECT CT were obtained after treatment to evaluate tumour and normal organ-at-risk dosimetry and to confirm the whole-body dose. All patients who had dosimetric analysis had 3–5 imaging sessions after each fraction of treatment. All images were acquired on a Discovery 670 Gamma Camera (GE Healthcare, Chicago, IL, USA). Whole-body planar scans were obtained with an emission window of 208 keV ± 10%. SPECT CT was performed with the same emission window and triple-energy scatter correction. A total of 120 projections were acquired at 30 s per projection. Iterative reconstruction was used (20 subsets) with attenuation and scatter correction. Dosimetry was performed by contouring on the CT transaxial slices and transposing to the SPECT dataset. Based on phantom SPECT sensitivity measurements, the activity in the organ at each time point was calculated so that a time-activity curve could be generated. Dose was calculated by multiplying the cumulated activity by the relevant s-factor.
Outcomes
The primary outcome measure was response (complete, very good partial, and partial) assessed by the International Neuroblastoma Response Criteria (INRC) [33,34] at 1 month after the completion of therapy, defined as the last administration of LuDO. Patients who did not have response assessment at 1 month after the completion of therapy were considered non-responders. The data were also analyzed by the recently published Revisions to the INRC [35].
Secondary outcome measures were as follows: toxicity, progression-free survival (PFS), overall survival (OS), and response by INRC at 4 months after the completion of therapy (defined as per primary outcome measure). Toxicity was assessed according to Common Terminology Criteria for Adverse Events (CTCAE) version 4.0. PFS time was defined as the time from trial inclusion until objective tumour progression, death without progression, or second malignancy. OS time was calculated from the date of inclusion into the trial until date of death. For all survival outcomes, patients who did not experience an event during trial follow-up were censored at their last assessment date.
Sample size
The sample size calculation was based on a Simon two-stage minimax design [36]. A rate of 40% or more was defined at the outset as the acceptable level of response. A response rate of 20% or less was considered unacceptably low. The probability of obtaining a false negative result, β (i.e. incorrectly rejecting for further study a treatment with a true response rate of ≥ 40%), was set at 20%. The probability of obtaining a false positive result, α (i.e. incorrectly accepting for further study a treatment that has a true response rate of ≤ 20%), was set at 10%. Stage 1 required 14 eligible and evaluable patients, with a minimum of three responders to proceed to stage 2. A further 10 patients were needed to be recruited in stage 2. A minimum of 8 out of 24 responses would indicate that the treatment schedule was active and should go onto further studies to evaluate efficacy (while taking account of toxicity). Sample size calculations were performed using Sample Size Tables for Clinical Trials [37].
Statistical methods
The analysis of the primary outcome measure was carried out on an eligible and evaluable patient basis. Patients who did not start treatment, and those who died due to any cause within 3 months from registration (i.e. time between death date and registration date is less than 92 days), were excluded from this analysis. All other analyses were carried out according to an intention-to-treat (ITT) principle, defined as all patients registered in the trial. Descriptive statistics were used to summarize baseline characteristics, treatment, report harms, and response outcomes. Data is expressed as mean (standard deviation (SD)), median (minimum, maximum), or number (percent). Kaplan-Meier plots with median survival and estimate at 6 and 12 months are presented. Two-sided confidence intervals (CIs) at 90% and 95% levels are reported; for the response outcome, one- and two-sided Wilson CIs were calculated.
Ethical and research governance considerations
This trial was conducted in accordance with the recommendations of guiding physicians in biomedical research involving human subjects, adopted by the 18th World Medical Association General Assembly, Helsinki, Finland, June 1964, amended at the 48th World Medical Association General Assembly, Somerset West, Republic of South Africa, October 1996; and with the Research Governance Framework for Health and Social Care, UK law, and the applicable UK Statutory Instruments. The trial received a favorable opinion from the National Research Ethics Service’s London – Hampstead Research Ethics Committee; a Clinical Trial Authorization from the Medicines and Healthcare products Regulatory Agency (MHRA); approval from the Administration of Radioactive Substances Advisory Committee (ARSAC); and local Research And Development approval. The Sponsor was the University of Birmingham, and data management and monitoring was undertaken by the Cancer Research UK Clinical Trials Unit at the University of Birmingham. Parents or legal guardians of patients, or patients if aged over 18 years, were given verbal and written information about the trial to consider for a period of at least 1 day, before being asked to give written informed consent for trial entry at a subsequent consultation.
An independent Data Monitoring Committee was established to review safety and outcome data at six monthly intervals. The trial terminated recruitment when the data for the primary outcome measure for the eligible and evaluable patient population was available on the database for stage 1 of the trial and the number of responders was insufficient to carry on to stage 2.
The trial was registered in the European Clinical Trials Database (EudraCT number 2012-000510-10), and in the International Standard Randomized Controlled Trials Number Registry (ISRCTN98918118).
Results
Patient population
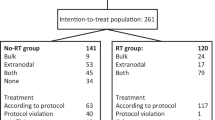
In total, 43 patients were screened. Between 19 September 2013 and 28 July 2017, 21 patients were registered in the trial; all had 123I-mIBG-positive disease on imaging. Participants were followed up after the end of treatment until progression of disease and death. At the date of data analysis, 14 September 2018, 18 patients had died from their disease. Median follow-up for alive patients was 17.3 months (13.1 to 37.4). The other 22 patients were not entered because of the following: low SUVmax on the GaDO scan (11); rapid disease progression, poor performance status or estimated life expectancy, or out of range organ function (6); a medical decision for alternative treatment (3); or parental wishes (2). Figure 1 is a CONSORT diagram of patients screened for this study.

CONSORT diagram of all patients enrolled in this study
Table 2 shows patient demographics and disease characteristics at baseline.
Treatment delivery
Of the 21 registered patients, 20 received at least one course of LuDO treatment. One patient was withdrawn before the start of treatment because of progressive disease. The median time between enrollment and start of treatment for the 20 patients was 3 days (range 0–26 days). Of the 20 treated patients, eight received four courses of LuDO, two received two courses, and ten received only one course. The reasons for early discontinuation were progression or death in 11 and severe haematological toxicity in one. A total of 46 courses of treatment were given.
The administered activity of LuDO was 75 MBq kg−1 for the first course, in all twenty treated patients. The administered activity in all subsequent courses was 100 MBq kg−1, because no patients needed dose modifications for toxicity, other than one patient who stopped treatment because of severe haematological toxicity.
Safety and toxicity
There was no treatment-related mortality. There were 151 adverse events (AE) among 19 of the 20 treated patients. The median AE number was 6.5 (range 0–20). The highest grade of AE was grade 1 in five patients, grade 3 in 12 patients, and grade 4 in 2 patients. Grade 3 and 4 toxicities are listed in Table 3. There were 10 serious adverse events (SAE) reported in six patients. Four patients experienced one SAE, and two patients experienced three SAE each. In each case, the reason for SAE reporting was unexpected hospitalization. In nine cases, the SAE was categorized as being unrelated to trial treatment. The one serious adverse reaction was caused by neutropenic sepsis. There were no serious unexpected serious adverse reactions. There was one case of dose-limiting haematological toxicity. This was after the first course of treatment, when an administered activity of 75 MBq kg−1 resulted in a whole-body dose of only 0.21 Gy, below the median. The observed haematological toxicity may have been due to concurrent use of myelosuppressive antibiotics, rather than an actual side effect of the trial treatment. No significant renal toxicity was observed.
SUVmax uptake levels and whole-body and tumour dosimetry
Only patients with a higher somatostatin receptor expression in tumour than in the normal liver were included in this trial. The median SUVmax for the liver measured on pre-treatment GaDO PET CT scans was 4.7 (range 2.1 to 9.0), and the median highest tumour SUVmax measurement for each patient was 11.2 (range 5.7 to 44.2).
The median whole-body radiation absorbed dose (as estimated from the ceiling-mounted monitor data) was 0.24 Gy (range 0.14 to 0.42 Gy) following the first course when the administered activity was 75 MBq kg−1, and 0.33 Gy (range 0.16 to 0.61 Gy) following subsequent courses when the administered activity was 100 MBq kg−1. The median cumulative whole-body absorbed dose in those patients who received all four courses for whom complete data were available was 1.26 Gy (range 0.97 to 1.48 Gy).
The median cumulative mean renal radiation dose in the six patients who received four courses of treatment and for whom full dosimetric data were recorded was 16.5 Gy (range 9.5 to 21.5 Gy).
The tumour dose per course was measured in 24 lesions, and the median was 2.43 Gy (range 0.04 to 13.50 Gy).
Response
Of the 20 treated patients, six were considered ineligible for the primary outcome assessment as they died within 92 days of enrollment: median 62 days, range 28–91 days. This left 14 evaluable patients, of whom none had a response either by the original or by the revised INRC criteria at 1 month after completion of treatment (Wilson 90% CI 0.0, 0.16; and 95% CI is 0.0, 0.22). Of these 14, eight patients had progressed and six were non-responders at that time point. In view of this, the trial was closed and did not move on to the second stage. Of 21 patients analyzed for the secondary outcome measure, response at 4 months post end of treatment, no patients responded (Wilson 90% CI 0.0, 0.11; and 95% CI is 0.0, 0.16): one was non-responder, one did not start treatment, and 19 had progressive disease. Eight patients had stable disease at reassessment after two cycles of treatment, and four had stable disease at reassessment after four cycles of treatment.
Survival
For all 21 patients, one (5%) patient had no event, two (10%) progressed without death, and 18 (85%) progressed and died. The median PFS was 2.96 months (95% CI 1.71, 7.66). The PFS at 6 months was 38% (95% CI 18%, 58%) and PFS at 12 months was 5% (95% CI 0%, 20%) (Fig. 2).

Progression-free survival
At the time of analysis, 18 of 21 patients had died. The median OS for the 21 patients was 13.0 months (95% CI 2.99, 21.52). The OS at 6 months was 62% (95% CI 38%, 79%) and OS at 12 months was 52% (95% CI 30%,71%) (Fig. 3).

Overall survival
At least ten of the patients went on to receive other treatments following progression.
An unplanned analysis assessed PFS and OS for 13 relapsed patients and for the eight refractory patients (Table 2). For relapsed patients, the median PFS was 1.87 months (95% CI 0.72, 82.96) and the median OS was 5.45 months (95% CI 2.23, 17.31). For refractory patients, the median PFS was 7.66 months (95% CI 1.71, 10.78) and the median OS was 24.74 (95% CI 1.87, 27.89) months.
Although no response by the INRC criteria was noted at 1 month after completion of treatment, an unplanned analysis showed a reduction in the SIOPEN semi-quantitative 123I-mIBG scan skeletal score in three patients between baseline and prior to the third administration of LuDO. The paired values were 9, 5; 19, 18; and 17, 12.
Discussion
This is the first formal phase II trial of LuDO molecular radiotherapy in children with neuroblastoma. As no objective responses by the INRC criteria were seen in 14 evaluable patients, we do not recommend the use of LuDO as a single agent at the dose schedule used in this study for the treatment of patients with neuroblastoma. This is disappointing, as we have seen responses in patients treated outside this trial [26,38], and an independent group observed objective responses in all of four patients treated similarly [39]. In this trial, we did observe an objective improvement on independent blinded 123I-mIBG semi-quantitative scoring after two courses of treatment in three patients, although this was insufficient to count as a partial response and was not sustained at the primary evaluation time point.
The poor survival observed in this trial is in keeping with that seen in similar patients enrolled in other early phase clinical trials. A meta-analysis of European phase II trials in neuroblastoma showed a median PFS of 5.7 months and a median OS of 11.0 months in relapsed disease, compared with our figures of 1.87 and 5.45 months respectively. In refractory disease, the median PFS was 12.5 months and median OS was 27.9 months, compared with our figures of 7.66 and 24.74 months respectively [40]. The better survival of refractory patients compared with relapsed patients is expected [41].
We feel it is important to report our data for these subgroups, even though the analysis was not planned at the outset, for several reasons. There is a growing recognition in the literature that results vary between refractory patients who have failed to respond adequately to induction chemotherapy, and move onto alternative treatments, and those who relapse after a good initial response chemotherapy [40,41]. Therefore, overall results may differ depending on the relative proportions of the two groups. We have previously observed that studies which fail to report the two groups separately limit the value of the data presented [19].
It is important to note (Fig. 1), as this is an imaging biomarker–driven study, that 11 of 43 patients screened did not have adequate uptake as measured by tumour SUVmax on the baseline GaDO PET CT scan to allow entry to the trial. This means that a significant proportion of patients will not be eligible for inclusion in any future trials of this agent. While all patients included in the trial had a higher tumour SUVmax than the liver, it is possible that those with a greater somatostatin receptor expression might have better outcomes; however, as no patients met the pre-defined response criteria, it is not possible to say from this patient population whether or not that was in fact the case.
There may be several reasons why, in this trial, LuDO did not prove to be as effective as anticipated:
The scheduling at two monthly intervals, based on adult neuroendocrine tumour experience, may have been too spaced out for a rapidly proliferating tumour like neuroblastoma, and permitted repopulation between treatments.
Only one patient was withdrawn from study treatment because of haematological toxicity. The measured cumulative renal radiation dose was in all cases lower than the 23-Gy objective, and at 16.5 Gy, the median value was about 70% of that. The measured cumulative whole-body dose was in all cases lower than the 2-Gy objective, and at 1.26, the median value was 63% of that. These factors indicate that with personalized dosing, the administered activity could have been substantially increased in many patients without excess toxicity.
Our observed whole-body doses are substantially less than the 4 Gy we use routinely in 131I-mIBG therapy with peripheral blood stem cell support [42]. Tumour doses measured after administration of 131I-mIBG to a whole-body dose of 4 Gy may vary by an order of magnitude—from about 10 to 100 Gy, depending on the avidity of the tumour and retention of the radiopharmaceutical over time [43]. Our tumour dose measurements using the described schedule of LuDO administration also vary widely, but with a median of 2.43 Gy per course, are substantially below those seen with 131I-mIBG therapy.
We have demonstrated heterogeneity in somatostatin receptor expression and a lack of concordance between somatostatin and norepinephrine transporter expression in tissue bank samples [25]. Using PET CT and scintigraphy, we have sometimes observed anatomical disparity in the distribution of neuroblastoma deposits taking up GaDO and 123I-mIBG; and another group has reported similar findings [39]. This microscopic and macroscopic diversity may be a factor contributing to a poor response to single-agent LuDO. We may hypothesize that a combination of LuDO and mIBG therapy may overcome this limitation and be more effective than either agent alone.
The concomitant use of radiation sensitizers has been demonstrated in laboratory models to potentiate mIBG therapy [44,45], and synergy between radiosensitizing drugs and LuDO has also been similarly shown [46].
Despite the negative result of this study, we believe that PRRT in general, and LuDO in particular, may have value as a treatment for neuroblastoma. Our view is supported by the fact that other groups are planning trials of different theranostic imaging and treatment radiopharmaceuticals targeting SSTR, for example 64-copper SARTATE PET imaging and 67-copper SARTATE molecular radiotherapy [47]. We propose further clinical trials bringing together increased activities of LuDO with mIBG in a dose-dense schedule, as this may potentially overcome some of the limitations discussed here. When an optimal dose schedule has been identified, further studies on the addition of radiation sensitizers may be considered.
Abbreviations
- AE:
-
Adverse event
- CT:
-
Computed tomography
- CTCAE:
-
Common Terminology Criteria for Adverse Events
- FDG:
-
Fluorodeoxyglucose
- GaDO:
-
68-Gallium DOTATATE
- GFR:
-
Glomerular filtration rate
- Gy:
-
Gray
- h:
-
Hour
- INRC:
-
International Neuroblastoma Response Criteria
- kg:
-
Kilogram
- kVp:
-
Kilovoltage peak
- l:
-
Litre
- L:
-
Levo
- LuDO:
-
177-Lutetium DOTATATE
- m:
-
Metre
- mA:
-
Milliamperes
- MBq:
-
Megabecquerel
- mIBG:
-
Meta-iodobenzylguanidine
- MRI:
-
Magnetic resonance imaging
- OS:
-
Overall survival
- PET:
-
Positron emission tomography
- PFS:
-
Progression-free survival
- PRRT:
-
Peptide receptor radionuclide therapy
- SAE:
-
Serious adverse event
- SIOPEN:
-
International Society of Paediatric Oncology European Neuroblastoma clinical research group
- SPECT:
-
Single-photon emission computed tomography
- SUVmax :
-
Maximum standardized uptake value
- UK:
-
United Kingdom
- USA:
-
United States of America
- USAN:
-
United States Adopted Name
References
Gains J, Mandeville H, Cork N, et al. Ten challenges in the management of neuroblastoma. Future Oncol. 2012;8:839–58.
Moroz V, Machin D, Faldum A, et al. Changes over three decades in outcome and the prognostic influence of age-at-diagnosis in young patients with neuroblastoma: a report from the International Neuroblastoma Risk Group Project. Eur J Cancer. 2011;47:561–71.
Monclair T, Brodeur GM, Ambros PF, et al. The International Neuroblastoma Risk Group (INRG) staging system: an INRG Task Force report. J Clin Oncol. 2009;27:298–303.
Bagatell R, Beck-Popovic M, London WB, et al. Significance of MYCN amplification in international neuroblastoma staging system stage 1 and 2 neuroblastoma: a report from the International Neuroblastoma Risk Group database. J Clin Oncol. 2009;27:365–70.
Cohn SL, Pearson ADJ, London WB, et al. The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. J Clin Oncol. 2009;27:289–97.
Pearson AD, Pinkerton CR, Lewis IJ, et al. High-dose rapid and standard induction chemotherapy for patients aged over 1 year with stage 4 neuroblastoma: a randomised trial. Lancet Oncol. 2008;9:247–56.
Matthay KK, Reynolds CP, Seeger RC, et al. Long-term results for children with high-risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13-cis-retinoic acid: a Children’s Oncology Group study. J Clin Oncol. 2009;27:1007–13.
Ladenstein R, Pötschger U, Pearson ADJ, et al. Busulfan and melphalan versus carboplatin, etoposide, and melphalan as high-dose chemotherapy for high-risk neuroblastoma (HR-NBL1/SIOPEN): an international, randomised, multi-arm, open-label, phase 3 trial. Lancet Oncol. 2017;18:500–14.
Yu AL, Gilman AL, Ozkaynak MF, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med. 2010;363:1324–34.
Ladenstein R, Pötschger U, Valteau-Couanet D, et al. Interleukin 2 with anti-GD2 antibody ch14.18/CHO (dinutuximab beta) in patients with high-risk neuroblastoma (HR-NBL1/SIOPEN): a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018. https://doi.org/10.1016/S1470-2045(18)30578-3.
von Allmen D, Davidoff AM, London WB, et al. Impact of extent of resection on local control and survival in patients from the COG A3973 study with high-risk neuroblastoma. J Clin Oncol. 2017;35:208–16.
Arumugam S, Manning-Cork NJ, Gains JE, et al. The evidence for external beam radiotherapy in high-risk neuroblastoma of childhood: a systematic review. Clin Oncol (R Coll Radiol). 2018. https://doi.org/10.1016/j.clon.2018.11.031.
Ladenstein R, Valteau-Couanet D, Brock P, et al. Randomized trial of prophylactic granulocyte colony-stimulating factor during rapid COJEC induction in pediatric patients with high-risk neuroblastoma: the European HR-NBL1/SIOPEN study. J Clin Oncol. 2010;28:3516–24.
Basta NO, Halliday GC, Makin G, et al. Factors associated with recurrence and survival length following relapse in patients with neuroblastoma. Br J Cancer. 2016;115:1048–57.
Wheldon TE, Livingstone A, Wilson L, et al. The radiosensitivity of human neuroblastoma cells estimated from regrowth curves of multicellular tumour spheroids. Br J Radiol. 1985;58:661–4.
Gaze MN, Boterberg T, Dieckmann K, et al. Results of a quality assurance review of external beam radiation therapy in the International Society of Paediatric Oncology (Europe) Neuroblastoma Group’s High-risk Neuroblastoma Trial: a SIOPEN study. Int J Radiat Oncol Biol Phys. 2013;85:170–4.
Gaze MN, Gains JE, Walker C, et al. Optimization of molecular radiotherapy with [131I]-meta iodobenzylguanidine for high-risk neuroblastoma. Q J Nucl Med Mol Imaging. 2013;57:66–78.
Glowniak JV, Kilty JE, Amara SG, et al. Evaluation of metaiodobenzylguanidine uptake by the norepinephrine, dopamine and serotonin transporters. J Nucl Med. 1993;34:1140–6.
Wilson JS, Gains JE, Moroz V, et al. A systematic review of 131I-meta iodobenzylguanidine molecular radiotherapy for neuroblastoma. Eur J Cancer. 2014;50:801–15.
Kwekkeboom DJ, Mueller-Brand J, Paganelli G, et al. Overview of results of peptide receptor radionuclide therapy with 3 radiolabeled somatostatin analogs. J Nucl Med. 2005;46:62S–6S.
Bodei L, Mueller-Brand J, Baum RP, et al. The joint IAEA, EANM, and SNMMI practical guidance on peptide receptor radionuclide therapy (PRRNT) in neuroendocrine tumours. Eur J Nucl Med Mol Imaging. 2013;40:800–16.
Strosberg J, El-Haddad G, Wolin E, et al. Phase 3 trial of 177Lu-Dotatate for midgut neuroendocrine tumors. N Engl J Med Jan. 2017;376:125–35.
Strosberg J, Wolin E, Chasen B, et al. Health-related quality of life in patients with progressive midgut neuroendocrine tumors treated with 177Lu-Dotatate in the phase III NETTER-1 trial. J Clin Oncol. 2018;36:2578–84.
Bodei L, Kidd M, Paganelli G, et al. Long-term tolerability of PRRT in 807 patients with neuroendocrine tumours: the value and limitations of clinical factors. Eur J Nucl Med Mol Imaging. 2015;42:5–19.
Gains JE, Sebire NJ, Moroz V, et al. Immunohistochemical evaluation of molecular radiotherapy target expression in neuroblastoma tissue. Eur J Nucl Med Mol Imaging. 2018;45:402–11.
Gains JE, Bomanji JB, Fersht NL, et al. 177Lu-DOTATATE molecular radiotherapy for childhood neuroblastoma. J Nucl Med. 2011;52:1041–7.
Alexander N, Vali R, Ahmadzadehfar H, et al. The role of radiolabeled DOTA-conjugated peptides for imaging and treatment of childhood neuroblastoma. Curr Radiopharm. 2018;11:14–21.
Gains JE, Walker C, Sullivan TM, et al. Radiation exposure to comforters and carers during paediatric molecular radiotherapy. Pediatr Blood Cancer. 2015;62:235–9.
Lewington V, Lambert B, Poetschger U, et al. 123I-mIBG scintigraphy in neuroblastoma: development of a SIOPEN semi-quantitative reporting method by an international panel. Eur J Nucl Med Mol Imaging. 2017;44:234–41.
Schwartz GJ, Work DF. Measurement and estimation of GFR in children and adolescents. Clin J Am Soc Nephrol. 2009;4:1832–43.
Buckley SE, Chittenden SJ, Saran FH, et al. Whole-body dosimetry for individualized treatment planning of 131I-MIBG radionuclide therapy for neuroblastoma. J Nucl Med. 2009;50:1518–24.
Trieu M, DuBois SG, Pon E, et al. Impact of whole-body radiation dose on response and toxicity in patients with neuroblastoma after therapy with 131 I-metaiodobenzylguanidine (MIBG). Pediatr Blood Cancer. 2016;63:436–42.
Brodeur GM, Seeger RC, Barrett A, et al. International criteria for diagnosis, staging, and response to treatment in patients with neuroblastoma. J Clin Oncol. 1988;6:1874–81.
Brodeur GM, Pritchard J, Berthold F, et al. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J Clin Oncol. 1993;11:1466–77.
Park JR, Bagatell R, Cohn SL, et al. Revisions to the International Neuroblastoma Response Criteria: a consensus statement from the National Cancer Institute Clinical Trials Planning Meeting. J Clin Oncol. 2017;35:2580–7.
Simon R. Optimal two-stage designs for phase II clinical trials. Control Clin Trials. 1989;10:1–10.
Machin D, Campbell MJ, Tan SB, et al. Sample size tables for clinical studies (ed 3). Chichester: Wiley-Blackwell; 2009.
Gaze MN, Gains JE, Bomanji JB. Current issues in molecular radiotherapy in children. In: Mansi L, Lopci E, Cuccurullo V, Chiti A, editors. Clinical nuclear medicine in pediatrics. Basel: Springer; 2016. p. 29–49.
Kong G, Hofman MS, Murray WK, et al. Initial experience with gallium-68 DOTA-octreotate PET/CT and peptide receptor radionuclide therapy for pediatric patients with refractory metastatic neuroblastoma. J Pediatr Hematol Oncol. 2016;38:87–96.
Moreno L, Rubie H, Varo A, et al. Outcome of children with relapsed or refractory neuroblastoma: a meta-analysis of ITCC/SIOPEN European phase II clinical trials. Pediatr Blood Cancer. 2017;64:25–31.
Zhou MJ, Doral MY, DuBois SG, et al. Different outcomes for relapsed versus refractory neuroblastoma after therapy with (131)I-metaiodobenzylguanidine ((131)I-MIBG). Eur J Cancer. 2015;51:2465–72.
Gaze MN, Chang YC, Flux GD, et al. Feasibility of dosimetry-based high-dose 131I-meta-iodobenzylguanidine with topotecan as a radiosensitizer in children with metastatic neuroblastoma. Cancer Biother Radiopharm. 2005;20:195–9.
Buckley SE, Saran FH, Gaze MN, et al. Dosimetry for fractionated (131)I-mIBG therapies in patients with primary resistant high-risk neuroblastoma: preliminary results. Cancer Biother Radiopharm. 2007;22:105–12.
McCluskey AG, Boyd M, Gaze MN, et al. [131I]MIBG and topotecan: a rationale for combination therapy for neuroblastoma. Cancer Lett. 2005;228:221–7.
McCluskey AG, Mairs RJ, Tesson M, et al. Inhibition of poly(ADP-Ribose) polymerase enhances the toxicity of 131I-metaiodobenzylguanidine/topotecan combination therapy to cells and xenografts that express the noradrenaline transporter. J Nucl Med. 2012;53:1146–54.
Tesson M, Vasan R, Hock A, et al. An evaluation in vitro of the efficacy of nutlin-3 and topotecan in combination with 177Lu-DOTATATE for the treatment of neuroblastoma. Oncotarget. 2018;9:29082–96.
https://clinicaltrials.gov/ct2/show/NCT04023331?term=sartate&rank=2 [last accessed 30 September 2019].
Funding
This trial was funded by the Cancer Research UK (reference C17807/A14091) and Joining Against Cancer in Kids, and supported by researchers at the National Institute for Health Research University College London Hospitals Biomedical Research Centre.
Author information
Authors and Affiliations
Contributions
Conception and design: Jennifer E. Gains, Veronica Moroz, Matthew D. Aldridge, Jamshed B. Bomanji, Keith Wheatley, Mark N. Gaze
Clinical care of patients: Jennifer E. Gains, Connie Peet, Matthew D. Aldridge, S Wan, Jamshed B. Bomanji, Mark N. Gaze
Collection and assembly of data: Connie Peet, Matthew D. Aldridge, Jennifer Laidler
Data analysis and interpretation: Jennifer E. Gains, Veronica Moroz, Simon Wan, Jamshed B. Bomanji, Matthew D. Aldridge, Keith Wheatley, Mark N. Gaze
Manuscript writing: all authors
Final approval of the manuscript: all authors
Accountable for all aspects of work: all authors
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Pediatric
Rights and permissions
About this article

Cite this article
Gains, J.E., Moroz, V., Aldridge, M.D. et al. A phase IIa trial of molecular radiotherapy with 177-lutetium DOTATATE in children with primary refractory or relapsed high-risk neuroblastoma. Eur J Nucl Med Mol Imaging 47, 2348–2357 (2020). https://doi.org/10.1007/s00259-020-04741-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00259-020-04741-x