
Abstract
Metarhizium species are the most abundant fungi that can be isolated from soil, with a well-known biopesticide capacity. Metarhizium recognizes their hosts when the conidium interacts with insects, where the fungi are in contact with the hydrocarbons of the outermost lipid layer cuticle. These cuticular hydrocarbons comprise a mixture of n-alkanes, n-alkenes, and methyl-branched chains. Metarhizium can degrade insect hydrocarbons and use these hydrocarbons for energy production and the biosynthesis of cellular components. The metabolism of nitroalkanes involves nitronate monooxygenase activity. In this work, we isolated a family of six genes with potential nitronate monooxygenase activity from Metarhizium brunneum. The six genes were expressed in Escherichia coli, and the nitronate monooxygenase activity was verified in the recombinant proteins. Additionally, when the conidia of M. brunneum were grown in medium with nitroalkanes, virulence against Plutella xylostella increased. Furthermore, we analyzed the expression of the six Npd genes during the infection to this insect, which showed differential expression of the six Npd genes during infection.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Nitroalkanes are toxic compounds that can be used as solvents, fuels for rockets, and explosives (Gadda and Fitzpatrick 1999). In nature, toxic nitro-compounds, such as nitroalkanes, are synthesized by some organisms. For example, 3-nitropropionate is produced by the fungi Penicillium atrovenetum and Aspergillus flavus (Porter and Bright 1987) and some leguminous plants such as Astragalus miser and Hippocrepis comosa (Salem et al. 1995). Chomcheon et al. (2005) reported that many legumes accumulate nitro toxins such as 3-nitro-1-propionic acid and may be produced by fungal endophyte association, such as the genus Phomosis. These nitroalkanes could be a plant defense mechanism against pathogens. This observation suggests that many organisms face toxic compounds, such as nitroalkanes, and depend on their ability to metabolize these compounds to survive. Presently, different enzymes from plants, fungi, and bacteria that can convert nitroalkanes to less harmful products have been purified and characterized (Gadda and Francis 2010): the 2-nitropropane dioxygenase, now named nitronate monooxygenase (NMO) (EC 1.13.12.16), from Hansenula mrakii (Kido et al. 1976) and Neurospora crassa (Gorlatova et al. 1998); the 3-nitropropionate oxidase from P. atrovenetum (Porter and Bright 1987) and H. comosa (Hipkin et al. 1999); the nitroalkane oxidase (NAO) (E.C. 1.7.3.1) from Fusarium oxysporum (Kido et al. 1978), Podospora anserina (Tormos et al. 2010), Streptomyces ansochromogenes (Zhang et al. 2002), and Pseudomonas aeruginosa (Lee et al. 2013), among others. Nitroalkane oxidase is a flavoprotein that catalyzes the oxidation of neutral nitroalkanes to their corresponding aldehyde or ketone, releasing nitrite and transferring electrons to O2 to form H2O2 (Fitzpatrick 2017). Nitronate monooxygenase is a flavoprotein enzyme that catalyzes the oxidative denitrification of nitroalkanes to their corresponding carbonyl compounds and nitrites (Gorlatova et al. 1998; Kido et al. 1984). NAO and NMO have differences, and their enzymatic mechanisms are distinct; additionally, NAO exclusively oxidizes neutral nitroalkanes, whereas NMO can oxidize anionic alkyl nitronates (Gadda and Francis 2010). Nitroalkane-oxidizing enzymes can convert nitroalkanes physiologically to obtain the nitrogen and carbon required for growth. In S. ansochromogenes, the nitroalkane oxidase NaoA is involved not only in the conversion of toxic nitroalkanes to less harmful compounds but also in growth (Li et al. 2008). In Ralstonia solanacearum, a putative nitropropane dioxygenase is involved in virulence (Zhang et al. 2017); in the same sense, in Magnaporthe oryzae, which contains five nitronate monooxygenase genes, only the NMO2 gene is essential for mitigating nitro-oxidative cellular damage and, in rice cells, maintaining a redox balance to avoid triggering plant defenses that impact M. oryzae growth (Marroquin-Guzman et al. 2017).
Metarhizium species are the most abundant fungi that can be isolated from soils (Lomer et al. 2001). In addition, Metarhizium spp. are well known as biopesticides by their entomopathogenic lifestyle (Roberts and St Leger 2004). Metarhizium recognizes their hosts when the conidium of the fungus contacts the cuticle of the insect. Later, the conidium germinates on the cuticle and penetrates this barrier of defense to reach the hemocoel where it faces the defense responses of the insects (immune response and toxins) and spreads, invading the tissues of the host and causing death (Arruda et al. 2005). In the early invasion stage of the host, the fungi are in contact with the hydrocarbons of the outermost lipid layer of the cuticle. These cuticular hydrocarbons comprise a mixture of n-alkanes, n-alkenes, and methyl-branched chains in more than 100 insect species (Pedrini et al. 2007). The entomopathogenic fungi Metarhizium and Beauveria can degrade insect hydrocarbons for utilization for energy production and the biosynthesis of cellular components (Huarte-Bonnet et al. 2015).
In entomopathogenic fungi, such as Beauveria bassiana and Metarhizium anisopliae, there is a relationship between the ability to catabolize very long-chain hydrocarbons and virulence parameters (Pedrini et al. 2007). The metabolism of nitroalkanes involves the nitronate monooxygenase enzyme. To investigate whether the Npd genes, putatively encoding this enzymatic activity, are expressed during the entomopathogenic lifestyle of Metarhizium brunneum, and potentially involved in the degradation or metabolism of hydrocarbons from the host cuticle, in this work: we isolated a family of six genes with potential nitronate monooxygenase activity in M. brunneum. The six genes were expressed in Escherichia coli, and the nitronate monooxygenase activity of the recombinant proteins verified. We evaluated the ability of M. brunneum to grow in nitroalkanes and the virulence of the newly formed conidia. M. brunneum conidia produced in the presence of nitroalkanes were more virulent than the conidia grown in the absence of nitroalkanes. Additionally, we measured the expression of the six Npd genes during the M. brunneum infection to Plutella xylostella. The six Npd genes were expressed throughout the infectious cycle, with the Npd1 gene having the highest expression.
Materials and methods
Strains and growth conditions
M. brunneum Ma10 (CNRCB MaPL10) was obtained from the entomopathogenic fungi collection from the “National Biological Control Reference Center” (WDCM 1034) and was originally isolated from Geraeus senilis (Coleoptera: Curculionidae) in the state of Colima, Mexico. The fungus was grown in minimal media (MM) consisting of 0.2% NH4NO3, 2% dextrose, 0.3% KH2PO4, and 2% 50× salt stock solution (25 g MgS04, 0.09 g ZnSO4, 0.05 g FeSO4, 0.015 g MnSO4, and 0.02 g CuSO4), or on Sabouraud dextrose media (SDM) to obtain the conidia or mycelium, respectively, as previously described (Morales Hernandez et al. 2010). To test the ability to grow on nitroalkanes, a drop of 5 × 104 conidia was inoculated in minimal medium (MM), supplemented with different concentrations (0.05%, 0.1%, 0.2%, 0.3%, 0.4%, and 0.5%) of nitroethane, 1-nitropropane, or 2-nitropropane (catalog numbers: 130206, N22851, and 30265, respectively, from Sigma-Aldrich, Toluca, Estado de Mexico, Mexico). The samples were incubated at 28 °C for 3 days to follow growth or 14 days to measure conidiation.
To isolate the Ncd-2 open reading frame (ORF) from N. crassa, strain 74-OR23-1A (FGSC#98) was employed, and the mycelium biomass was obtained by inoculating 5 × 104 conidia per milliliter in potato dextrose broth (PDB) medium and incubating at 30 °C for 48 h.
The E. coli DH5α strain (Invitrogen, Carlsbad, CA, USA) was used for DNA manipulations and transformations. It was maintained in Luria-Bertani medium at 37 °C (Sambrook and Russell 2001). Ampicillin (100 μg mL−1) was added as required. E. coli BL21(DE3) pLysS (Invitrogen, Carlsbad, CA, USA) was utilized for the heterologous expression of the Npd cDNAs.
Nucleic acid isolation and Npd gene cloning
Genomic DNA was isolated from M. brunneum Ma10 mycelium grown for 24 h in SDM. DNA was extracted by friction following standard protocols (Sambrook and Russell 2001).
Total RNA was extracted from cells of M. brunneum Ma10 grown in SDM culture media or from P. xylostella larvae previously inoculated with conidia of M. brunneum Ma10 using TRIzol® Reagent (Invitrogen, Accesolab, Queretaro, Mexico) according to the manufacturer’s instructions. The concentration and purity of both total RNA and DNA were determined by the ratio of the absorbencies at 260 and 280 nm. For cDNA synthesis, all samples were DNase-treated using RNase-Free DNase (Promega Co., Madison, WI, USA) according to the manufacturer’s instructions. Total RNA (1 μg) was used for cDNA synthesis, and RT-PCR was performed using the SuperScript® III One-Step RT-PCR System with the Platinum® Taq DNA Polymerase kit (Invitrogen, Accesolab, Queretaro, Mexico) according to the manufacturer’s instructions.
To amplify the six Npd genes, primers for each gene were designed according to the sequence of the encoding Npd genes from M. brunneum ARSEF 3297 (KID79125, KID72326.1, KID73333.1, KID65375.1, KID78097.1, and KID61795.1) (Supplementary Table S1). The reaction mixture contained total DNA or cDNA, each primer, and PCR SuperMix High Fidelity (Invitrogen, Accesolab, Queretaro, Mexico). The amplification product was cloned into the pCR®2.1-TOPO® (Invitrogen, Accesolab, Queretaro, Mexico) or pGEM®-T Easy (Promega, Quimica Valaner, Cd. de Mexico, Mexico) vectors and sequenced. Both strands of the nucleotide sequence of the clone were generated using universal primers and specific primers based on the Npd genomic sequence deposited into the database (www.ncbi.nlm.nih.gov). The DNA sequence was analyzed using the Lasergene 14 program (DNASTAR Inc., Madison, WI, USA), and its identity was confirmed by comparison with published nitronate monooxygenase sequences in the database (www.ebi.ac.uk).
Neighbor-joining tree based on percentage of identity of the Npd enzymes
The evolutionary history was inferred by using the maximum likelihood method based on the JTT matrix-based model (Jones et al. 1992). The tree with the highest log likelihood (− 8501.48) is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using a JTT model, and then selecting the topology with superior log likelihood value. A discrete gamma distribution was used to model evolutionary rate differences among sites (5 categories (+G, parameter = 1.6233)). The rate variation model allowed for some sites to be evolutionarily invariable ([+I], 1.44% sites). The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. Evolutionary analyses were conducted in MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets (Kumar et al. 2016; www.megasoftware.net).
Expression of the Npd genes from M. brunneum in infected larvae (P. xylostella) by RT-PCR analysis
The in vivo expression analysis of the six Npd genes was performed according to the protocol previously described (Morales Hernandez et al. 2010). Third-instar P. xylostella larvae were sprayed with a conidial suspension (500 μL of 1.0 × 107 conidia mL−1) at 10 lb plg−2, in a Potter spray tower (Potter-Precision Laboratory Spray Tower, Burkard Scientific, Uxbridge, Middlesex, UK). The larvae were fed with a leaf disk of Brassica oleracea var. italica and incubated at 25 °C. Every 48 h, the leaf disk was changed. Different stages of infection were monitored over 9 days. The stages of infection were categorized as previously described (Morales Hernandez et al. 2010). At each time point during the observed infection, 10–40 larvae were collected and stored at − 70 °C until RNA extraction. Upon death, conidia from the infected insect cadavers were collected with sterile 0.1% Triton X-100 solution. From a parallel control experiment, larvae treated only with 0.1% Triton X-100 solution were collected during the experiment. RT-PCR analysis of the Npd genes and AJ274118 (loading control, XM_014687121, ubiquitin-conjugating enzyme) was performed to compare transcript abundance. RT-PCR was performed with specific primers (Supplementary Table S2) using the SuperScript™ III One-step RT-PCR System with the Platinum® Taq DNA Polymerase kit (Invitrogen, Accesolab, Queretaro, Mexico) according to the manufacturer’s recommendations and as previously described (Morales Hernandez et al. 2010).
Expression of the Npd cDNA in E. coli cells
Expression of the M. brunneum Npd cDNAs in E. coli BL21(DE3) pLysS was accomplished by subcloning the Npd cDNA from each of the Npd genes into the pRSET A expression vector (Invitrogen, Accesolab, Queretaro, Mexico). The Npd cDNA was obtained from M. brunneum Ma10 amplified with the respective pair of primers from the cDNA of each one of the Npd genes (Supplementary Table S1) containing a BamHI restriction site in the direct primers and EcoRI or HindIII restriction site in the reverse primers in a reaction mixture with PCR SuperMix High Fidelity (Invitrogen, Accesolab, Queretaro, Mexico). The amplification products previously purified were digested with both restriction enzymes BamHI and EcoRI or BamHI and HindIII and ligated to the BamHI/EcoRI-digested pRSET A DNA plasmid or BamHI and HindIII digested pRSET A DNA plasmid. The resulting recombinant plasmids were designed: pGG468 (Npd1), pGG469 (Npd2), pGG673 (Npd3), pGG674 (Npd4), pGG675 (Npd5), and pGG654 (Npd6), which carry the open reading frame of each of the Npd genes, and pGG658, which contains the Ncd-2 from N. crassa. E. coli BL21(DE3) pLysS cells were transformed independently with each plasmid, and colony selection occurred in LB medium supplemented with ampicillin 100 μg mL−1 and chloramphenicol 34 μg mL−1. For recombinant protein expression and purification, bacterial cells were grown to an OD600 of 0.4 at 37 °C in LB liquid medium supplemented with 100 μg mL−1 ampicillin, 34 μg mL−1 chloramphenicol, and 0.1 mM IPTG for 4 h at 37 °C. Induced cells were collected by centrifugation (8000g for 5 min at 4 °C) and suspended in binding buffer (20 mM phosphate, 500 mM NaCl, 20 mM imidazole with 0.2 mg mL−1 lysozyme, and 20 μg mL−1 DNAse). The collected cells were lysed with a VCX 130 Vibra-Cell™ Ultrasonic Liquid Processor (CTR, Monterrey, NL, Mexico) at 130 W, 20 kHz, and amplitude of 95% over 1 min, repeating 5–6 times. Cell debris was separated by centrifugation (40,000g for 30 min at 4 °C), and the supernatant was passed through a 1-mL HisTrap FF column (G.E. Healthcare, Piscataway, NJ, USA) equilibrated with 50 mM phosphate buffer at pH 7.5 and 20 mM imidazole, according to the directions of the supplier. The proteins bound to the column were eluted with a gradient of 20 to 500 mM imidazole. The Npd proteins were detected at 300 mM imidazole. Fractions were stored at − 70 °C until use in nitronate monooxygenase enzymatic assays.
Enzymatic assays
Nitronate monooxygenase assays were performed according to the protocol previously described (Kido et al. 1984), with minor modifications as follows. The standard reaction mixture contained 20 mM nitroalkane (anionic form), 1.25 μL of 5 mM FMN (flavin mononucleotide), and 50 μL of the enzyme fraction, diluted to a final volume of 0.25 mL adding Britton-Robinson’s buffer (pH 6.5). After incubation at 28 °C for 10 min, the reaction was stopped by the addition of 250 μL of 95% ethanol. Immediately, 50 μL aliquots of the reaction mixture were mixed with 100 μL of deionized water and used for the determination of nitrite with 50 μL of sulfanilamide and 50 μL of N-(l-naphthyl) ethylenediamine dihydrochloride. The reaction mixture was incubated at room temperature for 10 min in the dark. The optical density at 540 nm of the standard solutions and samples were measured in a microplate reader (Varioskan Flash™ Thermo Scientific™, Αccesolab, Queretaro, Mexico). One unit of the enzyme was defined as the amount of enzyme required to produce 1 μmol of nitrite per minute. Specific activity was expressed as units per milligram of protein. Protein content was determined by the Lowry method (Lowry et al. 1951).
The Km values were calculated measuring the enzymatic activity with different concentrations of each substrate in the anionic form (1-nitropropane, 2.5, 5, 10, 20, 40, 60, 120, 140 mM; 2-nitropropane, 2.5, 5, 10, 20, 40, 60, 120 mM; nitroethane, 1.25, 2.5, 5, 10, 20, 40, 60 mM) fitting the experimental points to the Michaelis-Menten equation for one substrate.
Virulence assays
Virulence assays were performed according to the protocol previously described (Morales Hernandez et al. 2010). For each assay, ten third-instar P. xylostella larvae were sprayed with the LD90 (5 × 106 conidia) of M. brunneum Ma10 grown in minimal medium (MM) or MM supplemented with different concentrations (0.05%, 0.1%, 0.2%, 0.3%, 0.4%, or 0.5%) of nitroethane, 1-nitropropane, or 2-nitropropane. The larvae were fed with a leaf disk of B. oleracea var. italica and incubated at 25 °C. Every 48 h, the leaf disk was changed, and the survival was monitored every 12 h during the experiment. Three assays were performed with three replicates each. Statistical tests for survival analysis were performed using the log-rank test and the Gehan-Breslow-Wilcoxon test with the GraphPad Prism 8 software (version 8.3.0) (www.graphpad.com/scientific-software/prism).
Accession number of the Npd genes in the GenBank database
The accession numbers of the Npd genes from M. brunneum Ma10 are the following: Npd1 (FR776002.1); Npd2 (FR776003.1); Npd3 (MN548143); Npd4 (MN548144); Npd5 (MN546869); and Npd6 (MN548145).
Results
M. brunneum contains a nitronate monooxygenase gene family of six members
Analysis of the genome of M. brunneum ARSEF 3297 (Hu et al. 2014) revealed that it contains at least six genes with similarity to nitronate monooxygenase (nitropropane dioxygenase) genes. In this work, we isolated these six genes from the M. brunneum Ma10 strain. The genomic DNA and cDNA of each gene were amplified from M. brunneum Ma10 strain cells grown in the SDM medium using the six pairs of oligos described in Supplementary Table S1, cloned, and sequenced. The comparison between the genomic and cDNA sequences showed that the Npd1 gene contains two introns (110 bp and 160 bp in length, respectively), and Npd5 contains one intron (92 bp). The other genes, Npd2, Npd3, Npd4, and Npd6, do not contain introns. The Supplementary Table S3 shows the predicted amino acid number, molecular weight, and isoelectric point for each Npd protein. The DNA sequence of the Npd1, Npd2, and Npd4 genes from the M. brunneum Ma10 strain is identical to the reported sequences from M. brunneum ARSEF 3297. The DNA sequence of the Npd3, Npd5, and Npd6 genes from the M. brunneum Ma10 strain has small differences with those from the genome sequence of M. brunneum ARSEF 3297 (Hu et al. 2014) (Supplementary Fig. S1).
The sequences of the predicted protein were compared in the databases of the complete genomes from different filamentous fungi, observing that all the fungi analyzed contain between 3 and 8 putative Npd genes in their genomes (Fig. 1). This set included some beneficial microorganisms with different lifestyles, such as M. brunneum, M. anisopliae, Metarhizium robertsii, Metarhizium guizohuense, Metarhizium acridum, B. bassiana, Hypocrea jecorina (anamorph Trichoderma reesei), Trichoderma gamsii, Trichoderma harzianum, and Trichoderma guizohuense. Some phytopathogenic fungi were included, such as Colletotrichum graminicola and M. oryzae, as well as the opportunistic fungus Neosartorya fumigata and the yeast Williopsis saturnus. In the analysis, an outside group was included, the protein sequence, PA1024, from the bacteria P. aeruginosa, an opportunistic pathogen (Ha et al. 2006).

Phylogeny of fungal nitronate monooxygenase. The phylogenic tree of fungal nitronate monooxygenases was inferred by using the maximum likelihood method, with 1000 bootstrap replicates, utilizing MEGA7.0 software. Names of the species are followed by their GenBank accession number
In fungi, few nitronate monooxygenase activities have been described, in the saprophyte fungus N. crassa (Q01284) (Gorlatova et al. 1998) and in the phytopathogenic fungi F. oxysporum (Q8X1D8) (Gadda and Fitzpatrick 1999) and M. oryzae (MGG_02439) (Marroquin-Guzman et al. 2017). In the analyzed fungi, the putative and well-recognized nitronate monooxygenases are distributed in four main clades. Noting that the predicted proteins encoded by the six Npd genes of M. brunneum are highly conserved in the analyzed Metarhizium species (see Supplementary Table S4), and the two putative Npd (Npd2 and Npd3) proteins from M. brunneum are localized in the major clade. The second pair (Npd1 and Npd4) is localized together in a second clade. Npd1 is related to Npd4, and Npd2 is related to Npd3 with 72% identity in each pair (see Supplementary Table S5). Npd6 is related to the nitronate monooxygenase (Q01284) from N. crassa, with an identity between the two of them of 34% (see Supplementary Table S5). Additionally, Npd2 and Npd3 are the most related to Nmo2 from M. oryzae (Fig. 1), a nitronate monooxygenase involved in the nitrosative stress response during blast disease (Marroquin-Guzman et al. 2017).
Comparison of the Npd protein sequences with PA1024, which was initially classified as a nitronate monooxygenase but recently identified as an NADH:quinone reductase (Ball et al. 2016), and PA4202 from P. aeruginosa, which have both been crystallized (Ha et al. 2006; Salvi et al. 2014), showed that the Npd proteins from M. brunneum contain the motifs described for NMO class II (Supplementary Fig. S2): motif I 14P-I-X-Q-A-P-M-X-G-X-S-T-X-X-L-A-A30; motif II 130(V/I)-S-F-H-F-(G/N)-X-P137; motif III 174(V/I)-X-Q-G-X-E-A-G-G-H-R-G-X-F187; motif IV 297(P/A)-(D/E/P)-Y-P-X-X-Y-D-X-X-K-X-L309 (Salvi et al. 2014); and motif V 77ASGGhAD(A/G)RGLhAALALGA(D/E)GhXMGTRF204, described by Ha et al. (2006). Similarly, the catalytic residue His196 from Ncd-2 from N. crassa (Q01284) is conserved in the six Npd proteins from M. brunneum, corresponding to His168 of Npd1, Npd2, Npd3, and Npd4, and in Npd5 and Npd6, corresponding to His153 and His 174, respectively. This catalytic His in all these strains is inside signature motif III, the most conserved motif of the nitronate monooxygenase enzymes.
The six Npd genes encode proteins with nitronate monooxygenase activity
To determine whether the proteins encoded by the Npd genes with similarity to nitronate monooxygenases have this activity, the ORFs of the six genes (Npd1, Npd2, Npd3, Npd4, Npd5, Npd6) and the Ncd-2 from N. crassa were amplified by RT-PCR from RNA from M. brunneum Ma10 cells grown in MDS and N. crassa cells grown in PDB medium and cloned in the pRSET A expression vector and expressed in E. coli cells. The recombinant proteins were overexpressed and purified (Supplementary Fig. S3). The activity of the fractions eluted with 300 mM imidazole, using anionic nitroalkanes nitroethane, 1-nitropropane, and 2-nitropropane as substrates, was measured. The six Npd proteins show activity with at least one of the substrates. All these proteins showed a low Km, suggesting a high affinity for the substrates tested. The Npd1, Npd2, Npd4, and Npd5 proteins showed Km values for 2-nitropropane similar to those previously reported for Ncd-2 of N. crassa (Gorlatova et al. 1998) and 2Npd from H. mrakii (Kido et al. 1984) (Table 1). The M. brunneum Npd1, Npd2, and Npd4 enzymes recognize 2-nitropropane and nitroethane as substrates and have a lower affinity for 1-nitropropane. For Npd3 and Npd6, the preferred substrate is 1-nitropropane. Additionally, Npd6 recognizes nitroethane as a substrate, and Npd3 and Npd6 have a lower affinity for 2-nitropropane. Moreover, for the Npd5 enzyme, its preferred substrates are 1-nitropropane and 2-nitropropane. These results indicate that the proteins encoded by the Npd1, Npd2, Npd3, Npd4, Npd5, and Npd6 genes display nitronate monooxygenase activity, showing differences in the preference for nitroalkane species, and these preferences are independent of the amino acid sequence similarity between them.
M. brunneum Ma10 is capable of growing in nitroalkanes
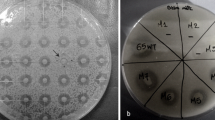
Once it had been determined that this fungus has nitronate monooxygenase activity, Metarhizium was grown in MM supplemented with nitroalkanes at different concentrations. Figure 2 a shows that M. brunneum can grow on nitroalkanes, with 2-nitropropane being the most toxic for the growth of the fungus. Therefore, the fungus can grow on nitroethane and 1-nitropropane at the tested concentrations. Moreover, growth in nitroethane and 2-nitropropane negatively affected the conidiation (Fig. 2b). The conidiation was almost ten times lower in nitroethane 0.3–0.5% compared with the conidiation in the absence of nitroalkane. The conidiation in 2-nitropropane was minimal. Additionally, this adverse effect on conidiation was absent when the fungus grew in 1-nitropropane.

Effect of the nitroalkanes nitroethane, 1-nitropropane, and 2-nitropropane on Metarhizium conidiation. M. brunneum Ma10 conidia were grown in MM supplemented with nitroethane, 1-nitropropane, or 2-nitropropane at 0.05, 01, 0.2, 0.3, 0.4, and 0.5%. aM. brunneum Ma10 colony morphology after 3 days of incubation at 28 °C. bM. brunneum Ma10 conidia number after 14 days of incubation
Conidia obtained from M. brunneum cells grown in nitroalkanes reduce the median lethal time (LT50) in P. xylostella larvae
Since the relationship between the fungal ability to catabolize long-chain hydrocarbons and virulence parameters has been described (Pedrini et al. 2007), we analyzed the capacity of the conidia obtained when M. brunneum was grown in nitroalkanes to infect P. xylostella larvae. In this experiment, the lethal dose 90 (LD90) of conidia was applied to third-instar larvae (Morales Hernandez et al. 2010) and followed the survival of the larvae during the experiment. Figure 3 (Supplementary Fig. S4 and Supplementary Table S6) shows that in the population of larvae treated with conidia from the fungus growth on MM, it kills half of the larvae (LT50) in 96 h. Moreover, the conidia obtained from MM supplemented with nitroethane (0.2, 0.3, 0.4%) or 1-nitropropane (0.05 and 0.2%) showed a statistically significant reduction of the LT50 (between 66 and 84 h; 31 to 12.5% reduction). Therefore, conidia obtained from M. brunneum cells grown in nitroalkanes increased their infective capacity.

Insect bioassays. Survival of P. xylostella larvae after the application of conidia from M. brunneum Ma10 grown in MM, or MM supplemented with nitroethane (0.2, 0.3, and 0.4%) or 1-nitropropane (0.05 and 0.2%)
The six Npd genes are expressed during M. brunneum infection of P. xylostella
Some plants, insects, and microorganisms contain nitroalkanes, potentially as defense mechanisms (Chomcheon et al. 2005; Niknam et al. 2003; Porter and Bright 1987; Salem et al. 1995); therefore, it is essential to follow Npd gene expression during M. brunneum infection in insects. We analyzed the relative expression of the six Npd genes over the course of the infection process to third-instar larvae of P. xylostella inoculated with conidia (Fig. 4) using the ubiquitin gene as a constitutive expression gene. This experiment showed that in early infection, the first twelve hours post-inoculation, the Npd1 gene is the most highly expressed, followed by Npd3 and Npd5. Meanwhile, the expression of the Npd2, Npd4, and Npd6 genes is at basal levels. During the invasion process, from 24 to 72 h post-inoculation, the Npd1 gene has the highest level of expression, followed by the genes Npd5, Npd3, and Npd2; meanwhile, expression of the genes Npd4 and Npd6 is at basal levels. Finally, during extrusion of the mycelium from the body insect and conidiation, the next 96–168 h, the Npd genes again increase their expression; the Npd1 gene is the most expressed and, to a lesser extent, the Npd3, Npd5, Npd4, and Npd2 genes, while the Npd6 gene is not expressed at this stage.

Relative Npd gene expression during the infection process P. xylostella. a RT-PCR analysis of the six Npd genes during the infection of third-instar P. xylostella larvae with M. brunneum Ma10. Lane M corresponds to the DNA marker, and lanes 0 to 206 correspond to RT-PCR amplification products from the total RNA of P. xylostella larvae infected with M. brunneum Ma10 at different time points post-infection. Lane I corresponds to the RT-PCR amplification products from the total RNA from uninfected P. xylostella larvae. b Relative expression analysis measured by densitometric analysis
The meaning of these fluctuations in the levels of gene expression of the six Npd genes during the life cycle of the fungus during infection to the insect is not clear. However, the expression of the Npd1, Npd5, and Npd3 genes during this process is notorious. To understand their physiological role in the lifestyle of this fungus, we are currently constructing null mutants of each of these six Npd genes.
Discussion
According to Ball et al. (2016), there are over 5000 genes in the GenBank currently annotated as nitronate monooxygenases (NMOs). Nitronate monooxygenase activity was described many years ago. In the filamentous fungus N. crassa, it was described for the first time as nitroalkane oxidase (Little 1951), afterward as nitropropane dioxygenase (Gorlatova et al. 1998), and now as a nitronate monooxygenase (https://www.uniprot.org/uniprot/Q01284). This Ncd-2 gene from N. crassa was cloned and expressed in E. coli, the resulting protein purified, and kinetic parameters analyzed (Francis et al. 2005). The nitroalkane oxidase (NAO) from F. oxysporum was expressed in E. coli, and the recombinant enzyme characterized. Recombinant NAO has identical kinetic parameters to the native enzyme isolated from F. oxysporum (Daubner et al. 2002).
In this sense, gene expression in E. coli and analysis of the kinetic parameters of the recombinant enzymes is an appropriate tool for the characterization of the gene products that potentially encode for this annotated nitronate monooxygenases.
In this work, the expression of six genes that encode nitronate monooxygenases in M. brunneum was isolated and studied. This work is the first report about the nitronate monooxygenase activity and the respective genes in the genus Metarhizium that display at least four lifestyles in nature: entomopathogenic, mycorrhiza, promoter of growth of plants, and saprophyte. The nitronate monooxygenase enzymes from M. brunneum encoded by the genes Npd1, Npd2, Npd3, Npd4, Npd5, and Npd6 are active on primary and secondary short-length nitroalkanes. These enzymes display Km values similar to the nitroalkane oxidase from F. oxysporum (Gadda and Fitzpatrick 1999) and the nitronate monooxygenases from H. mrakii (Kido et al. 1984) and N. crassa (Gorlatova et al. 1998).
Interestingly, the analyzed fungi, saprophytes, entomopathogens, phytopathogens, and fungi with plant growth-promoting activity contain 3 or more putative nitronate monooxygenase genes. Moreover, microorganisms such as Saccharomyces cerevisiae, Schizosaccharomyces pombe, Kluyveromyces marxianus and, similarly, animal pathogens such as Candida albicans, Sporothrix schenckii, Phaeoacremonium minimum, Trichophyton equinum, and Trichophyton verrucosum, as far as we know, do not contain nitronate monooxygenase genes in their genomes (http://fungi.ensembl.org). This distribution of nitronate monooxygenase genes could be related to the environment in which each microorganism lives, considering that phytopathogens, entomopathogens, and saprophytes have a greater probability to be in contact with nitroalkanes in their niche (Chomcheon et al. 2005; Porter and Bright 1987; Salem et al. 1995).
According to the presence of nitronate monooxygenase activity in Metarhizium spp., the fungus was able to grow in nitroalkanes. Interestingly, we observed an adverse effect of 1-nitropropane and 2-nitropropane on conidiation, with 2-nitropropane being the most toxic, most likely because of their nitroalkane conversion into their corresponding carbonyl and nitrite increases the nitrosative stress by nitric oxide (NO) production, altering the balance of reactive oxygen species (ROS) necessary for conidia differentiation. Nitrite can be a vital source of NO, which can modify the levels of oxidative stress, acting as a signaling molecule in some biological processes, such as responses to abiotic or biotic stresses and development in plants and reproduction in mammals (Canovas et al. 2016).
As mentioned previously, in R. solanacearum, a putative nitropropane dioxygenase is involved in virulence (Zhang et al. 2017); in this sense, M. brunneum conidia produced in the presence of nitroalkanes were more virulent than the conidia of mycelium grown in the absence of nitroalkanes. They display a shorter LT50 than that observed in the larvae of P. xylostella treated with conidia from MM without nitroalkanes. This result suggests that the conidia produced in the presence of these compounds can kill the insects faster, probably by the same mechanism suggested by Pedrini et al. (2007); the newly formed conidia contain high levels of enzymes involved in nitroalkane catabolism, which helps the fungus to increase its virulence and/or perhaps the conidia are prepared with a higher level of expression of the Npd genes to confront the defense mechanisms of the insect. Although the Npd gene expression assays on those conidia have not been performed, the genes Npd1 and Npd5 are the most expressed Npd genes in M. brunneum mycelium grown in minimal medium with 2-nitropropane (data not shown).
The predicted protein sequence of nitronate monooxygenases of the Npd1, Npd2, Npd3, Npd4, Npd5, and Npd6 genes from M. brunneum contains the signature sequence motifs observed in other nitronate monooxygenases, i.e., Ncd2 (Q01284) from N. crassa (Gorlatova et al. 1998) and PA4204 from P. aeruginosa (Salvi et al. 2014). These motifs contain residues involved in binding to FMN, and importantly, signature motif III is the most conserved of all the motifs and contains the catalytic His described for the 2-nitropropane dioxygenases (Ha et al. 2006). Interestingly, the six M. brunneum Npd proteins contain the carboxy-terminal peroxisomal targeting signal 1 (PTS-1, (S/A/C)(K/R/H)L), and there are three sequence variations between the six Npd proteins: AKL, SKL, and AHL, suggesting their peroxisomal localization. The M. oryzae genome carries five NMO genes encoding putative nitronate monooxygenase enzymes: MGG_07261 (NMO1); MGG_02439 (NMO2); MGG_02593 (NMO3); MGG_09511 (NMO4); and MGG_01473 (NMO5) (Marroquin-Guzman et al. 2017), and only NMO4 and NMO5 have PTS1 targeting signals (SKL and AKL, respectively). The presence of the SKL tripeptide is not sufficient, and amino acids upstream of this sequence also contribute to the targeting, and more complete PTS1 definitions include at least ~ 10 amino acids of a protein (Notzel et al. 2016). Nevertheless, in our preliminary results by GFP fusion and localization, all the Npd proteins have peroxisomal localization in M. brunneum (unpublished). In fungi, peroxisomes have diverse functions: the biosynthesis of antibiotics, toxins, lysine, biotin, and secondary metabolites; the degradation of amino acids, polyamines, and H2O2; the oxidation of fatty acids; methanol degradation; the glyoxylate cycle; and maintenance of cellular integrity (Smith and Aitchison 2013). The presence of the six Npd proteins in the M. brunneum peroxisomes and the possible role in its life cycle is the next step in our research.
M. brunneum contains at least six Npd functional genes in its genome and is expressed differentially during infection in insect, suggesting that at least the genes Npd1, Npd5, and Npd3 could be essential for this lifestyle. The genes Npd1, Npd5, and Npd3 display a peak of expression during the first 12 h post-inoculation of the third-instar larvae of P. xylostella. This peak coincides with earlier invasion, including germination and appressorium formation. During the invasion of the insect body, the genes Npd1, Npd5, Npd3, and Npd2 are expressed at different levels. Finally, during mycelial merge and conidiation over the insect body, Npd1 and Npd3 increase their expression levels, while Npd5, Npd4, and Npd2 maintain a low level of expression. Only the Npd6 gene is practically not expressed during the lifecycle of this fungus in the insect host. In this fungus, other genes, such as the Odc1 gene (Madrigal Pulido et al. 2011), Cat1 (Morales Hernandez et al. 2010), and Mad1 and Mad2 (Barelli et al. 2011), show fluctuating expression during the invasion process.
We do not know the reason why M. brunneum expresses these genes differentially, and if these variations are different in the Metarhizium-plant interaction. The deletion of each gene will be necessary to analyze their participation in the lifestyles of the fungus.
In conclusion, in this work, we demonstrated that the genes Npd1, Npd2, Npd3, Npd4, Npd5, and Npd6 from M. brunneum encode proteins with nitronate monooxygenase activity that are differentially expressed during invasion of the host P. xylostella, the Npd1 gene with the highest expression during this process. Now, we are working with the null construction of these Npd genes to study their participation in the different lifestyles of this fungus.
References
Arruda W, Lubeck I, Schrank A, Vainstein MH (2005) Morphological alterations of Metarhizium anisopliae during penetration of Boophilus microplus ticks. Exp Appl Acarol 37(3–4):231–244. https://doi.org/10.1007/s10493-005-3818-6
Ball J, Salvi F, Gadda G (2016) Functional annotation of a presumed nitronate monoxygenase reveals a new class of NADH:Quinone reductases. J Biol Chem 291(40):21160–21170. https://doi.org/10.1074/jbc.M116.739151
Barelli L, Padilla-Guerrero IE, Bidochka MJ (2011) Differential expression of insect and plant specific adhesin genes, Mad1 and Mad2, in Metarhizium robertsii. Fungal Biol 115(11):1174–1185. https://doi.org/10.1016/j.funbio.2011.08.003
Canovas D, Marcos JF, Marcos AT, Strauss J (2016) Nitric oxide in fungi: is there NO light at the end of the tunnel? Curr Genet 62(3):513–518. https://doi.org/10.1007/s00294-016-0574-6
Chomcheon P, Wiyakrutta S, Sriubolmas N, Ngamrojanavanich N, Isarangkul D, Kittakoop P (2005) 3-Nitropropionic acid (3-NPA), a potent antimycobacterial agent from endophytic fungi: is 3-NPA in some plants produced by endophytes? J Nat Prod 68(7):1103–1105. https://doi.org/10.1021/np050036a
Daubner SC, Gadda G, Valley MP, Fitzpatrick PF (2002) Cloning of nitroalkane oxidase form Fusarium oxysporum identifies a new member of the acyl-CoA dehydrogenase superfamily. Proc Natl Acad Sci U S A 99(5):2702–2707
Fitzpatrick PF (2017) Nitroalkane oxidase: structure and mechanism. Arch Biochem Biophys 632:41–46. https://doi.org/10.1016/j.abb.2017.05.012
Francis K, Russell B, Gadda G (2005) Involvement of a flavosemiquinone in the enzymatic oxidation of nitroalkanes catalyzed by 2-nitropropane dioxygenase. J Biol Chem 280(7):5195–51204
Gadda G, Fitzpatrick PF (1999) Substrate specificity of a nitroalkane-oxidizing enzyme. Arch Biochem Biophys 363(2):309–313. https://doi.org/10.1006/abbi.1998.1081
Gadda G, Francis K (2010) Nitronate monooxygenase, a model for anionic flavin semiquinone intermediates in oxidative catalysis. Arch Biochem Biophys 493(1):53–61. https://doi.org/10.1016/j.abb.2009.06.018
Gorlatova N, Tchorzewski M, Kurihara T, Soda K, Esaki N (1998) Purification, characterization, and mechanism of a flavin mononucleotide-dependent 2-nitropropane dioxygenase from Neurospora crassa. Appl Environ Microbiol 64(3):1029–1033
Ha JY, Min JY, Lee SK, Kim HS, Kim DJ, Kim KH, Lee HH, Kim HK, Yoon HJ, Suh SW (2006) Crystal structure of 2-nitropropane dioxygenase complexed with FMN and substrate. Identification of the catalytic base. J Biol Chem 281(27):18660–18667. https://doi.org/10.1074/jbc.M601658200
Hipkin CR, Salem MA, Simpson D, Wainwright SJ (1999) 3-Nitropropionic acid oxidase from horseshoe vetch (Hippocrepis comosa): a novel plant enzyme. Biochem J 340(Pt 2):491–495. https://doi.org/10.1042/bj3400491
Hu X, Xiao G, Zheng P, Shang Y, Su Y, Zhang X, Liu X, Zhang S, St Leger RJ, Wang C (2014) Trajectory and genomic determinants of fungal-pathogen speciation and host adaptation. Proc Natl Acad Sci U S A 111(47):16796–16801. https://doi.org/10.1073/pnas.1412662111
Huarte-Bonnet C, Juarez MP, Pedrini N (2015) Oxidative stress in entomopathogenic fungi grown on insect-like hydrocarbons. Curr Genet 61(3):289–297. https://doi.org/10.1007/s00294-014-0452-z
Jones DT, Taylor WR, Thornton JM (1992) The rapid generation of mutation data matrices form protein sequences. Comput Appl Biosci 8:275–282
Kido T, Soda K, Suzuki T, Asada K (1976) A new oxygenase, 2-nitropropane dioxygenase of Hansenula mrakii. Enzymologic and spectrophotometric properties. J Biol Chem 251(22):6994–7000
Kido T, Hashizume K, Soda K (1978) Purification and properties of nitroalkane oxidase from Fusarium oxysporum. J Bacteriol 133(1):53–58
Kido T, Tanizawa K, Inagaki K, Yoshimura T, Ishida K, Soda K (1984) 2-Nitropropane dioxygenase from Hansenula mrakii: re-characterization of the enzyme and oxidation of anionic nitroalkanes. Agric Biol Chem 48(10):2549–2554. https://doi.org/10.1271/bbb1961.48.2549
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33(7):1870–1874. https://doi.org/10.1093/molbev/msw054
Lee JH, Park AK, Oh JS, Lee KS, Chi YM (2013) Expression, purification and preliminary X-ray crystallographic analysis of nitroalkane oxidase (NAO) from Pseudomonas aeruginosa. Acta Crystallogr Sect F Struct Biol Cryst Commun 69(Pt 8):888–890. https://doi.org/10.1107/S1744309113017235
Li Y, Zhang J, Tan H (2008) Identification of a nitroalkane oxidase gene: naoA related to the growth of Streptomyces ansochromogenes. Curr Microbiol 57(6):588–592. https://doi.org/10.1007/s00284-008-9247-0
Little HN (1951) Oxidation of nitroethane by extracts from Neurospora. J Biol Chem 193:347–359
Lomer CJ, Bateman RP, Johnson DL, Langewald J, Thomas M (2001) Biological control of locusts and grasshoppers. Annu Rev Entomol 46:667–702. https://doi.org/10.1146/annurev.ento.46.1.667
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193(1):265–275
Madrigal Pulido J, Padilla Guerrero I, Magana Martinez Ide J, Cacho Valadez B, Torres Guzman JC, Salazar Solis E, Felix Gutierrez Corona J, Schrank A, Jimenez Bremont F, Gonzalez Hernandez A (2011) Isolation, characterization and expression analysis of the ornithine decarboxylase gene (ODC1) of the entomopathogenic fungus, Metarhizium anisopliae. Microbiol Res 166(6):494–507. https://doi.org/10.1016/j.micres.2010.10.002
Marroquin-Guzman M, Hartline D, Wright JD, Elowsky C, Bourret TJ, Wilson RA (2017) The Magnaporthe oryzae nitrooxidative stress response suppresses rice innate immunity during blast disease. Nat Microbiol 2:17054. https://doi.org/10.1038/nmicrobiol.2017.54
Morales Hernandez CE, Padilla Guerrero IE, Gonzalez Hernandez GA, Salazar Solis E, Torres Guzman JC (2010) Catalase overexpression reduces the germination time and increases the pathogenicity of the fungus Metarhizium anisopliae. Appl Microbiol Biotechnol 87(3):1033–1044. https://doi.org/10.1007/s00253-010-2517-3
Niknam V, Ebrahimzadeh H, Maassoumi AA (2003) Toxic nitro compounds in Astragalus species. Biochem Syst Ecol 31:557–562. https://doi.org/10.1016/S0305-1978(02)00179-5
Notzel C, Lingner T, Klingenberg H, Thoms S (2016) Identification of new fungal peroxisomal matrix proteins and revision of the PTS1 consensus. Traffic 17(10):1110–1124. https://doi.org/10.1111/tra.12426
Pedrini N, Crespo R, Juarez MP (2007) Biochemistry of insect epicuticle degradation by entomopathogenic fungi. Comp Biochem Physiol C Toxicol Pharmacol 146(1–2):124–137. https://doi.org/10.1016/j.cbpc.2006.08.003
Porter DJ, Bright HJ (1987) Propionate-3-nitronate oxidase from Penicillium atrovenetum is a flavoprotein which initiates the autoxidation of its substrate by O2. J Biol Chem 262(30):14428–14434
Roberts DW, St Leger RJ (2004) Metarhizium spp., cosmopolitan insect-pathogenic fungi: mycological aspects. Adv Appl Microbiol 54:1–70. https://doi.org/10.1016/S0065-2164(04)54001-7
Salem MA, Williams JM, Wainwright SJ, Hipkin CR (1995) Nitroaliphatic compounds in Hippocrepis comosa and other legumes in the European flora. Phytochemistry 40(1):89–91. https://doi.org/10.1016/0031-9422(95)00346-9
Salvi F, Agniswamy J, Yuan H, Vercammen K, Pelicaen R, Cornelis P, Spain JC, Weber IT, Gadda G (2014) The combined structural and kinetic characterization of a bacterial nitronate monooxygenase from Pseudomonas aeruginosa PAO1 establishes NMO class I and II. J Biol Chem 289(34):23764–23775. https://doi.org/10.1074/jbc.M114.577791
Sambrook JG, Russell R (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Smith JJ, Aitchison JD (2013) Peroxisomes take shape. Nat Rev Mol Cell Biol 14(12):803–817. https://doi.org/10.1038/nrm3700
Tormos JR, Taylor AB, Daubner SC, Hart PJ, Fitzpatrick PF (2010) Identification of a hypothetical protein from Podospora anserina as a nitroalkane oxidase. Biochemistry 49(24):5035–5041. https://doi.org/10.1021/bi100610e
Zhang J, Ma W, Tan H (2002) Cloning, expression and characterization of a gene encoding nitroalkane-oxidizing enzyme from Streptomyces ansochromogenes. Eur J Biochem 269(24):6302–6307. https://doi.org/10.1046/j.1432-1033.2002.03350.x
Zhang W, Li J, Tang Y, Chen K, Shi X, Ohnishi K, Zhang Y (2017) Involvement of NpdA, a putative 2-nitropropane dioxygenase, in the T3SS expression and full virulence in Ralstonia solanacearum OE1-1. Front Microbiol 8:1990. https://doi.org/10.3389/fmicb.2017.01990
Funding
This work was funded by the Consejo Nacional de Ciencia y Tecnología, CONACYT (grant nos. 220780 and 103173), and Universidad de Guanajuato (grant nos. 000014/11 and 511/2015). CQKY, VMBG, and VFA received a fellowship from CONACYT.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
This article does not contain any studies with human participants or vertebrates, performed by any of the authors.
Conflict of interest
All authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 3156 kb)
Rights and permissions
About this article

Cite this article
Cervantes Quintero, K.Y., Padilla Guerrero, I.E., Torres Guzmán, J.C. et al. Members of the nitronate monooxygenase gene family from Metarhizium brunneum are induced during the process of infection to Plutella xylostella. Appl Microbiol Biotechnol 104, 2987–2997 (2020). https://doi.org/10.1007/s00253-020-10450-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-020-10450-0