
Abstract
Purpose
Late-life depression even in subsyndromal stages is strongly associated with Alzheimer’s disease (AD). Furthermore, brain amyloidosis is an early biomarker in subjects who subsequently suffer from AD and can be sensitively detected by amyloid PET. Therefore, we aimed to compare amyloid load and glucose metabolism in subsyndromally depressed subjects with mild cognitive impairment (MCI).
Methods
[18F]AV45 PET, [18F]FDG PET and MRI were performed in 371 MCI subjects from the Alzheimer’s Disease Neuroimaging Initiative Subjects were judged β-amyloid-positive (Aβ+; 206 patients) or β-amyloid-negative (Aβ−; 165 patients) according to [18F]AV45 PET. Depressive symptoms were assessed by the Neuropsychiatric Inventory Questionnaire depression item 4. Subjects with depressive symptoms (65 Aβ+, 41 Aβ−) were compared with their nondepressed counterparts. Conversion rates to AD were analysed (mean follow-up time 21.5 ± 9.1 months) with regard to coexisting depressive symptoms and brain amyloid load.
Results
Aβ+ depressed subjects showed large clusters with a higher amyloid load in the frontotemporal and insular cortices (p < 0.001) with coincident hypermetabolism (p < 0.001) in the frontal cortices than nondepressed subjects. Faster progression to AD was observed in subjects with depressive symptoms (p < 0.005) and in Aβ+ subjects (p < 0.001). Coincident depressive symptoms additionally shortened the conversion time in all Aβ+ subjects (p < 0.005) and to a greater extent in those with a high amyloid load (p < 0.001).
Conclusion
Our results clearly indicate that Aβ+ MCI subjects with depressive symptoms have an elevated amyloid load together with relative hypermetabolism of connected brain areas compared with cognitively matched nondepressed individuals. MCI subjects with high amyloid load and coexistent depressive symptoms are at high risk of faster conversion to AD.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is imposing an onerous burden on health-care systems in societies with ageing populations [1]. Neurofibrillary tangles and amyloid plaques together comprise the hallmark neuropathology of AD [2]. Elevated brain amyloid burden is clearly associated with cognitive decline in the healthy elderly [3] and in individuals with mild cognitive impairment (MCI) [4]. Recent studies have shown that late-life depression is associated with MCI [5] and with AD [6]. A higher relative risk of conversion from cognitively normal to MCI and to a lesser degree from MCI to AD [7] has been demonstrated in subsyndromally depressed elderly patients (rated by the Neuropsychiatric Inventory Questionnaire, NPI-Q). However, the literature remains inconclusive as to whether late-life depression is a risk factor for emergence of AD, or whether late-life depression, as an early symptom of AD, is implicated in the pathophysiology of Alzheimer’s type dementia. Previous investigations regarding the link between depression and brain amyloidosis have mainly focused on subjects who had previously had depressive episodes [8–10], and have mostly found elevated β-amyloid (Aβ) levels. Despite these roughly consistent amyloid PET findings, there is less concordance among [18F]FDG PET studies in depressed subjects, which have shown regions of hypermetabolism [11–13] or hypometabolism [14–17].
Given this background, we aimed in the present study to investigate brain amyloidosis in conjunction with studies of brain glucose metabolism in the presence or absence of depressive symptoms (defined by NPI-Q) in a large cohort of MCI subjects from the Alzheimer’s Disease Neuroimaging Initiative (ADNI). We also investigated the impact of depressive symptoms at baseline on the progression of dementia from clinical follow-up data.
Methods
Alzheimer’s Disease Neuroimaging Initiative
The data used in the preparation of this article were obtained from the ADNI database (adni.loni.usc.edu). The ADNI was launched in 2003 by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, the Food and Drug Administration, private pharmaceutical companies and nonprofit organizations, as a 5-year public–private partnership with a US$60 million budget. The primary goal of ADNI is to identify the optimal combinations of serial MRI, PET and other biological markers, in conjunction with clinical and neuropsychological assessments to predict and measure the progression of MCI and early AD. The objective is to determine sensitive and specific markers of very early AD progression that will aid researchers and clinicians in developing new treatments and monitoring their effectiveness, while lessening the expense and duration of clinical trials.
The Principal Investigator of this initiative is Michael W. Weiner, MD, VA Medical Center and University of California – San Francisco, but ADNI is the fruit of the efforts of many coinvestigators from diverse academic institutions and private corporations; subjects have been recruited from over 50 sites across the US and Canada. ADNI studies are conducted in accordance with the Good Clinical Practice guidelines, the principles of the Declaration of Helsinki, and US 21 CFR Part 50 (Protection of Human Subjects), and Part 56 (Institutional Review Boards). This study was approved by the Institutional Review Boards of all of the participating institutions. Written informed consent was obtained from all participants at each site. The initial goal of ADNI was to recruit 800 subjects, but with the project extensions ADNI-GO and ADNI-2 has recruited over 1500 subjects aged 55 to 90 years. The research population consists of cognitively normal older individuals, individuals with early or late MCI, or patients with early AD. The follow-up duration of each group is specified in the protocols for ADNI-1, ADNI-2 and ADNI-GO. Subjects originally recruited for ADNI-1 and ADNI-GO had the option to be followed in ADNI-2. For up-to-date information, see www.adni-info.org.
Data from ADNI-GO/ADNI2 are included in the present work. Preprocessed brain PET recordings, images and corresponding T1-weighted MPRAGE MR images (T1-W MRI) were downloaded from the ADNI database as on 30 August 2013.
Patient selection and study design
On the database cut-off date, 371 clinically rated subjects with MCI had received [18F]AV45 PET, FDG PET and T1-W MRI at baseline within ADNI-GO/ADNI2. In addition, apolipoprotein Eε4 (APOE ε4) status was assessed, and the NPI-Q score, Mini Mental State Examination (MMSE) score and education level were recorded at the time of the PET scans.
All subjects were categorized according to their depressive symptoms and brain Aβ status. Subsyndromal depression was diagnosed according to item 4 (depressive symptoms) of the NPI-Q [18] at the time (±2 months) of PET scanning where negative on item 4 indicated no depression and positive indicated depression. Aβ-positive (Aβ+) and Aβ-negative (Aβ−) [18F]AV45 PET status was defined according to the threshold of ≥1.10, a criterion derived from the ADNI database for the composite volume of interest (VOI) standardized uptake value ratio (SUVR) providing the highest diagnostic discrimination between cognitively normal individuals and AD patients [19]. As the proportions of Aβ+ subjects were unequal between nondepressed subjects (53 %, 141/265) and depressed subjects (61 %, 65/106; p = 0.16), analyses were performed separately in Aβ− subjects (N = 165) and Aβ+ subjects (N = 206).
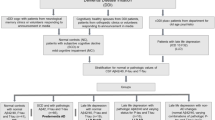
A mean follow-up of 21.5 ± 9.1 months with regard to conversion to dementia was available in 366 subjects (database to 2 June 2014). Each of these subjects was identified as a nonconverter when MCI was stable over the whole observation time, or as a converter when MCI had progressed to AD. A detailed overview of all study groups including demographics is provided in Fig. 1 and Table 1.

Stratification of 371 MCI subjects with contemporaneous [18F]AV45 PET, FDG PET and T1-W MRI at the ADNI-GO/2 baseline assessment. All subjects were first categorized as positive or negative according to their amyloid PET status [19] for the voxel-wise analysis (left branch). Subsequently, the Neuropsychiatric Inventory Questionnaire (NPI-Q; depression item 4) was used to identify subclinically depressed (DEP) and nondepressed (NON-DEP) study groups. The 366 subjects who received clinical follow-up were used for the conversion analysis (right branch) with respect to Aβ and depression status
Image data
ADNI [18F]AV45 and FDG PET acquisition and preprocessing
[18F]AV45 and FDG PET images had been acquired using Siemens, GE and Philips PET scanners (http://adni.loni.usc.edu/wp-content/uploads/2010/05/ADNI2_PET_Tech_Manual_0142011.pdf) and were preprocessed as described in: http://adni.loni.usc.edu/methods/pet-analysis/pre-processing/.
ADNI MRI acquisition and preprocessing
T1-W MRI scans had been acquired using Siemens, GE or Philips MRI scanners followed by MRI preprocessing according to a standard protocol [20].
Image processing
MRI coregistration and segmentation
All coregistration procedures were performed using the PMOD FUSION tool (v. 3.407 PMOD Technologies). First, T1-W MRI images were rigidly coregistered to the corresponding PET images to provide linear MRI-to-PET and inverted PET-to-MRI transformations, which were saved in MATLAB format. Next, T1-W MRI images were nonlinearly coregistered to the standard Montreal Neurological Institute (MNI) space T1-W template, and the calculated transformations were also saved in MATLAB format (MRI-to-MNI). T1-W MRI images were segmented into grey matter (GM), white matter (WM) and cerebrospinal fluid (CSF) within native MRI space using the PMOD PNEURO tool [21]. All segmentations were visually checked for correctness and extracerebral artefacts. When artefacts were present, masking through the individual’s whole-brain FDG PET image binarization (in T1-W MRI space) was applied to the segmentation.
Partial volume effect correction of [18F]AV45 PET and FDG PET
T1-W MRI segmentations were coregistered to the corresponding PET images by MRI-to-PET transformation in order to perform partial volume effect (PVE) correction in noninterpolated PET space. Voxel-wise PVE correction [22] was executed in PET space by PMOD with a GM threshold of 0.3, WM regression of 0.95 and isotropic full-width at half-maximum (FWHM) of 8 mm. PET-to-MRI and MRI-to-MNI transformations were finally combined and applied to the atrophy-corrected PET images to achieve spatially normalized PET images with minimum interpolation and maximum accuracy.
Image analysis
Voxel-wise [18F]AV45 and FDG PET analysis and statistics
Group comparisons were performed voxel-wise using two-sample t tests in SPM8 (Wellcome Department of Cognitive Neurology) implemented in MATLAB (R 2011a; MathWorks Inc.). APOE ε4 allelic status, age, gender, education and MMSE scores were entered as covariates. For SPM analysis, all images were Gaussian-filtered with 8 mm FWHM to minimize interimage variability. Intensity was subsequently normalized by scaling of [18F]AV45 and FDG activities to the cerebellum as defined by the Hammers atlas [23]. Implicit masking was used to compare only those voxels with valid values in all subjects after PVE correction.
Images from depressed and nondepressed subjects were compared using a significance threshold of p < 0.005, uncorrected for multiple comparisons and a cluster size of >100 voxels, while Aβ and FDG differences in parametric images were analysed separately with two-tailed tests.
VOI-based [18F]AV45 PET analysis
Whole-brain composite VOI values were assessed using the Hammers atlas and PVE-corrected SUVRs for whole cerebellum (SUVRCBL) as a reference region. SUVRCBL values were compared between nonconverters and converters and between nondepressed and depressed subjects. APOE ε4 alleles, age, gender, MMSE score and education level were used as covariates (SPSS, version 21.0; IBM, Chicago, IL). P values <0.05 were deemed to be significant after Bonferroni correction.
Conversion analysis
Kaplan-Meier plots were used to compare conversion rates between nondepressed and depressed subjects and between Aβ+ and Aβ− subjects. Additionally conversion rates were compared between nondepressed and depressed Aβ+ subjects separately and a subgroup of subjects with a high amyloid load (SUVRCBL >1.7). The multivariate Cox proportional hazards model was used to obtain hazard ratio estimates and 95 % confidence intervals for Aβ+, depression and the presence of established risk factors (APOE ε4 alleles, age, gender, MMSE score and education level).
Results
The rates of depression were 32 % among Aβ+ and 25 % among Aβ− subjects. There were no significant differences in MMSE score, gender, age or education level within the Aβ+ and Aβ− groups. APOE ε4 status significantly differed between Aβ+ and Aβ− subjects (Table 1).
Voxel-wise [18F]AV45 and FDG PET analysis
In Aβ+ subjects
Among all Aβ+ subjects, those with depression showed higher amyloid deposition in the left superior temporal gyrus, left uncus and gyrus parahippocampalis, left insula and the left cingulate gyrus (p < 0.001) as well as in the left medial frontal and rectal gyrus (p < 0.005) compared with those without depression. Significantly lower levels of amyloid were found in a small cluster of the right cuneal cortex (p < 0.001; Fig. 2a). Corresponding FDG data showed relative hypermetabolism in the bilateral frontal lobes as well as in the left fusiform gyrus (p < 0.001) in depressed subjects compared with nondepressed subjects. Hypometabolism was found in a small cluster of the left cuneal cortex (p < 0.001; Fig. 2b). All cluster sizes, localizations and T-scores are presented in Supplementary Table 1.

Statistical parametric mapping for [18F]AV45 PET (a) and FDG PET (b) in Aβ+ subjects corrected for MMSE score, age, gender, APOE ε4 allelic status and years of education. Subsyndromally depressed subjects (N = 65) are contrasted with nondepressed subjects (N = 141). Voxels exceeding a significance threshold of p < 0.005 (uncorrected for multiple comparisons, cluster size >100) for increased amyloid levels or FDG hypermetabolism in depressed subjects are indicated in red, while voxels of decreased amyloid levels or FDG hypometabolism in subjects with depressive symptoms are indicated in green. Both contrasts are rendered on the surface of the standard SPM8 template
in Aβ− subjects
Among Aβ− subjects, [18F]AV45 PET in the depressed subjects showed small clusters with lower amyloid deposition in bilateral temporal, left precentral and right inferior frontal gyri (p < 0.001) compared with the nondepressed subjects, and no increases were seen (Supplementary Fig. S1a). Corresponding FDG data did not show any metabolic differences among the whole group of depressed subjects (Supplementary Fig. S1b). All cluster sizes, localizations and T-scores are presented in Supplementary Table 2.
VOI-based [18F]AV45 PET analysis
Significant differences in PVE-corrected whole-brain SUVRCBL were found between depressed converters, who had the highest amyloid load (SUVRCBL 2.09 ± 0.48), and depressed nonconverters (SUVRCBL 1.77 ± 0.34; p < 0.001), and between depressed converters and nondepressed nonconverters (SUVRCBL 1.82 ± 0.30; p < 0.01). Nondepressed converters also showed a significantly higher amyloid load (SUVRCBL 2.05 ± 0.42) than depressed nonconverters (SUVRCBL 1.77 ± 0.34; p < 0.01). No significant differences were found between subgroups of Aβ− subjects (SUVRCBL range 1.15 – 1.23).
Conversion analysis
Of 366 MCI subjects with a mean clinical follow-up of 21.9 ± 9.1 months, 50 (14 %) progressed to AD. Considered separately, of 105 depressed subjects and 261 nondepressed subjects, 24 (23 %) and 26 (10 %), respectively, converted during this time. Of 205 Aβ+ subjects and 161 Aβ− subjects, 44 (21 %) and 6 (4 %), respectively, converted to AD. Of the 44 Aβ+ converters, 20 (45 %) were also depressed.
Both the depressed subjects (log-rank p < 0.005; Fig. 3a) and the Aβ+ subjects (log-rank p < 0.001; Fig. 3b) showed significantly faster progression to AD than their respective counterparts. Further categorization of Aβ+ subjects into depressed and nondepressed showed significantly faster progression in depressed subjects (log-rank p < 0.005; Fig. 3c). In subjects with a high amyloid load (SUVRCBL > 1.7), the effect of coexistent depressive symptoms was even more pronounced (log-rank p < 0.001): 100 % of these subjects converted during follow-up, compared with 45 % of the nondepressed subjects (Fig. 3d).

Kaplan-Meier analyses of time to progression to dementia. Conversion to AD at a consecutive ADNI follow-up visit was used as the specific end-point. Progression-free survival in a depressed versus nondepressed subjects, b Aβ+ versus Aβ− subjects, c depressed versus nondepressed Aβ+ subjects, and d depressed versus nondepressed subjects with a high amyloid load (SUVRCBL > 1.7) . **p < 0.005, ***p < 0.001 (log rank test)
Hazard ratios for conversion to AD were 4.5 (95 % CI 1.7 – 11.4, p < 0.005) for Aβ+ subjects, 2.9 (95 % CI 1.6 – 5.3, p < 0.001) for depressed subjects, and 0.8 (95 % CI 0.7 – 0.9, p < 0.005) for nondepressed subjects. Considering Aβ+ subjects separately, those who were also depressed showed a hazard ratio of 8.1 (95 % CI 3.1 – 21.6, p < 0.001) in contrast to 3.0 (95 % CI: 1.1 – 8.2, p < 0.05) in those who were not depressed.
Discussion
We present the results of the largest analysis so far of combined amyloid and FDG PET assessment in MCI subjects with respect to coexisting depressive symptoms. In addition, we provide the first longitudinal evaluation of progression to dementia in subsyndromal but depressed subjects using amyloid PET as an integral biomarker. Our results clearly indicate that Aβ+ MCI subjects with depressive symptoms suffer from elevated amyloid load compared with nondepressed individuals, with adjustment for various factors influencing cognition. The pronounced frontotemporal amyloid deposition in these patients occurred in association with relative hypermetabolism of connected brain areas on FDG PET. This finding may be related to active inflammation, or represent a form of metabolic compensation in MCI subjects. Subjects with elevated amyloid load and coexisting depressive symptoms were at high risk of faster progression to AD.
Amyloid-positive subjects
[18F]AV45 PET revealed elevated amyloid load in depressed Aβ+ subjects in the left superior temporal, parahippocampal, insular and medial frontal gyri, brain regions comprising or connected with the mood disorder-related medial prefrontal network [24]. The link between depression and dementia in Aβ+ subjects therefore might be related to the region-specific deposition of amyloid, when mood-related neurocircuits happen to be particularly affected by amyloid pathology. Our findings are in line with previously reported associations of region-specific lateral and medial temporal amyloid/neurofibrillary tangle deposition with depression and anxiety in MCI subjects shown by [18F]FDDNP PET [25]. Previous [11C]PiB PET findings in a small group of patients have also provided evidence that amyloid load is elevated in late-life depressed MCI subjects [8]. CSF levels of Aβ42 are concordantly reduced in MCI subjects suffering from late-life depression and are correlated with cognitive status [26]. Post-mortem analyses have indicated a pronounced deposition of amyloid plaques and tangles in the hippocampus in the brain of AD patients with a history of major depression, while subjects with concurrent depressive symptoms at diagnosis of AD exhibit even higher levels of neuropathological change [27, 28]. The elevated frontotemporal levels of amyloid in depressed subjects found in this study are in line with these earlier histological findings. Another study revealed that AD pathology is prominent in the majority of patients with dementia and coexistent depression [29]. This is also in line with our data showing a higher proportion of Aβ+ subjects among MCI subjects with depression (61 %) than among MCI subjects without depression (53 %).
The question still remains as to whether late-life depression constitutes part of the dementia prodrome and/or represents an individual risk factor for AD. From our data we can conclude that depressive symptoms in Aβ+ MCI subjects were clearly associated with higher frontotemporal amyloid levels. Recently published results of the prospective Australian Imaging, Biomarkers and Lifestyle (AIBL) study indicate notably slow increases in brain amyloidosis with age, with an estimated 12 years from absence to [11C]PiB positivity and another 19 years from [11C]PiB positivity to AD-like [11C]PiB levels [30]. Therefore, we can speculate that late-onset depression triggered by significant amyloid deposition may be part of the dementia prodrome, presenting as much as two decades before manifestation of AD. Indeed, Wu et al. found that patients with life-time occurrence of major depression showed increased [18F]AV45 uptake (and elevation in Hamilton Depression Rating Scale score) compared with depression-free MCI subjects [10] in similar brain regions as in the present investigation. However, a previous [11C]PiB study of similar design did not support this association [9]. This may reflect the fact that in the latter study the cognitively normal subjects were significantly younger (mean age 61 years) with onset of depression six or more years previously and with complete remission from depression at the time of PET scanning. Overall, findings are consistent with a model in which present depressive episodes in older subjects could constitute a prodrome and risk factor for AD development.
Analysis of our longitudinal follow-up data showed that subsyndromally depressed MCI subjects (mean age 72.1 years) with a high amyloid load had a high risk of rapid conversion to AD. Recent findings in the AIBL population indicate that cognitively intact subjects with abnormal amyloid burden assessed by [11C]PiB PET show a greater decline in episodic and verbal memory at 3 years especially those with comorbid anxiety [31]. Hence, mood-related neuropsychiatric symptoms are associated with more aggressive AD pathology in Aβ+ subjects. Although the pathophysiological mechanism underlying this association cannot be resolved by this or other studies, present data confirm a link among rapid cognitive decline, amyloid burden and depressive symptoms, which may have important implications for new treatment targets [32].
Amyloid-negative subjects
Following the conjecture of higher amyloid deposition in subsyndromally depressed subjects, one might expect that amyloidosis in Aβ− MCI subjects with depressive symptoms would all have amyloid levels close to the range or threshold for subjects with Aβ+ status. Surprisingly, we found significantly lower levels of amyloid in the frontotemporal areas of subsyndromally depressed Aβ− subjects. These preliminary findings suggest that two different “dementia” pathways can occur in late-life depressed subjects: (1) depression as a prodrome in the “normal” amyloid pathway, and (2) depression without amyloid accumulation, in which subjective memory complaints may play an important role [33].
Amyloid levels in relation to FDG metabolism
Our results indicate that relative hypermetabolism is present in subsyndromally depressed Aβ+ MCI subjects within a similar domain of frontal brain areas revealing elevated amyloidosis in contrast to the findings in nondepressed subjects. Increased glucose metabolism may reveal an inflammatory process in the brain [34], possibly caused by the amyloid pathology, or alternatively reflecting a metabolic compensation in still healthy neurons embedded in amyloid-affected networks [35]. A correlation between binding of ligands such as [11C]PBR28 for the 18-kDa translocator protein and AD severity has recently been documented, lending support to the inflammation hypothesis [36]. It is noteworthy that [11C]PBR28 and [11C]PiB binding also correlate with clinical scores after PVE correction, indicating that they reveal pathology rather than simply atrophy. In Aβ− MCI subjects with depressive symptoms FDG metabolism was generally inconspicuous. This finding probably explains the inconsistent results among previous FDG PET studies regarding late-life depression where Aβ status was not hitherto considered. Both hypermetabolism [11–13, 24] and hypometabolism [14–17] have been reported in late-life depressed subjects in cognitively preserved and impaired subjects and AD patients. Moreover, PVE correction was not applied in these FDG PET studies, so that atrophy, as may occur in depressed subjects [37, 38], may have resulted in spurious findings of hypometabolism.
Limitations
The NPI-Q was used to diagnose late-life depression because there was no clinical diagnosis of depression, and especially no gold standard structured clinical interview data available. Therefore, the low sensitivity of the single item has to be considered as a limitation. The geriatric depression scale (GDS) might have been a reasonable alternative. However, a GDS score >5 was defined as an exclusion criterion for ADNI enrolment. Therefore, we think that defining clinical groups by the GDS might have led to more selection bias due to this exclusion criterion, and we consequently focused on NPI-Q. In addition, late-life depression as a clinical syndrome may include heterogeneous subtypes, including common vascular depression [39], which are probably different from amyloid pathology. Subjects with depressive symptoms often suffer from additional neuropsychiatric symptoms such as anxiety and apathy [40, 41], which may be independent factors contributing to amyloid or metabolic status. However, had we restricted our search to patients positive for depressive symptoms only, the group size would have been too small to allow statistical comparison. Therefore, we compared our depressed subjects consistently against nondepressed controls irrespective of the presence of other NPI-Q categories to minimize this bias.
Finally, antidepressant medication was documented only from the time of the baseline PET scan irrespective of prior duration, and 62 % of depressed subjects and 5 % of nondepressed subjects were treated with mostly serotonin reuptake inhibitors (SSRI). Therefore, no final conclusions can be drawn as to the effect of previous SSRI treatment on amyloid levels in our subjects. However, according to a recent report in cognitively normal subjects [32], amyloid levels in SSRI-treated subjects would be expected to be decreased. Therefore, the significance of our findings is more likely to have been attenuated by this circumstance than positively biased. Longitudinal amyloid PET imaging in a prospective study of SSRI-treated and untreated late-life depressed subjects would be of great value.
References
Ziegler-Graham K, Brookmeyer R, Johnson E, Arrighi HM. Worldwide variation in the doubling time of Alzheimer’s disease incidence rates. Alzheimers Dement. 2008;4:316–23. doi:10.1016/j.jalz.2008.05.2479.
Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–59.
Lim YY, Ellis KA, Pietrzak RH, Ames D, Darby D, Harrington K, et al. Stronger effect of amyloid load than APOE genotype on cognitive decline in healthy older adults. Neurology. 2012;79:1645–52. doi:10.1212/WNL.0b013e31826e9ae6.
Lim YY, Maruff P, Pietrzak RH, Ellis KA, Darby D, Ames D, et al. Abeta and cognitive change: examining the preclinical and prodromal stages of Alzheimer’s disease. Alzheimer’s Dement. 2014;10:743–51. doi:10.1016/j.jalz.2013.11.005.
Ownby RL, Crocco E, Acevedo A, John V, Loewenstein D. Depression and risk for Alzheimer disease: systematic review, meta-analysis, and metaregression analysis. Arch Gen Psychiatry. 2006;63:530–8. doi:10.1001/archpsyc.63.5.530.
Olin JT, Katz IR, Meyers BS, Schneider LS, Lebowitz BD. Provisional diagnostic criteria for depression of Alzheimer disease: rationale and background. Am J Geriatr Psychiatry. 2002;10:129–41.
Steenland K, Karnes C, Seals R, Carnevale C, Hermida A, Levey A. Late-life depression as a risk factor for mild cognitive impairment or Alzheimer’s disease in 30 US Alzheimer’s disease centers. J Alzheimer’s Dis. 2012;31:265–75. doi:10.3233/JAD-2012-111922.
Butters MA, Klunk WE, Mathis CA, Price JC, Ziolko SK, Hoge JA, et al. Imaging Alzheimer pathology in late-life depression with PET and Pittsburgh compound-B. Alzheimer Dis Assoc Disord. 2008;22:261–8. doi:10.1097/WAD.0b013e31816c92bf.
Madsen K, Hasselbalch BJ, Frederiksen KS, Haahr ME, Gade A, Law I, et al. Lack of association between prior depressive episodes and cerebral [11C]PiB binding. Neurobiol Aging. 2012;33:2334–42. doi:10.1016/j.neurobiolaging.2011.11.021.
Wu KY, Hsiao IT, Chen CS, Chen CH, Hsieh CJ, Wai YY, et al. Increased brain amyloid deposition in patients with a lifetime history of major depression: evidenced on F-florbetapir (AV-45/Amyvid) positron emission tomography. Eur J Nucl Med Mol Imaging. 2014;41:714–22. doi:10.1007/s00259-013-2627-0.
Smith GS, Kramer E, Hermann C, Ma Y, Dhawan V, Chaly T, et al. Serotonin modulation of cerebral glucose metabolism in depressed older adults. Biol Psychiatry. 2009;66:259–66. doi:10.1016/j.biopsych.2009.02.012.
Smith GS, Kramer E, Ma Y, Kingsley P, Dhawan V, Chaly T, et al. The functional neuroanatomy of geriatric depression. Int J Geriatr Psychiatr. 2009;24:798–808. doi:10.1002/gps.2185.
Marano CM, Workman CI, Kramer E, Hermann CR, Ma Y, Dhawan V, et al. Longitudinal studies of cerebral glucose metabolism in late-life depression and normal aging. Int J Geriatr Psychiatr. 2013;28:417–23. doi:10.1002/gps.3840.
Hirono N, Mori E, Ishii K, Ikejiri Y, Imamura T, Shimomura T, et al. Frontal lobe hypometabolism and depression in Alzheimer’s disease. Neurology. 1998;50:380–3.
Holthoff VA, Beuthien-Baumann B, Kalbe E, Ludecke S, Lenz O, Zundorf G, et al. Regional cerebral metabolism in early Alzheimer’s disease with clinically significant apathy or depression. Biol Psychiatry. 2005;57:412–21. doi:10.1016/j.biopsych.2004.11.035.
Lee DY, Choo IH, Jhoo JH, Kim KW, Youn JC, Lee DS, et al. Frontal dysfunction underlies depressive syndrome in Alzheimer disease: a FDG-PET study. Am J Geriatr Psychiatry. 2006;14:625–8. doi:10.1097/01.JGP.0000214541.79965.2d.
Lee HS, Choo IH, Lee DY, Kim JW, Seo EH, Kim SG, et al. Frontal dysfunction underlies depression in mild cognitive impairment: a FDG-PET study. Psychiatr Investig. 2010;7:208–14. doi:10.4306/pi.2010.7.3.208.
Kaufer DI, Cummings JL, Ketchel P, Smith V, MacMillan A, Shelley T, et al. Validation of the NPI-Q, a brief clinical form of the neuropsychiatric inventory. J Neuropsychiatry Clin Neurosci. 2000;12:233–9.
Johnson KA, Minoshima S, Bohnen NI, Donohoe KJ, Foster NL, Herscovitch P, et al. Appropriate use criteria for amyloid PET: a report of the amyloid imaging task force, the society of nuclear medicine and molecular imaging, and the Alzheimer’s association. Alzheimer’s Dement. 2013;9:e-1–16. doi:10.1016/j.jalz.2013.01.002.
Jack Jr CR, Bernstein MA, Fox NC, Thompson P, Alexander G, Harvey D, et al. The Alzheimer's Disease Neuroimaging Initiative (ADNI): MRI methods. J Magn Reson Imaging. 2008;27:685–91. doi:10.1002/jmri.21049.
Ashburner J, Friston KJ. Unified segmentation. Neuroimage. 2005;26:839–51. doi:10.1016/j.neuroimage.2005.02.018.
Muller-Gartner HW, Links JM, Prince JL, Bryan RN, McVeigh E, Leal JP, et al. Measurement of radiotracer concentration in brain gray matter using positron emission tomography: MRI-based correction for partial volume effects. J Cereb Blood Flow Metab. 1992;12:571–83. doi:10.1038/jcbfm.1992.81.
Hammers A, Allom R, Koepp MJ, Free SL, Myers R, Lemieux L, et al. Three dimensional maximum probability atlas of the human brain, with particular reference to the temporal lobe. Hum Brain Mapp. 2003;19:224–247.
Price JL, Drevets WC. Neurocircuitry of mood disorders. Neuropsychopharmacology. 2010;35:192–216. doi:10.1038/npp.2009.104.
Lavretsky H, Siddarth P, Kepe V, Ercoli LM, Miller KJ, Burggren AC, et al. Depression and anxiety symptoms are associated with cerebral FDDNP-PET binding in middle-aged and older nondemented adults. Am J Geriatr Psychiatry. 2009;17:493–502.
Sun X, Steffens DC, Au R, Folstein M, Summergrad P, Yee J, et al. Amyloid-associated depression: a prodromal depression of Alzheimer disease? Arch Gen Psychiatry. 2008;65:542–50. doi:10.1001/archpsyc.65.5.542.
Rapp MA, Schnaider-Beeri M, Grossman HT, Sano M, Perl DP, Purohit DP, et al. Increased hippocampal plaques and tangles in patients with Alzheimer disease with a lifetime history of major depression. Arch Gen Psychiatry. 2006;63:161–7. doi:10.1001/archpsyc.63.2.161.
Rapp MA, Schnaider-Beeri M, Purohit DP, Perl DP, Haroutunian V, Sano M. Increased neurofibrillary tangles in patients with Alzheimer disease with comorbid depression. Am J Geriatr Psychiatry. 2008;16:168–74. doi:10.1097/JGP.0b013e31816029ec.
Sweet RA, Hamilton RL, Butters MA, Mulsant BH, Pollock BG, Lewis DA, et al. Neuropathologic correlates of late-onset major depression. Neuropsychopharmacology. 2004;29:2242–50. doi:10.1038/sj.npp.1300554.
Villemagne VL, Burnham S, Bourgeat P, Brown B, Ellis KA, Salvado O, et al. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol. 2013;12:357–67. doi:10.1016/S1474-4422(13)70044-9.
Pietrzak RH, Scott JC, Neumeister A, Lim YY, Ames D, Ellis KA, et al. Anxiety symptoms, cerebral amyloid burden and memory decline in healthy older adults without dementia: 3-year prospective cohort study. Br J Psychiatry. 2014;204:400–1. doi:10.1192/bjp.bp.113.134239.
Sheline YI, West T, Yarasheski K, Swarm R, Jasielec MS, Fisher JR, et al. An antidepressant decreases CSF abeta production in healthy individuals and in transgenic AD mice. Sci Transl Med. 2014;6:236re4. doi:10.1126/scitranslmed.3008169.
Buckley RF, Saling MM, Irish M, Ames D, Rowe CC, Lautenschlager NT, et al. Personal memory function in mild cognitive impairment and subjective memory complaints: results from the Australian Imaging, Biomarkers, and Lifestyle (AIBL) Study of Ageing. J Alzheimers Dis. 2014;40:551–61. doi:10.3233/JAD-131820.
Hermida AP, McDonald WM, Steenland K, Levey A. The association between late-life depression, mild cognitive impairment and dementia: is inflammation the missing link? Expert Rev Neurother. 2012;12:1339–50. doi:10.1586/ern.12.127.
Caroli A, Lorenzi M, Geroldi C, Nobili F, Paghera B, Bonetti M, et al. Metabolic compensation and depression in Alzheimer’s disease. Dement Geriatr Cogn Disord. 2010;29:37–45. doi:10.1159/000257761.
Kreisl WC, Lyoo CH, McGwier M, Snow J, Jenko KJ, Kimura N, et al. In vivo radioligand binding to translocator protein correlates with severity of Alzheimer’s disease. Brain. 2013;136:2228–38. doi:10.1093/brain/awt145.
Lebedeva A, Westman E, Lebedev AV, Li X, Winblad B, Simmons A, et al. Structural brain changes associated with depressive symptoms in the elderly with Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2014;85:930–5. doi:10.1136/jnnp-2013-307110.
Lee GJ, Lu PH, Hua X, Lee S, Wu S, Nguyen K, et al. Depressive symptoms in mild cognitive impairment predict greater atrophy in Alzheimer’s disease-related regions. Biol Psychiatry. 2012;71:814–21. doi:10.1016/j.biopsych.2011.12.024.
Alexopoulos GS, Meyers BS, Young RC, Campbell S, Silbersweig D, Charlson M. ‘Vascular depression’ hypothesis. Arch Gen Psychiatry. 1997;54:915–22.
Marshall GA, Donovan NJ, Lorius N, Gidicsin CM, Maye J, Pepin LC, et al. Apathy is associated with increased amyloid burden in mild cognitive impairment. J Neuropsychiatr Clin Neurosci. 2013;25:302–7. doi:10.1176/appi.neuropsych.12060156.
Mori T, Shimada H, Shinotoh H, Hirano S, Eguchi Y, Yamada M, et al. Apathy correlates with prefrontal amyloid beta deposition in Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2014;85:449–55. doi:10.1136/jnnp-2013-306110.
Acknowledgments
Data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen Idec Inc.; Bristol-Myers Squibb Company; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; ; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Medpace, Inc.; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Synarc Inc.; and Takeda Pharmaceutical Company. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. The authors acknowledge Inglewood Biomedical Editing for professional editing of the manuscript.
Conflicts of interest
Matthias Brendel reports no disclosures.
Oliver Pogarell reports no disclosures.
Guoming Xiong reports no disclosures.
Andreas Delker reports no disclosures.
Peter Bartenstein received research support from the Federal Ministry of Education and Science (BMBF).
Axel Rominger received research support from the Friedrich-Baur Foundation and SyNergy cluster.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
The data used in the preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). The ADNI investigators contributed to the design and implementation of ADNI and/or provided data but did not participate in the analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf
Rights and permissions
About this article

Cite this article
Brendel, M., Pogarell, O., Xiong, G. et al. Depressive symptoms accelerate cognitive decline in amyloid-positive MCI patients. Eur J Nucl Med Mol Imaging 42, 716–724 (2015). https://doi.org/10.1007/s00259-014-2975-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00259-014-2975-4