
Abstract
Glycerol dehydratase (gdrAB-dhaB123) operon from Klebsiella pneumoniae and NADPH-dependent 1,3-propanediol oxidoreductase (yqhD) from Escherichia coli were stably integrated on the chromosomal DNA of E. coli under the control of the native-host ldhA and pflB constitutive promoters, respectively. The developed E. coli NSK015 (∆ldhA::gdrAB-dhaB123 ∆ackA::FRT ∆pflB::yqhD ∆frdABCD::cat-sacB) produced 1,3-propanediol (1,3-PDO) at the level of 36.8 g/L with a yield of 0.99 mol/mol of glycerol consumed when glucose was used as a co-substrate with glycerol. Co-substrate of glycerol and cassava starch was also utilized for 1,3-PDO production with the concentration and yield of 31.9 g/L and 0.84 mol/mol of glycerol respectively. This represents a work for efficient 1,3-PDO production in which the overexpression of heterologous genes on the E. coli host genome devoid of plasmid expression systems. Plasmids, antibiotics, IPTG, and rich nutrients were omitted during 1,3-PDO production. This may allow a further application of E. coli NSK015 for the efficient 1,3-PDO production in an economically industrial scale.
Key points
• gdrAB-dhaB123 and yqhD were overexpressed in E. coli devoid of a plasmid system
• E. coli NSK015 produced a high yield of 1,3-PDO at 99% theoretical maximum
• Cassava starch was alternatively used as substrate for economical 1,3-PDO production
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
1,3-propanediol (1,3-PDO) can be used in many applications, such as the manufacture of cosmetics, lubricants, adhesives, laminates, coating materials, and personal care products. It is also a monomer for producing poly(trimethylene terephthalate) (PTT) that has the combined property of poly(ethylene terephthalate) (PET) and poly(butyrene terephthalate) (PBT), thus making PTT a suitable fiber for application in carpet and textile industries. Conventionally, 1,3-PDO has been chemically synthesized from acrolein and ethylene oxide derived from crude oil (Zhu et al. 2002). As an awareness on global warming and environmental challenges increase, researchers are attracted to focusing on an alternative bio-based production of 1,3-PDO from wasted glycerol from biodiesel plants using microbes including Klebsiella pneumoniae (Lee et al. 2018a; Oh et al. 2018) and Clostridium sp. (Liu et al. 2010). However, a disadvantage for the use of K. pneumoniae is that it is a potential pathogen that is dangerous to operators and may contaminate the surroundings. Additionally, the growth of Clostridium sp. greatly relies on strict anaerobic conditions that are quite difficult to control during 1,3-PDO production and may cause an increase in capital investments and operational expenses in the industry (Liu et al. 2010). So far, metabolically engineered E. coli strains producing 1,3-PDO have been previously reported. The 1,3-PDO production by developed E. coli strains was extensively studied through an overexpression of heterologous genes for glycerol utilization in E. coli strains. However, the overexpression of these genes was normally employed using plasmid-dependent systems while the low production yield of 1,3-PDO was then reported (Table 1).
In this study, the artificially re-arranged gdrAB-dhaB123 (glycerol dehydratase) operon from K. pneumoniae and the native E. coli NADPH-dependent 1,3-propanediol oxidoreductase (yqhD) gene were simultaneously introduced into E. coli host cell. To the best of our knowledge, this is a pioneer work for 1,3-PDO production in which our constructed strains possessed genes that were integrated and constitutively expressed on the host E. coli genome under the control of the native host ldhA and pflB promoters respectively, devoid of plasmid-dependent conditions. Concerns about plasmid instability and plasmid induction for the heterologous gene expression in the developed E. coli strain and the occurrence of pathogenicity by K. pneumoniae for 1,3-PDO production were thus excluded. Additionally, some anaerobic fermentative genes were also deleted from the host genome to conserve the maximum carbon flow through 1,3-PDO production route. The developed E. coli NSK015 strain capable of the over-expression of necessary enzymes involved in the reduction of glycerol through 1,3-PDO biosynthesis pathway is resemble to that of K. pneumoniae, a native 1,3-PDO producer (Fig. 1). The strain allowed producing a high yield of 1,3-PDO from glycerol closed to theoretical maximum (1.0 mol/mol glycerol) when glucose was provided as a co-substrate under both microaerobic conditions (shaking flasks) and aerobic conditions (1–2 vvm aeration). The strain was able to utilize mineral salts medium (4 g/L total salts) regardless of yeast extract or any rich and expensive nutrients during fermentation, thus reducing production costs related to medium preparation, product recovery, purification, and waste disposal. E. coli NSK015 strain may be industrially applied for the economically and environmentally friendly production of 1,3-PDO.

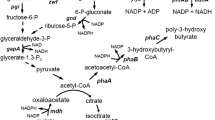
Proposed metabolism for 1,3-propanediol production in a metabolically engineered Escherichia coli NSK015. Genes involving in the pathway are summarized in the box. Red cross signs represent the gene deletion. Genes gdrAB-dhaB123 and yqhD were expressed under the control of native ldhA and pflB promoters
Materials and methods
Microbial strains, plasmid, media, primers, and growth conditions
Table 2 shows microbial strains used in this study. E. coli C was used as a parental strain for engineering its metabolic pathway. E. coli TOP 10 was used to maintain constructed plasmids. Microbial strains were cultivated and maintained in LB broth or agar, at 37 \(\mathrm{^\circ{\rm C} }\). Apramycin (Apra), chloramphenicol (Cm), ampicillin (Amp), and kanamycin (Km) antibiotics were supplemented into media as appropriated. An LB aldehyde indicator plate containing 3% (w/v) glycerol, 0.5% (w/v) sodium bisulfite, and 0.05% (w/v) pararosaniline in ethanol was modified from the report of Conway et al. (1987) and used to identify the expression of glycerol dehydratase operon under the native ldhA promoter. Coenzyme-B12 was also provided at the concentration of 15 µM during 1,3-PDO production. The low salt AM1 production medium (Martinez et al. 2007) was used throughout this study. The medium is composed of 19.9 mM (NH4)2HPO4, 7.6 mM (NH4)2HPO4, 1.5 mM MgSO4.7H2O, 1 mM betaine HCl and trace metal (8.9 µM FeCl3.6H2O, 1.3 µM CoCl2.6H2O, 0.9 µM CuCl2.2H2O, 2.2 µM ZnCl2, 1.2 µM Na2MoO4.2H2O, 1.2 µM H3BO3, and 2.5 µM MnCl2.4H2O2). Yeast extract (5 g/L) was only provided in AM1 if required to determine its effect over fermentation.
Construction of an artificially expressing operon of glycerol dehydratase with its reactivating factors
Table S1 shows primers and plasmids used in this study. Briefly, gdrB gene and its surrounding sequences were amplified using Comp-gdrA(down)-gdrB-R (forward) and DH-ldhAp-gdrAB-dhaB123-F (reverse) primers. The amplified PCR fragment (gdrB fragment) was cloned into pCR2.1-TOPO, and the plasmid was designated to be pKJ1010. The plasmid pKJ1010 was further used as the template for PCR amplification in an inside-out direction using dhaB1-M13-R/F (forward) and Comp-gdrA(down)-gdrB-R (reverse) primers. The resulting PCR fragment was a linearized pKJ1010 plasmid missing surrounding sequences of gdrB gene. The linearized DpnI-treated pKJ1010 plasmid was used as the template to generate sticky ends at both sides using either dhaB1-M13-R/F or Comp-gdrA(down)-gdrB-R primers in separated PCR reactions. Both single-stranded amplified PCR fragments were recombined, and the double-stranded PCR product was CIP treated.
On the other hand, dhaB123 and gdrA genes and their surrounding sequences were also amplified using Comp-gdrB(up)-gdrA-F (forward) and DH-ldhAp-gdrAB-dhaB123-R (reverse) primers. The resulting PCR fragment contained 5’-gdrA-dhaB123-3’ sequences lying in the same orientation of gdrB gene on the plasmid pKJ1010. Consequently, this fragment was amplified using either Comp-gdrB(up)-gdrA-F and M13-dhaB1-R primers in separate PCR reactions followed by CIP treatment in the same manner as previously mentioned. The resulting double-stranded PCR fragment contained sequences derived from the gdrA-dhaB123 with sticky ends at both sides. Finally, the CIP-treated linearized pKJ1010 plasmid was ligated with the T4-PNK treated gdrA-dhaB123 insert cassette facilitated by the complementary sequences at overhangs on their both ends. The ligated plasmid contained the artificially expressing cassette gdrAB-dhaB123 and was designated to pKJ1011.
Construction of an expressing cassette encoding the native E. coli yqhD
The upstream of pflB was first amplified by polymerase chain reaction (PCR) using yqhD-pflB-F (forward) and upfocA-R (reverse) primers. Next, a pair of primers, pflB-yqhD-F and pflB-yqhD-R, was further used to initiate the amplification of the yqhD gene and its own promoter sequences while the downstream of pflB gene was also amplified by PCR using pflA-F and yqhD-pflB-R primers. Both PCR fragments were then combined as a molar ratio of 1:1 in the reaction containing DNA polymerase without primers using a slow annealing procedure. The resulting fragment was further amplified using pflA-F and pflB-yqhD-R primers. The amplified fragment was then joined to the upstream fragment of pflB as previously amplified. The annealed fragment was used as a template for the amplification of the shorter fragment containing focA’-yqhD-pflB” sequences using pflA-F and focA-R primers. The focA’-yqhD-pflB”, an expressing cassette encoding the native E. coli yqhD, was thus cloned into pCR2.1-TOPO, generating the plasmid pKJ1014.
Deletions of ldhA, ackA, and frdABCD genes and chromosomal integrations of the artificial gdrAB-dhaB123 operon and yqhD gene in E. coli C
Gene encoding lactate dehydrogenase (ldhA) was initially deleted in E. coli C wild type. Briefly, the cat-sacB (chloramphenicol acetyltransferase-levan sucrase) cassette was amplified by PCR using the genomic DNA of K. oxytoca KMS005 (Jantama et al. 2015) as a template and DH-ldhAp-cat-sacB-F and DH-ldhAp-cat-sacB-R as primers. The amplified PCR fragment (ldhA’-cat-sacB-ldhA”) was then transformed into E. coli C carrying the plasmid pLOI3420. The homologous recombination of the fragment on the chromosomal ldhA region of E. coli C wildtype was confirmed using the colony PCR technique. The verified clone was renamed as E. coli NSK001 (ΔldhA::cat-sacB).
Gene encoding acetate kinase (ackA) was also interrupted using techniques previously developed by Datsenko and Wanner (2000). The PCR fragments containing kanamycin resistant gene flanked by parts of upstream and downstream sequences encoding ackA genes were amplified by PCR reaction using the pKD4 plasmid as a template with ackA-pKD4-F and ackA-pKD4-R primers. The resulting PCR fragments (ackA’-FRT-km-FRT-ackA”) were transformed into E. coli NSK001 carrying the plasmid pLOI3420. The integration of the fragment on the chromosomal ackA region of E. coli NSK001 was verified and the clone was renamed as E. coli NSK002 (ΔldhA::cat-sacB ΔackA::FRT-km-FRT). Furthermore, E. coli NSK002 strain carrying pLOI3420 was transformed with the fragment of gdrAB-dhaB123 cassette derived from pKJ1011 by electroporation. A specific integration of the artificially expressing gdrAB-dhaB123 cassette at E. coli ldhA region occurred via a double homologous recombination event. The gdrAB-dhaB123 cassette was designed to integrate in frame with the start codon at the ldhA promoter. The confirmed clone was designated E. coli NSK003 (ΔldhA::gdrAB-dhaB123ΔackA::FRT-km-FRT). To remove the km gene, a plasmid pFT-A expressing a recombinase was transformed into E. coli NSK003. E. coli NSK003 strain carrying pFT-A was further cultured in LB medium containing chlortetracycline (20 µg/mL) at 30 °C for 8 h to allow self-recombining between FRT sites facilitated by the recombinase. The verified clone was renamed as E. coli NSK012 (ΔldhA::gdrAB-dhaB123ΔackA::FRT).
Gene encoding pyruvate formate lyase (pflB) was deleted in E. coli NSK012. The PCR fragment (pflB’-cat-sacB-pflB”) amplified using WMpflBA-F and WMpflBA-R as primers and the genomic DNA of K. oxytoca KMS005 as template was then transformed into E. coli NSK012 carrying the plasmid pLOI3420. The verified clone was re-named as E. coli NSK013 (ΔldhA::gdrAB-dhaB123ΔackA::FRTΔpflB::cat-sacB). E. coli NSK013 strain carrying pLOI3420 was further transformed with the fragment of focA’-yqhD-pflB” derived from pKJ1014. The confirmed clone was designated E. coli NSK014 (ΔldhAB::gdrAB-dhaB123ΔackA::FRTΔpflB::yqhD).
Gene encoding fumarate reductase (frdABCD) was further deleted in E. coli NSK014 following the same strategy of Jantama et al. (2008). The frdABCD fragments were amplified by frdABCD-F and frdABCD-R primers and cloned into pCR2.1-TOPO. The cloned plasmid was used as the template by IOfrd-F and IOfrd-R primers. The PCR product contained sequences derived from pCR2.1-TOPO with the flanking regions of frdABCD. The cat-sacB cassette amplified from K. oxytoca KMS005 by catfrd-F and catfrd-R primers was ligated into the PCR product to generate pKJ1012. The amplified PCR fragment (frd’-cat-sacB-frd”) derived from pKJ1012 was then transformed into E. coli NSK014 carrying pLOI3420. The verified clone was re-named E. coli NSK015 (ΔldhA::gdrAB-dhaB123ΔackA::FRTΔpflB::yqhDΔfrdABCD::cat-sacB).
Enzyme assay
Cells at the late-log phase cultures were harvested by centrifugation of 4000 g at 4 ℃ for 10 min. The pellets were washed using 0.05 M PBS buffer (pH 8.0) twice then re-suspended with a final volume of 2 mL. The cell was further disrupted by an ultrasonic disintegrator at 40 kHz with alternate pulses of sonication for 10 min. The cell debris in the crude lysate were separated. The supernatant was used to determine the total protein concentration by the Bradford (1976) and its enzymatic activities.
To determine activities of glycerol dehydratase and 1,3-propanediol oxidoreductase, the method of Knietsch et al. (2003) was applied. The apparent molar extinction coefficient derived from 3-hydroxypropionaldehyde (3-HPA) used for the calculation of the specific enzyme activities was 5.23*103 M−1 cm−1 determined at the absorbance of 670 nm. The specific glycerol dehydratase and 1,3-propanediol oxidoreductase activities were reported as amounts of aldehydes in the unit of micromole formed (U) in 1 min per mg total protein.
Fermentation experiment
In shake flask experiments, the seed cultures at an initial OD550 of 0.1 were inoculated in 100 mL AM1 medium containing 20 g/L glycerol and 100 mM KHCO3 with or without either glucose or yeast extract in a 500 mL Erlenmeyer flask at 37 ℃, 200 rpm for 72 h. Samples were intermittently taken every 12 h for analyses. In 2 L fermenter experiments, E. coli NSK015 strain was initially grown in 100 mL LB supplemented with 2% (w/v) glucose in a 250 mL shaking flask at 37 °C, 200 rpm for 12 h. The inoculum was transferred into AM1 medium containing substrates with the initial OD550 of 0.1 with specified volumes. The solution of 3 M KOH was automatically added into the broth to maintain pH of cultures at 7.0. The aeration rate of 1 or 2 vvm and agitation of 200 rpm were provided according to experiments. One milliliter of sample was taken every 12 h for analyses for 108 h.
The mixed cassava starch (70 g/L) and glycerol (50 g/L) in AM1 medium containing coenzyme B12 were used as substrates for studying 1,3-PDO production in a 2 L fermenter. After sterilization, the solution mixture was cooled down to 60 °C. The crude lysate enzyme containing amylase (Amy) and amylo-glucosidase (AMG) at concentrations of 100 U and 600 U per gram cassava starch, respectively, was used to hydrolyze the cassava starch (Khor et al. 2016; Khunnonkwao et al. 2020). The cassava hydrolysis was allowed for 4 h at 50 °C for 2 h. After hydrolysis, the seed of E. coli NSK015 strain was inoculated into the medium with the initial OD550 of 0.1 and temperature was maintained at 37 °C during fermentation.
Analytical method
Fermentation broth was collected and separated by centrifugation at 15,000 rpm for 5 min. The supernatant was analyzed with HPLC while cell pellet was mixed with 1 mL DI water and determined the cell biomass. The biomass concentration was determined when three OD550 are approximately 1 g/L cell dry weight (Khunnonkwao et al. 2018). HPLC analysis coupled with an anion exclusion column (Bio-RAD, Aminex, HPX-87H, USA) was used to analyze sugars and fermentative products. The column was controlled at the temperature of 45 °C. The solution of 4 mM H2SO4 was used as a mobile phase with a flow rate of 0.4 mL/min.
Statistical analysis
All experiments were performed in triplicate. All data represent the averages of three fermentations with standard deviations. Data were analyzed with the SPSS program (version 13.0). Duncan’s multiple-ranges test (DMRT) was used to determine differences among mean values at 95% significance level (P < 0.05).
Results
Functional expression of the gdrAB-dhaB123 operon for 1,3-PDO pathway in E. coli
In this study, the glycerol dehydratase operon (dhaB123), catalyzing the conversion of glycerol to 3-HPA, from K. pneumoniae subsp. pneumoniae TISTR1867 was overexpressed with its re-activating factors (gdrAB) in E. coli NSK002. Since dhaB123 and gdrA genes in K. pneumoniae are arranged in the opposite direction to gdrB gene (Rathnasingh et al. 2009), the re-arranged gdrAB-dhaB123 cassette was constructed (pKJ1011). The structural ldhA gene of E. coli NSK002 was simultaneously deleted and replaced by the artificially constructed gdrAB-dhaB123 operon (Fig. S1A). The operon was then stably integrated in frame with the E. coli ldhA promoter. The automated sequencing analysis confirmed that no mutations at the ldhA promoter region and gdrAB-dhaB123 gene were found (Fig. S1B) on the genome of recombinant clone named E. coli NSK012. The aldehyde indicator plate was used to preliminarily screen clones owning an ability to produce aldehydes. The produced aldehyde can react with sodium bisulfite and pararosaniline in ethanol thus developing a dark pink color. Out of 150 clones, only two positive clones of E. coli NSK012 accumulated 3-HPA from glycerol since they developed the dark pink colonies on the aldehyde indicator plate containing glycerol, thus resemble to that of K. pneumoniae while E. coli C wild type did not (Fig. S1C). This result indicated that the artificially constructed gdrAB-dhaB123 operon was successfully functioned in the E. coli NSK012 clones (and its derivatives) under the control of ldhA promoter as a minimally inducible promoter thus sufficiently allowing a glycerol conversion to 3-HPA. All developed strains were also summarized in Fig. S1D.
E. coli NSK012 grew well in mineral salts AM1 medium containing glycerol as a sole carbon source (Fig. 2A) and produced 1,3-PDO from glycerol at the level of 4.63 g/L within 72 h incubation in a shaking flask. Whereas, no 1,3-PDO production was observed by E. coli NSK002 lacking glycerol dehydratase activity (Fig. 2B and C). This demonstrated that an overexpressed glycerol dehydratase in E. coli NSK012 was mandatory to convert glycerol to 3-HPA, an intermediate of 1,3-PDO. Together, results of the enzymatic activity assay showed that E. coli NSK012 possessed the significantly higher activity of glycerol dehydratase at the level of 0.38 ± 0.07 U/min/mg protein compared with that of its parental E. coli NSK002 strain (0.26 ± 0.05 U/min/mg protein). Though, the background activity detected in E. coli NSK002 strain could arise from the presence of other aldehydes (not 3-HPA) in the crude lysate. These aldehydes may include glyceraldehyde-3-phosphate, one of the intermediates in glycolysis that is partially diverted from the consumed glycerol (Fig. 1).


Comparison of growth (A), maximum 1,3-PDO production yield (B), and fermentation profiles (C) in AM1 medium containing 20 g/L glycerol and 100 mM KHCO3 of developed E. coli NSK derivatives in shaking flasks
Improved 1,3-PDO yield caused by deletion of pflB but not the over-expression of yqhD
Acetate up to 2.0 g/L with a few formate was accumulated during 1,3-PDO production by E. coli NSK012 strain (Fig. 2C) even though the strain possessed the deletion of ackA (encoding acetate kinase enzyme). Previous studies demonstrated that the elimination of pflB gene in E. coli and K. oxytoca led the strain lacking the ability to produce acetyl-CoA and formic acid from pyruvate in the central metabolism, thus conserving the carbon flux towards production pathways of desired biochemicals (In et al. 2020; Jampatesh et al. 2019; Sawisit et al. 2018). After the deletion of pflB, E. coli NSK013 considerably increased the level of 1,3-PDO up to 5.8 g/L (Fig. 2C), which was about a 26.1% increase, compared to that of E. coli NSK012 (4.6 g/L). The 1,3-PDO production yield by E. coli NSK013 was significantly improved to be 0.38 mol/mol glycerol used in shaking flask conditions (Fig. 2B). Acetate production declined by three folds in E. coli NSK013 (0.8 g/L), compared to that of E. coli NSK012 (2.4 g/L).
With the hope to increase 1,3-PDO production, pflB gene was simultaneously replaced by the native E. coli yqhD gene thus the 1,3-propanediol oxidoreductase (YqhD) was overexpressed. Results of the enzymatic activity analysis revealed that both E. coli NSK013 and E. coli NSK014 strains showed comparable levels of glycerol dehydratase activity (0.44 ± 0.04 and 0.42 ± 0.04 U/min/mg protein, respectively) compared to that of E. coli NSK012 (0.38 ± 0.07 U/min/mg protein). Additionally, E. coli NSK013 (0.47 ± 0.03 U/min/mg protein) and E. coli NSK014 (0.50 ± 0.07 U/min/mg protein) possessed the YqhD activity at a significantly higher level (about 2 folds increase) than that of E. coli NSK012 (0.25 ± 0.02 U/min/mg protein). Surprisingly, the over-expression of yqhD gene did not result in an increase in 1,3-PDO concentration and yield in E. coli NSK014 compared to those of E. coli NSK013 (Fig. 2B and C). The result indicated that the deletion of pflB provided more effects on improving 1,3-PDO yield even though the YqhD activity was enhanced in E. coli NSK014.
Effect of frdABCD deletion on 1,3-PDO production
E. coli NSK014 produced up to 4.3 g/L succinate as a major by-product (Fig. 2C), which accounted for about 20.7% of the carbon recovery during fermentation. This was likely that E. coli NSK014 as an E. coli C derivative preferably diverted glycerol through the succinate-producing pathway. To further conserve more carbon flux and NADH through 1,3-PDO route, the frdABCD genes (encoding fumarate dehydrogenase; FRD) were therefore deleted from E. coli NSK014. The resulting strain, E. coli NSK015, did not exhibit an impaired growth while the 1,3-PDO yield of 0.43 mol/mol was improved about 7.5% compared to that of E. coli NSK014 (Fig. 2B). Succinate was also abolished in E. coli NSK015 (Fig. 2C). Considering the enzymatic activities, it was surprised that both activities of glycerol dehydratase and YqhD were about two-folds decreases in E. coli NSK015 (0.27 ± 0.06 and 0.24 ± 0.07 U/min/mg protein, respectively) compared to those of E. coli NSK014.
Improved 1,3-PDO yield by the supplementation of glucose but not yeast extract
The yield of 1,3-PDO production reached 0.43 mol/mol glycerol used by E. coli NSK015. Tong and Cameron (1992) suggested that the maximum theoretical yield of 1,3-PDO varied in the range of 0.67–1.0 mol/mol depending on carbon substrates used. The maximum theoretical yield of 1,3-PDO (1.0 mol/mol glycerol used) also depended on the availability of external sources of reducing powers (derived from glucose, xylose, or even H2) and nutritional constraints during fermentation (Tong et al. 1991). An addition of yeast extract (5 g/L) into AM1 medium resulted in a two folds increase of biomass up to 3 g/L (OD550 ≈ 9.0) in both E. coli NSK014 and NSK015 strains compared to those without yeast extract (Fig. 2A). However, the 1,3-PDO yields and concentrations by both strains were not improved when compared to those without yeast extract (Fig. 3A and B). Further investigation was done by the addition of both yeast extract and glucose (20 g/L) into AM1 medium containing glycerol. The 1,3-PDO concentrations and yields (Fig. 3C and D) were dramatically improved up to 0.94 mol/mol glycerol used for both strains, accounting for two folds increase (Fig. 2B). Furthermore, yeast extract was removed from the medium to observe the sole effect of glucose on 1,3-PDO biosynthesis. The yields from both strains (0.90–0.92 mol/mol glycerol used) in the medium containing both glycerol and glucose as co-substrate were comparable to those with the sole supplementation of yeast extract (Figs. 2B and 3E and F). This clearly demonstrated that an only addition of glucose as a co-substrate with glycerol was enough to improve the production yield of 1,3-PDO. Noticeably, our achieved 1,3-PDO yields and even proposed theoretical maximum yield were calculated based on a sole utilization of glycerol by the strains, not a combined glucose and glycerol. This was due to fact that no production of 1,3-PDO by E. coli NSK015 was observed in the medium containing only glucose as a sole carbon source (Fig. S2).

Fermentation profiles of developed E. coli NSK014 and NSK015 strains in AM1 medium containing only 20 g/L glycerol (Gly) and 100 mM KHCO3 with or without glucose (Glu) and yeast extract (YE) in shaking flasks
Improved 1,3-PDO production in 2L fermenter by E. coli NSK015
Most previously reported studies in 1,3-PDO production by natural producers and other engineered E. coli strains were done under anaerobic or even microaerobic conditions (Chen et al., 2003; Tong and Cameron 1992). Though, Tang et al. (2009) reported 1,3-PDO production by an engineered E. coli under dual aerobic/anaerobic phases while Emptage et al. (2003) produced 1,3-PDO under fully aerated conditions. In our experiments, growth and glycerol assimilation of our engineered strains were impaired in AM1 medium containing glycerol (Fig. S3) in shaking flasks (resemble to microaerobic conditions). On the other hand, glycerol was efficiently consumed by all strains when bicarbonate (source of CO2) was supplemented (Fig. 2C) and growths were then not impaired. This was likely that bicarbonate was essential for the growth and utilization of glycerol during 1,3-PDO production for the strains when conditions were not fully aerated as in shaking flasks. To see effects of oxygen on 1,3-PDO production, the fully aerated condition at 2 vvm aeration was allowed when E. coli NSK015 was cultivated in 2 L fermenter in AM1 medium containing co-substrates of glucose and glycerol without the bicarbonate supplementation. The result showed that the growth and utilizations of glucose and glycerol by E. coli NSK015 were repaired even though bicarbonate was not provided (Fig. 4A). Glucose and glycerol were exhausted within 10 and 16 h incubation, respectively. 1,3-PDO was produced at the level of 7.2 g/L with the yield and productivity of 0.82 mol/mol glycerol used and 0.45 g/(L∙h), respectively. Results suggested that the complete aeration was mandatory for 1,3-PDO production by E. coli NSK015 while bicarbonate prevented suboptimal growth and substrate utilization during glycerol fermentation under oxygen-limited conditions. Cintolesi et al. (2012) suggested that E. coli possessed higher activities of glycerol dehydrogenase and dihydroxyacetone kinase when glycerol was supplied together with CO2. This resulted in increasing rates of glycolytic flux and glycerol utilization under microaerobic or anaerobic conditions.

Preliminary optimization of 1,3-PDO production by E. coli NSK015 in 2 L fermenter
Glucose concentrations affected the rate of glycerol utilization
To see the effect of glucose levels on the rate of glycerol utilization, glucose at different concentrations was supplied with 50 g/L glycerol for 1,3-PDO production by E. coli NSK015. Results revealed that the glucose supplementation at levels of 20 g/L detrimentally affected on the glycerol utilization of the strain. Even though the strain grew well and biomass was produced at 2.5 g/L (OD550 ≈ 7.5), the utilization of glycerol was completely stalled after glucose was exhausted at 36 h incubation. This clearly demonstrated that glycerol was co-transported with glucose into cells. The production of 1,3-PDO was only 15.5 ± 0.2 g/L with the productivity of 0.14 ± 0.02 g/(L∙h) (Fig. 4B). With 70 g/L glucose, the high osmotic pressure caused from high combined concentrations of glucose and glycerol also provided adverse effects on substrates utilization by the strain. In Fig. 4C, glucose and glycerol were remained at concentrations of 20.0 and 16.6 g/L, respectively, even the incubation time was prolonged till 108 h. 1,3-PDO was produced at the level of 33.2 ± 0.1 g/L with the productivity of 0.31 ± 0.01 g/(L∙h). On the other hand, 1,3-PDO was produced at the best level of 35.3 ± 0.9 g/L with the yield reaching 1.03 ± 0.05 mol/mol glycerol used and productivity of 0.42 ± 0.08 g/(L∙h) when the co-substrate of glucose and glycerol at 50 g/L each was provided. Glucose and glycerol were simultaneously utilized at comparable rates during the first 48 h incubation. The overall utilization rate of glycerol was thus improved. Glucose was then completely consumed within 72 h followed by a slow consumption of glycerol after that (Fig. 4D). Our results suggested that the suitable ratio of glucose and glycerol at the level of 1:1 allowed an efficient utilization of glycerol for 1,3-PDO production. The further improvement in 1,3-PDO production was attempted at the aeration rate of 1.0 vvm to reduce the power requirement during fermentation. The result showed that glucose was exhausted after 96 h incubation while glycerol remained at the level less than 1 g/L (Fig. 4E). The concentration and yield of 1,3-PDO at 36.8 ± 0.7 g/L and 0.99 ± 0.04 mol/mol glycerol utilized respectively were observed with acetate as a sole by-product of 5.0 g/L. Even though 1,3-PDO production obtained at 1 vvm was comparable to that at 2 vvm, the 1,3-PDO productivity was lower. This may cause from that the overall utilization rates of glucose and glycerol were lower than those at the higher aeration. Based on our experiments, 1,3-PDO yield by E. coli NSK015 was approached the theoretical maximum (1.0 mol/mol glycerol used) proposed by Tong and Cameron (1992).
Valorization of cassava starch for 1,3-PDO production by E. coli NSK015
E. coli NSK015 was further elucidated an ability to co-metabolize glycerol and cassava starch as a source of glucose for the biosynthesis 1,3-PDO to reduce production cost related to the use of a pure glucose. The result revealed that the use of hydrolyzed cassava starch with glycerol did not affect the microbial growth. The cell growth was resembled to that observed when a pure glucose was used in which the maximum biomass was reached 2 g/L after 36 h. Glucose was exhausted within 84 h while about 10 g/L of glycerol remained. However, 1,3-PDO concentration was gradually reduced to 31.9 ± 0.1 g/L with the yield of 0.84 ± 0.01 mol/mol glycerol used. This result suggested that the sustainable production of 1,3-PDO from renewable and cheap agricultural substrates by E. coli NSK015 is promising. Though, acetate was produced with a two-fold increase as a main by-product (11.4 g/L) compared to that of a pure glucose (5.5 g/L) (Fig. 4F). The overall consumption rates of glucose and glycerol by the strain in the condition of cassava starch seemed higher than those of pure glucose. This may be the effect of nitrogen sources in the cassava starch thus accelerating the growth and substrate consumptions. The rapid consumption of glycerol and glucose derived from cassava starch caused an imbalance ratio of NADH/NAD due to a high glycolytic flux. The strain channeled more carbon fluxes through the acetate-producing pathway to prevent detrimental effects to cells due to a high accumulation of NADH. This resulted in more carbon wasting to an acetate excretion rather than 1,3-PDO biosynthesis.
Discussion
Removal of some fermentative genes contributed to an improvement of 1,3-PDO production yield
It is well known that 1,3-PDO production by natural 1,3-PDO producers like K. pneumoniae occurs under anerobic or microaerobic conditions. The maximum theoretical yield under anaerobic conditions has been proposed in range of 0.67–0.875 mol/mol glycerol when some of the glycerol fluxes are wasted to acetate and/or formate during the mixed-acid fermentation (Tong and Cameron 1992). Several groups firmly reported that 1,3-PDO production yield was in the range only of 0.5–0.82 mol/mol glycerol when cultivating their constructed strains under anaerobic or microaerobic conditions (Table 1). To improve the 1,3-PDO yield, the carbon flux should be conserved by deleting some genes encoding enzymes involved in fermentative metabolism. E. coli NSK012 produced acetate and formate at the total level up to 6 g/L during the late log and stationary phases (Figs. 2C and 3) under shaking flasks conditions. This suggested that pyruvate dehydrogenase was likely to be responsible for supplementing acetyl-CoA at the initial growth in the presence of oxygen by an oxidative decarboxylation of pyruvate while the acetate/formate accumulated at later stages of growth could arise from PflB activity resulting from an oxygen limitation due to increasing cell density. The deletion of pflB in E. coli NSK013 showed a significant reduction of acetate production about 75% without the formation of formate. This clearly confirmed that E. coli NSK013 could conserve more carbon fluxes through 1,3-PDO pathway resulting in a significant increase in the 1,3-PDO yield about 41% compared to that of E. coli NSK012 (Fig. 2B).
In addition to pflB, E. coli C possesses relatively high expression of frdABCD genes regardless of level of oxygen and generally converts glycerol to succinate due to its highly reduced nature of glycerol (Dharmadi et al. 2006; Jantama et al. 2008; McCloskey et al. 2018). Since our developed strains were derived from E. coli C, E. coli NSK014 also concomitantly produced 1,3-PDO with succinate as one of the major byproducts to maintain the overall redox balance and to synthesize macromolecules for biomass. The deletion of frdABCD genes from E. coli NSK014 caused an improved 1,3-PDO yield about 7.5% and succinate production was abolished in E. coli NSK015. The results suggested that E. coli NSK015 lacking FRD activity diverted more glycerol flux through 1,3-PDO biosynthesis as a major route for NADH or NADPH reoxidation during glycerol fermentation.
E. coli NSK015 produced 1,3-PDO production approaching the theoretical maximum
In previous studies, the high activity of glycerol dehydratase caused an accumulation of 3-HPA from glycerol. This inhibited cell growth and ceased 1,3-PDO production (Chen et al. 2011; Kim et al. 2014). To overcome this obstacle, overexpression of glycerol dehydratase on low-copy number plasmids and codon optimization were performed to ensure that the glycerol conversion to 3-HPA was not too rapid. However, these attempts caused an imbalance expression and an instability of glycerol dehydratase enzyme thus resulting in the low 1,3-PDO yield and concentration (Lim et al. 2016; Rathnasingh et al. 2009). It has been suggested that the overexpression of glycerol dehydratase should be controlled by appropriate promoters to maintain its suitable enzymatic activity inside cells. Based on the results in our study, the ldhA promoter allowed the suitable employment of a constitutive expression of the gdrAB-dhaB123 operon without the growth retardation in E. coli NSK015 and provided 1,3-PDO production with the high yield up to the theoretical maximum (1.0 mol/mol glycerol used) from glycerol in the presence of glucose. These results indicated that the control expression of glycerol dehydratase by the native host ldhA promoter resulted in a balance between its enzymatic activity suiting the conversion of glycerol to 3-HPA and the intact YqhD activity facilitating the reduction of 3-HPA to 1,3-PDO in the strain.
In E. coli, an endogenous NADPH-dependent aldehyde dehydrogenase/reductase family including YqhD, YjgB, and YahK contributed to the degradation of several aldehydes to desired alcohols (Koma et al. 2012; Rodriguez and Atsumi 2012). Additionally, the NADH-dependent aldo–keto reductase (AKR) superfamily including methylglyoxal reductase had been also demonstrated to catalyze the reduction of carbonyl-containing aldehyde and/or ketone containing compounds to their corresponding alcohols (Di Laccio et al. 2006). In our study, the deletion of yqhD in E. coli NSKD2 caused about 89% reduction of 1,3-PDO (Fig. S4) compared to that of E. coli NSK012. This result suggested that YqhD played a major and specific role in the reduction of 3-HPA to 1,3-PDO to relieve the inhibitory growth effect caused by the accumulation of 3-HPA. Other aldehyde dehydrogenases or reductases may only involve in the non-specific reduction of 3-HPA to 1,3-PDO with a limited extent. Perez et al. (2008) and Li et al. (2008) previously reported the Michalis-Menten constant (Km) and the catalytic constant (Kcat) of a purified YqhD from E. coli. They also confirmed that the purified YqhD catalyzed the in vitro reduction of short-chain aldehydes to alcohols in the NADPH-dependent reaction. They both have suggested that YqhD may have a physiological function by protecting the cell against the detrimental effect of aldehydes derived from lipid oxidation.
Many attempts for overexpressing E. coli yqhD gene were then applied in many microorganisms to accelerate the reduction reaction of 3-HPA to 1,3-PDO. The overexpression of the E. coli yqhD in wildtype K. pnuemoniae increased 1,3-PDO oxidoreductase activity about 100 folds and enhanced the production of 1,3-PDO about 125% (Zhu et al. 2009). An introduction of multiple copies of the E. coli yqhD could also restore 1,3-PDO production in the 1,3-PDO oxidoreductase (DhaT) deficient K. pnuemoniae (Seo et al. 2010). A simultaneous overexpression of K. pneumoniae glycerol dehydratase (dhaB) and E. coli yqhD was performed in Saccharomyces cerevisiae to directly convert glucose to 1,3-PDO. The 1,3-PDO concentration of 0.4 g/L was only obtained from D-glucose though it introduced a new strategy to produce 1,3-PDO from low-cost feedstock (Rao et al. 2008). Additionally, E. coli overexpressing K. pneumoniae dhaB and E. coli yqhD using incompatible plasmids system was developed thus increasing 1,3-PDO level to 13.2 g/L compare to 8.6 g/L of E. coli overexpressing K. pneumoniae dhaT (Wang et al. 2007). Due to many successful works to enhance 1,3-PDO by overexpressing E. coli yqhD, an increased level of YqhD was therefore attempted in E. coli NSK014 to enhance 1,3-PDO production yield. However, no further increase in 1,3-PDO yield was observed in E. coli NSK014 compared to that of E. coli NSK013. These results suggested that the native YqhD activity was sufficient to detoxify the toxicity of 3-HPA and to complete the 1,3-PDO biosynthesis. Our results were in an accordance with the study of Emptage et al. (2003) in which they also demonstrated that E. coli strain was preferable to utilize its intact YqhD to efficiently convert 3-HPA to 1,3-PDO. The further overexpression of 1,3-propanediol oxidoreductase (encoded by dhaT from Klebsiella sp.) in E. coli did not substantially enhance the 1,3-PDO yield.
The rate of 3-HPA reduction by YqhD activity depends upon the availability of reducing powers, especially NADPH or NADH, inside cells thus governing the reaction. The availability of reducing equivalents subjecting to topological constraints owning to different metabolic stages of cells determines the maximum theoretical yield of 1,3-PDO from glycerol (Tong and Cameron 1992). Then, the conversion of 3-HPA by the activity of YqhD is considered a rate-limiting step in the 1,3-PDO biosynthesis in E. coli (Jiang et al. 2015). Several studies successfully enhanced the metabolic shift toward reduction of 3-HPA to 1,3-PDO by supplying an exogenous NADH pool (Yun et al. 2018) or increasing NADPH pools by overexpressing the NADP+-dependent glyceroldehyse-3-phosphate dehydrogenase (Yang et al. 2018). In our experiments, the maximum theoretical yield of 1,3-PDO at 1.0 mol/mol glycerol used was achieved when glucose was used as a co-substrate with glycerol and served as an additional source of reducing equivalents. The rate of glycerol utilization by E. coli NSK015 was greatly improved and 1,3-PDO production was enhanced in the presence of glucose. It was likely that glycerol was simultaneously consumed with glucose even though glucose is a preferred catabolite (Fig. 4). When glucose was only provided as a sole carbon in the medium, 1,3-PDO was not detected (Fig. S2). This confirmed that glucose only served as a source of carbon intermediates during glycolysis to generate ATP and NADH for biosynthesis and cell maintenance but did not involve in 1,3-PDO biosynthesis. Not only glycolysis, but glucose was also partially utilized by hexose monophosphate (HMP) shunt to generate NADPH as a preferred reducing equivalent coupled in the reduction of 3-HPA to 1,3-PDO by the activity of YqhD (Fig. 1). The additional reducing equivalents derived from glucose contributed to the high 1,3-PDO yield (0.99–1.03 mol/mol glycerol) closed to the theoretical maximum by E. coli NSK015. Without glucose, the glycerol flux was channeled through both 1,3-PDO biosynthesis and glycolysis thus limiting 1,3-PDO yield up to only 0.43 mol/mol glycerol used.
Future improvement of 1,3-PDO production by E. coli NSK015
Chromosomal integration of glycerol dehydratase and 1,3-PDO oxidoreductase genes may be optimal for 1,3-PDO production in the industrial scale by E. coli NSK015, as Rathnasingh et al. (2009) have suggested that plasmid-based fermentation causes extra metabolic burden and genetic instabilities. Together, deletion of genes encoding enzymes involving in mixed acid fermentation could prevent the carbon wasting and preserved carbon fluxes through 1,3-PDO biosynthetic pathway. To achieve the theoretical maximum of 1,3-PDO production by E. coli NSK015, the aeration was also required to maximize the cell growth and to efficiently co-transport glycerol and glucose. Even the theoretical maximum 1,3-PDO yield was achieved, the 1,3-PDO concentration and productivity by E. coli NSK015 have not yet been satisfied the industrial criterion for its commercialized scales. The 1,3-PDO production by the strain should be further improved. Since the rate of glycerol utilization depends upon the availability of glucose (Fig. 4), glucose seems to be a more preferred carbon catabolite and may be provided as a sole substrate for 1,3-PDO production. Genes encoding glycerol 3-phosphate dehydrogenase (encoded by dar) and glycerol 3-phosphate phosphatase (encoded by gpp) may be overexpressed for converting glucose to an internal glycerol thus directly channeling through 1,3-PDO production route. Galactose permease (encoded by galP) and glucokinase (encoded by glk) may be also overexpressed to enhance the rate of glucose transportation in E. coli NSK015. By fermentation optimization, fed-batch fermentation with different loading patterns of substrates should be further assessed to improve 1,3-PDO concentration and productivity by E. coli NSK015 thus satisfying its industrial needs.
Data availability
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
References
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Chen Z, Liu H, Liu D (2011) Metabolic pathway analysis of 1,3-propanediol production with a genetically modified Klebsiella pneumoniae by overexpressing an endogenous NADPH-dependent alcohol dehydrogenase. Biochem Eng J 54:151–157
Cintolesi A, Clomburg JM, Rigou V, Zygourakis K, Gonzalez R (2012) Quantitative analysis of the fermentative metabolism of glycerol in Escherichia coli. Biotechnol Bioeng 109:187–198
Conway T, Sewell GW, Osman YA, Ingram LO (1987) Cloning and sequencing of the alcohol dehydrogenase II gene from Zymomonas mobilis. J Bacteriol 169:2591–2597
Datsenko KA, Wanner BL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA 97:6640–6645
Dharmadi Y, Murarka A, Gonzalez R (2006) Anaerobic fermentation of glycerol by Escherichia coli: a new platform for metabolic engineering. Biotechnol Bioeng 94:821–829
Di Laccio E, Elling RA, Wilson DK (2006) Identification of a novel NADH-specific aldo-keto reductase using sequence and structural homologies. Biochem J 400:105–114
Emptage M, Haynie SL, Laffend LA, Pucci JP, Whited G (2003) Process for the biological production of 1,3-propanediol with high titer. United States Patent, US6514733B1
Hong E, Kim J, Ha S, Ryu Y (2015) Improved 1,3-propanediol production by Escherichia coli from glycerol due to Co-expression of glycerol dehydratase reactivation factors and succinate addition. Biotechnol Bioprocess Eng 20:849–855
In S, Khunnonkwao P, Wong N, Phosiran C, Jantama SS, Jantama K (2020) Combining metabolic engineering and evolutionary adaptation in Klebsiella oxytoca KMS004 to significantly improve optically pure D-(−)-lactic acid yield and specific productivity in low nutrient medium. Appl Microbiol Biotechnol 104:9565–9579
Jampatesh S, Sawisit A, Wong N, Jantama SS, Jantama K (2019) Evaluation of inhibitory effect and feasible utilization of dilute acid-pretreated rice straws on succinate production by metabolically engineered Escherichia coli AS1600a. Bioresourc Technol 273:93–102
Jantama K, Zhang X, Moore JC, Shanmugam KT, Svoronos SA, Ingram LO (2008) Eliminating side products and increasing succinate yields in engineered strains of Escherichia coli C. Biotechnol Bioeng 101:881–893
Jantama K, Polyiam P, Khunnonkwao P, Chan S, Sangproo M, Khor K, Jantama SS, Kanchanatawee S (2015) Efficient reduction of the formation of by-products and improvement of production yield of 2,3-butanediol by a combined deletion of alcohol dehydrogenase, acetate kinase-phosphotransacetylase, and lactate dehydrogenase genes in metabolically engineered Klebsiella oxytoca in mineral salts medium. Metab Eng 30:16–26
Jiang W, Zhuang Y, Wang S, Fang B (2015) Directed evolution and resolution mechanism of 1, 3-propanediol oxidoreductase from Klebsiella pneumoniae toward higher activity by error-prone PCR and bioinformatics. PLoS ONE 10:1–10. https://doi.org/10.1371/journal.pone.0141837
Khor K, Sawisit A, Chan S, Kanchanatawee S, Jantama SS, Jantama K (2016) High production yield and specific productivity of succinate from cassava starch by metabolically engineered Escherichia coli KJ122. J Chem Technol Biotechnol 91:2834–2841
Khunnonkwao P, Jantama SS, Kanchanatawee S, Jantama K (2018) Re-engineering Escherichia coli KJ122 to enhance the utilization of xylose and xylose/glucose mixture for efficient succinate production in mineral salt medium. Appl Microbiol Biotechnol 102:127–141
Khunnonkwao P, Jantama SS, Jantama K, Joannis-Cassan C, Taillandier P (2020) Sequential coupling of enzymatic hydrolysis and fermentation platform for high yield and economical production of 2,3-butanediol from cassava by metabolically engineered Klebsiella oxytoca. J Chem Technol Biotechnol 96:1292–1301
Kim K, Kim SK, Park YC, Seo JH (2014) Enhanced production of 3-hydroxypropionic acid from glycerol by modulation of glycerol metabolism in recombinant Escherichia coli. Bioresour Technol 156:170–175
Knietsch A, Bowien S, Whited G, Gottschalk G, Daniell R (2003) Identification and characterization of coenzyme B12-dependent glycerol dehydratase- and diol dehydratase-encoding genes from metagenomic DNA libraries derived from enrichment cultures. Appl Environ Microbiol 69:3048–3060
Koma D, Yamanaka H, Moriyoshi K, Ohmoto SK (2012) Production of aromatic compounds by metabolically engineered Escherichia coli with an expanded shikimate pathway. Appl Environ Microbiol 78:6203–6216
Lee JH, Jung MY, Oh MK (2018a) High-yield production of 1,3-propanediol from glycerol by metabolically engineered Klebsiella pneumoniae. Biotechnol Biofuels 11:1–13
Lee JH, Lama S, Kim JR, Park SH (2018b) Production of 1,3-propanediol from glucose by recombinant Escherichia coli BL21(DE3). Biotechnol Bioprocess Eng 23:250–258
Li H, Chen J, Li Y (2008) Enhanced activity of yqhD oxidoreductase in synthesis of 1,3-propanediol by error-prone PCR. Prog Nat Sci 18:1519–1524
Liang Q, Zhang H, Li S, Qi Q (2011) Construction of stress-induced metabolic pathway from glucose to 1,3-propanediol in Escherichia coli. Appl Microbiol Biotechnol 89:57–62
Lim HG, Noh MH, Jeong JH, Park S, Jung GY (2016) Optimum rebalancing of the 3-hydroxypropionic acid production pathway from glycerol in Escherichia coli. ACS Synth Biol 5:1247–1255
Liu H, Xu Y, Zheng Z, Liu D (2010) 1,3-Propanediol and its copolymers: research, development and industrialization. Biotechnol J 5:1137–1148
Ma Z, Rao Z, Xu L, Liao X, Fang H, Zhuge B, Zhuge J (2009) Production of 1,3-propanediol from glycerol by engineered Escherichia coli using a novel co-expression vector. African J Biotechnol 8:5489–5494
Martinez A, Grabar TB, Shanmugam KT, Yomano LP, York SW, Ingram LO (2007) Low salt medium for lactate and ethanol production by recombinant Escherichia coli B. Biotechnol Lett 29:397–404
McCloskey D, Xu J, Schrübbers L, Christensen HB, Herrgård MJ (2018) RapidRIP quantifies the intracellular metabolome of 7 industrial strains of E. coli. Metab Eng 47:383–392
Oh BR, Lee SM, Heo SY, Seo JW, Kim CH (2018) Efficient production of 1,3-propanediol from crude glycerol by repeated fed-batch fermentation strategy of a lactate and 2,3-butanediol deficient mutant of Klebsiella pneumoniae. Microb Cell Fact 17:1–9. https://doi.org/10.1186/s12934-018-0921-z
Perez JM, Arenas FA, Pradenas GA, Sandoval JM, Vasquez CC (2008) Escherichia coli YqhD exhibits aldehyde reductase activity and protects from the harmful effect of lipid peroxidation-derived aldehydes. J Bio Chem 283:7346–7353
Przystałowska H, Zeyland J, Szymanowska-Powałowska D, Szalata M, Słomski R, Lipiński D (2015) 1,3-propanediol production by new recombinant Escherichia coli containing genes from pathogenic bacteria. Microbiol Res 171:1–7
Rao Z, Ma Z, Shen W, Fang H, Zhuge J, Wang X (2008) Engineered Saccharomyces cerevisiae that produces 1,3-propanediol from D-glucose. J Appl Microbiol 105:1768–1776
Rathnasingh C, Raj SM, Jo JE, Park S (2009) Development and evaluation of efficient recombinant Escherichia coli strains for the production of 3-hydroxypropionic acid from glycerol. Biotechnol Bioeng 104:729–739
Rodriguez G, Atsumi S (2012) Isobutyraldehyde production from Escherichia coli by removing aldehyde reductase activity. Microb Cell Fact 11:90
Sawisit A, Jampatesh S, Jantama SS, Jantama K (2018) Optimization of sodium hydroxide pretreatment and enzyme loading for efficient hydrolysis of rice straw to improve succinate production by metabolically engineered Escherichia coli KJ122 under simultaneous saccharification and fermentation. Bioresourc Technol 260:348–356
Seo JW, Seo MY, Oh BR, Heo SY, Baek JO, Rairakhwada D, Luo LH, Hong WK, Kim CH (2010) Identification and utilization of a 1,3-propanediol oxidoreductase isozyme for production of 1,3-propanediol from glycerol in Klebsiella pneumoniae. Appl Microbiol Biotechnol 85:659–666
Skraly FA, Lytle BL, Cameron DC (1998) Construction and characterization of a 1,3-propanediol operon. Appl Environ Microbiol 64:98–105
Tang X, Tan Y, Zhu H, Zhao K, Shen W (2009) Microbial conversion of glycerol to 1,3-propanediol by an engineered strain of Escherichia coli. Appl Environ Microbiol 75:1628–1634
Tong IT, Cameron DC (1992) Enhancement of 1,3-propanediol production by cofermentation in Escherichia coli expressing Klebsiella pneumoniae dha regulon genes. Appl Biochem Biotechnol 34:149–159
Tong IT, Liao HH, Cameron DC (1991) 1,3-Propanediol production by Escherichia coli expressing genes from the Klebsiella pneumoniae dha regulon. Appl Environ Microbiol 57:3541–3546
Wang F, Qu H, Zhang D, Tian P, Tan T (2007) Production of 1,3-propanediol from glycerol by recombinant Escherichia coli using incompatible plasmids system. Mol Biotechnol 37:112–119
Yang B, Liang S, Liu H, Liu J, Cui Z, Wen J (2018) Metabolic engineering of Escherichia coli for 1,3-propanediol biosynthesis from glycerol. Bioresour Technol 267:599–607
Yun J, Yang M, Magocha TA, Zhang H, Xue Y, Zhang G, Qi X, Sun W (2018) Production of 1,3-propanediol using a novel 1,3-propanediol dehydrogenase from isolated Clostridium butyricum and co-biotransformation of whole cells. Bioresour Technol 247:838–843
Yun J, Zabed HM, Zhang Y, Parvez A, Zhang G, Qi X (2021) Co-fermentation of glycerol and glucose by a co-culture system of engineered Escherichia coli strains for 1,3-propanediol production without vitamin B12 supplementation. Bioresour Technol 319:124218
Zhu MM, Lawman PD, Cameron DC (2002) Improving 1,3-propanediol production from glycerol in a metabolically engineered Escherichia coli by reducing accumulation of sn-glycerol-3-phosphate. Biotechnol Prog 18:694–699
Zhu JG, Li S, Ji XJ, Huang H, Hu N (2009) Enhanced 1,3-propanediol production in recombinant Klebsiella pneumoniae carrying the gene ydhD encoding 1,3-propanediol oxidoreductase isozyme. World J Microbiol Biotechnol 25:1217–1223
Acknowledgements
Author thanks the Thailand Research Fund (TRF) under the Royal Golden Jubilee PhD scholarship (Grant No. PHD/0125/2556) that provided a financial support for this work.
Funding
This study was funded by Thailand Research Fund (TRF) under the Royal Golden Jubilee PhD scholarship (Grant No. PHD/0125/2556).
Author information
Authors and Affiliations
Contributions
KJ conceived, designed research, and performed project administration and funding acquisition. NW conducted experiments and wrote an original draft of the manuscript. KJ also analyzed data, provided comments, and edited and reviewed the final manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wong, N., Jantama, K. Engineering Escherichia coli for a high yield of 1,3-propanediol near the theoretical maximum through chromosomal integration and gene deletion. Appl Microbiol Biotechnol 106, 2937–2951 (2022). https://doi.org/10.1007/s00253-022-11898-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-022-11898-y