
Abstract
Bacitracin is a cyclic dodecyl peptide antibiotic that is an effective bacteriocide against Gram-positive and some Gram-negative bacteria. Bacitracin has been widely used as an antibacterial feed additive for livestock since it is not absorbed easily by the intestine and is easily excreted. Precursor availability has been proven to be one of the core factors for bacitracin production by many previous studies. In this study, we focused on enhancing the supply of the precursor amino acid l-ornithine to enhance bacitracin production by Bacillus licheniformis DW2 through systematic metabolic pathway modification. Several genes encoding rate-limiting enzymes for l-ornithine biosynthesis were episomally overexpressed, including argB, rocF, ppnk1, and ppnk2. The results showed that the overexpression of ppnK1 was the most effective for both l-ornithine and bacitracin biosynthesis. Secondly, the competitive branch pathways for l-ornithine biosynthesis were blocked, and the repressor was also deleted to boost l-ornithine biosynthesis. The results suggested that the deletion of genes proB and proJ to prevent proline biosynthesis and the disruption of the gene encoding the arginine repressor ArgR could enhance the intracellular concentration of l-ornithine by 49% and 2.1 times respectively, and the bacitracin production also increased accordingly by 6.6% and 11.9% respectively. Finally, several most effective efforts were combined to construct the optimal strain DW2ΔproBΔproJΔargR::ppnk1. In the optimal strain, the NADPH availability was improved and the expression levels of several essential genes for l-ornithine biosynthesis were upregulated, resulting in the enhancement of both l-ornithine and bacitracin production by 71.4% and 16.5% respectively. The final bacitracin production titer was 950 U/mL, which reached the level for industrial production.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Bacitracin is a commercially important peptide antibiotic produced by microorganism Bacillus licheniformis and Bacillus subtilis (Haavik and Froyshov 1975; Johnson et al. 1945). Like many other peptides produced from Bacillus, bacitracin is nonribosomally synthesized by a large multienzyme complex through a thiotemplate mechanism (Konz et al. 1997). It works by inhibiting cell wall biosynthesis in Gram-positive and some Gram-negative bacteria and has been used as an antibacterial feed additive for over 50 years (Shalak 1971). Bacitracin contains at least ten different dodecapeptides that vary by one or two amino acids, and bacitracin A is the most abundant and active among them (Morris 1994). It is comprised of four amino acids in the d-configuration, including the nonproteinogenic residue l-ornithine (Drablos et al. 1999). Soybean powder is used as the primary organic nitrogen source for industrial production of bacitracin since this inexpensive and commercially available source has the advantage of a slow rate of hydrolysis during the fermentation process (Elander 2003). Supplementation of the key bacitracin amino acids, l-Cys, l-Lys, l-Ile, and l-Glu, into soybean powder-based growth media has resulted in significant enhancement of bacitracin production in B. licheniformis (Wang et al. 2017b). Increased amino acid biosynthesis and uptake in B. licheniformis through improved flux through the TCA cycle resulted in an enlarged intracellular pool of bacitracin-containing amino acids, which led to an increase of bacitracin production (Liu et al. 2018). These previous studies suggest that the supply of precursor amino acids is a crucial factor for bacitracin production. Furthermore, the addition of l-ornithine to the media could improve the yield of bacitracin by 5.8% in a 10-L fermentor (Chen et al. 2013). However, the extracellular addition of l-ornithine is costly and presents considerable challenges for large-scale industrial fermentation of B. licheniformis. In this study, we systematically increased the intracellular pool of l-ornithine by enhancing the biosynthesis capacity of the commercial bacitracin production strain B. licheniformis DW2.
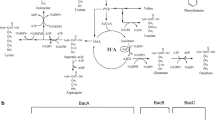
l-ornithine is an essential intermediate of the urea cycle and can be further converted to l-citrulline and l-arginine. In B. licheniformis, l-ornithine is converted from l-glutamate through a cyclic pathway including four enzymes, ArgC, ArgJ, ArgB, and ArgD, which are expressed as an operon and regulated by the arginine repressor, ArgR (Fig. 1). Recently, several microorganisms have been metabolically engineered in order to enhance the production of l-ornithine. For industrial-scale production of l-ornithine, most previous work has been done using Corynebacterium glutamicum as the production strain. Kim et al. developed a C. glutamicum strain capable of producing high levels of l-ornithine by deleting the proB and argF genes to block the competitive branch pathways and regulatory repressor argR of the operon argCJBD (Kim et al. 2015). Hwang and Cho improved the yield of l-ornithine up to 14 g/L by modulating the NADPH supply for l-ornithine production in C. glutamicum (Hwang and Cho 2014). A high l-ornithine-producing C. glutamicum strain was generated by disrupting argF, argR, glutamate transporter Ncgl1221, and proline transporter PutP (Zhang et al. 2017). Zhang et al. overexpressed cg3035 encoding N-acetylglutamate synthase (NAGS) and manipulated the central metabolic pathway, resulting in a recombinant C. glutamicum with a titer of 32.3 g/L l-ornithine (Zhang et al. 2018).

Metabolic pathway and modification strategies for l-ornithine biosynthesis pathway
In this report, we manipulated the metabolic pathway in the industrial bacitracin-producing strain B. licheniformis DW2 (Fig. 1). In order to enhance the intracellular pool of l-ornithine, we deleted proB and proJ to block the carbon flux toward proline, argF and arcB to block the conversion of l-ornithine to citrulline, and the arginine repressor gene argR to enhance expression levels of l-ornithine synthesis genes argCJBD. In addition to the aforementioned deletions, ArgB, the enzyme catalyzing the rate-limiting step to convert glutamate to l-ornithine, and RocF, the enzyme that converts arginine to l-ornithine, were overexpressed. Finally, the enzymes PpnK1 and PpnK2 were overexpressed to increase the level of NADPH available in the cells. Compared with the original strain DW2, the titer of bacitracin increased by 16.5% in the optimal strain DW2ΔproBΔproJΔargR::ppnk1. This study provides an alternative strategy to improve bacitracin production by enhancing de novo production of essential bacitracin amino acids.
Materials and methods
Strains, plasmids, primers, and growth media
Strains and plasmids used in this study are listed in Table 1. Primers were designed based on sequence information from the B. licheniformis ATCC 14580 genome (www.kegg.jp). Escherichia coli DH5α was used as the host for cloning and construction of recombinant plasmids. LB medium was used for starter inocula for both E. coli and B. licheniformis. The bacitracin fermentation medium included 10% soybean meal, 4.5% starch, 0.1% (NH4)2SO4, and 0.6% CaCO3. Starter seed cultures (1 mL) were inoculated into 20 mL of the bacitracin production medium in 250-mL flasks and then cultivated at 37 °C and 230 rpm for 48 h. All the fermentation experiments were repeated at least three replicates.
Construction of episomal expression plasmids
The rocF (GenBank accession no. 3029661), argB (GenBank accession no. 3030197), ppnK1 (GenBank accession no. 3098767), and ppnK2 (GenBank accession no. 3098741 ) genes were independently cloned into the expression vector pHY300PLK (Liu et al.2018). The P43 promoter from B. subtilis 168, the target genes from B. licheniformis ATCC 14580, and the amyL terminator were amplified by PCR using the primer pairs listed in the supplementary materials (Table S1). These three fragments were ligated via splicing overlap-extension PCR (SOE-PCR) with the primers P43-F and amyl-R (Table S1) with about 20 bp homologous ends of the vector. The linearized vector was obtained by PCR using the primers pHY-t5-F/R. Both the vector and the insert were treated by T5 exonuclease and ligated using the ClonExpress One Step Cloning Kit (Vazyme Biotech Co., Ltd, Nanjing, China), according to the manufacturer’s instructions. The recombinant plasmids were verified by sequencing.
Gene deletion in B. licheniformis
The method for gene deletion in B. licheniformis was referred to in our previous study (Liu et al. 2018). Here we describe the procedure using this method for constructing the proB-deficient strain as an example. Briefly, the upstream and downstream homology arms of proB were amplified using the corresponding primers (proB -AF/AR and proB -BF/BR) and fused by SOE-PCR. Then, the fused fragments were digested by restriction enzymes XbaI and SacI, and inserted into the temperature sensitive vector T2(2)-Ori. The resulting recombinant plasmid was confirmed by DNA sequence and named T2- proB. Then, the vector T2- proB was transformed into B. licheniformis DW2 by electroporation, and verified by PCR. Positive transformants were cultivated in LB liquid medium with 20 μg/mL kanamycin at 45 °C, sub-cultured for three generations, and transferred to kanamycin-free LB medium at 37 °C for another five generations. The cells were then plated on LB agar medium with or without kanamycin and incubated for 12 h, and the proB deletion strain was verified by PCR and DNA sequence, named as DW2∆ proB.
Construction of integrant recombinant strains
The expression cassette including the P43 promoter, ppnK1 gene, and amyL terminator were amplified from the plasmid of 300- ppnK1 using primers of PppnK1-F/R (Table 2). Two ~ 500-bp homologous arms comprised of the 5′ and 3′ coding regions of the insertion sites located in a prophage region were amplified by PCR from B. licheniformis DW2 using primer pairs ppnK1-A-F/R and ppnK1-B-F/R, respectively. The three DNA fragments were ligated by SOE-PCR with primer pairs ppnK1-A-F and ppnK1-B-R. Then, the fragment was subcloned into vector T2(2)-ori (Qi et al. 2014) joined by Spe I and Bgl II restriction sites. The resulting plasmids were verified by DNA sequencing. The plasmid T2(2)-ori- ppnK1 was transformed into B. licheniformis DW2∆proB∆proJ cells by electroporation. The integration of ppnK1 was performed following the same procedure as gene deletion described above. The ppnK1 integration strain DW2∆proB∆proJ::ppnK1 was confirmed by PCR with primers ppnK1 -Y-F/R.
Analytical methods
The bacitracin yields were determined using an Agilent 1260 HPLC equipped with Eclipse Plus C18 column (4.6 mm × 150 mm, 3.5 μm) using a previously described method (Wang et al. 2017c). The cell biomass was measured by the dilution agar plate method (Liu et al. 2018). The concentration of l-ornithine was measured using an Agilent 7890b GC equipped with an Agilent HP-5 column (30 m × 0.32 mm × 0.25 μm), according to previous method (Wang et al. 2017c).
Analysis of gene transcription levels by quantitative real-time PCR
The cells were harvested at the middle stage of the exponential phase for total RNA extraction using a FORE GENE kit (Chengdu Fuji Biotechnology Co., Ltd.). First, the insoluble soybean meal was removed from the fermentation broth by low-speed centrifugation at 629×g at 4 °C for 10 min (Liu et al. 2018). A volume of 200 μL supernatant was extracted for cell collection, and 100 μL of lysozyme was added to a final concentration of 15 g/L. The mixture was treated at room temperature for 10–15 min, and 100 μL of the mixture was transferred to a 1.5-mL centrifuge tube. To produce cDNA for qRTPCR from the cell extracts, the following procedures were followed according to the FORE GENE kit instructions (Chengdu Fuji Biotechnology Co., Ltd.). After digestion with RNase-free DNaseI (NEB, USA), the total RNA was used as the template to synthesize cDNA at 42 °C with PrimeScript II 1st Strand cDNA Synthesis Kit (Takara, Japan) according to the manufacturer’s instructions. The resulting cDNA was used as the template for qRT-PCR with the primers listed in Table S2 using the iTaqTM Universal SYBR® Green Supermix (BIO-RAD, USA). The gene adk was served as the reference gene (Zuo et al. 2018), and all assays were performed in triplicate.
Statistics
The transcription levels of genes are presented as a mean value of three replicates. One-way ANOVA was performed, and multiple comparisons were made by Dunnett’s t tests. The significant levels were indicated by * (P < 0.05), ** (P < 0.01).
Results
The addition of l-ornithine can effectively enhance bacitracin production
In order to investigate the effect of l-ornithine on bacitracin production, the titers of bacitracin were measured upon addition of different concentrations of exogenous l-ornithine to the growth media. The results showed that the supplement of l-ornithine could effectively improve bacitracin production (Fig. 2). The titer of bacitracin increased by 10% compared with the control when 40 mg/L L-ornithine was added into the medium. This result was consistent with the previous research which showed that the yield of bacitracin increased by 5.8% with the addition of l-ornithine in 10-L fermentor (Chen et al. 2013). Therefore, we concluded that the endogenous supply of l-ornithine was a limiting factor for bacitracin production. Since the addition of exogenous amino acids was costly and presented great challenges for large-scale industrial fermentation, we planned to increase the de novo biosynthesis of l-ornithine through metabolic modification.

The comparison of bacitracin production in wild type strain DW2 by addition of different concentrations of l-ornithine. At the end of the fermentation, the bacitracin and intracellular ornithine content were sampled at 48 h. The significant levels were indicated by *(P < 0.05), **(P < 0.01)
Effect of overexpression of rate-limiting enzymes involved in l-ornithine biosynthesis
In order to increase l-ornithine biosynthesis, the rate-limiting enzyme ArgB (Jiang et al. 2013a) and an arginase (RocF) that catalyze the conversion of arginine to l-ornithine were overexpressed in B. licheniformis DW2. These two genes were individually inserted into the shuttle vector pHY300plk, and the strain harboring the empty pHY300plk was used as the control. The results showed that the bacitracin production increased by 13.2% with the overexpression of argB, whereas the overexpression of rocF had no significant effect on bacitracin production (Fig. 3a). Compared with the control, the concentration of l-ornithine increased by 33.3% in DW2/300-argB (Fig. 3b), which was consistent with the increase observed for bacitracin production. Since the biosynthesis of l-ornithine and many other amino acids requires NADPH, the genes encoding ATP-NAD kinase, ppnK1 and ppnK2, were overexpressed to enhance the intracellular pool of NADPH. The results showed that only overexpression of ppnK1 could increase the production of l-ornithine (increase of 25.9%) and bacitracin (increase of 31.9%) compared with the control strain (Fig. 3). On the other hand, the concentration of both bacitracin and l-ornithine declined in DW2/300-ppnK2.

The effect of episomal overexpression of rate-limiting genes on l-ornithine and bacitracin production. a The production of l-ornithine; b he production of bacitracin. At the end of the fermentation, the bacitracin and intracellular ornithine content were sampled at 48 h. The significant levels were indicated by *(P < 0.05), **(P < 0.01)
Improvement of l-ornithine production by gene deletions
The competitive branch pathway for biosynthesis of proline was blocked by deleting the genes proB and proJ, and two putative l-ornithine carbamoyltransferase genes argF and arcB were disrupted for l-ornithine accumulation. The successful deletions were confirmed by PCR (Fig. S1). The growth of these two double knock-out strains had no significant differences with the wild type strain DW2 (data not shown), indicating that the supply of proline and arginine from the media was sufficient for the cell growth. Compared with the wild type strain DW2, the bacitracin production increased by 6.6% in the proB and proJ double deletion strain DW2ΔproBΔproJ, but decreased by 7.0% in the argF and arcB double deletion strain DW2 ΔargFΔarcB (Fig. 4a). The intracellular concentration of l-ornithine followed a similar trend as bacitracin production in these two strains, increasing by 49.2% and showed no significant difference respectively (Fig. 4b). The expression of the arg operon for l-ornithine biosynthesis is controlled by the arginine repressor ArgR, and the removal of ArgR has been shown to be an effective strategy for the enhancement of l-ornithine production (Zhang et al. 2017). As expected, the l-ornithine concentration enhanced by 2.1-fold, and the bacitracin production improved by 11.9% in the argR deletion strain DW2ΔargR, when compared with bacitracin production by the wild type strain DW2.

The effect of gene deletions on l-ornithine and bacitracin production. A. The production of l-ornithine; B. The production of bacitracin. At the end of the fermentation, the bacitracin and intracellular ornithine content were sampled at 48 h. The significant levels were indicated by *(P < 0.05), **(P < 0.01)
Construction of the optimal strain for l-ornithine and bacitracin production
We demonstrated that several different B. licheniformis strain modifications, including overexpression of ppnK1 to enhance the NADPH pool, deletion of proB and proJ to block the branch pathway, and deletion of the repressor gene argR, led to significant increases in bacitracin production. Therefore, these strategies were combined to construct the optimal B. licheniformis strain for bacitracin production. Moreover, since harboring the plasmid pHY300PLK could diminish bacitracin production, and episomal expression is not suitable for industrial production, the gene ppnK1 was integrated into the genomic DNA (Fig. S2). The modification included the deletion of argR, proB, and proJ, as well as the integration of ppnK1, and the final strain was named as DW2ΔproBΔproJΔargR::ppnk1. Based on the results, the production of bacitracin and intracellular l-ornithine concentration improved by 16.5% and 71.4% respectively compared with the wild type strain DW2 (Table 2). While the total concentration of NADPH increased in DW2ΔproBΔproJΔargR::ppnk1 compared with the wild type strain, the ratio of NADPH to NADP+ was reduced compared with the wild type strain (Table 2), indicating high flux (NADPH use) for the production of the target products. Monitoring the change in growth rates and bacitracin production for DW2 and DW2ΔproBΔproJΔargR::ppnk1 showed that the strains grew at similar rates for the first 24 h in the exponential phase, and entered the death phase after 24 h (Fig. 5a). The maximal growth yields were achieved at 24 h with the slightly higher cell densities recorded for DW2ΔproBΔproJΔargR::ppnk1 compared with the wild type strain DW2. Bacitracin production of both strains increased gradually until the end of the fermentation (Fig. 5b). Although there were no significant differences in bacitracin production between DW2 and DW2ΔproBΔproJΔargR::ppnk1 during the first 24 h, the DW2ΔproBΔproJΔargR::ppnk1 strain optimized for bacitracin production started to produce more bacitracin (P < 0.05) from 24 to 48 h, which was consistent with the previous study that the biosynthesis of bacitracin mainly occurred in the stationary phase (Wang et al. 2017a).

The growth curve and the time profile of bacitracin production. a Growth curve; b bacitracin production
Transcription analysis of the DW2ΔproBΔproJΔargR::ppnk1
We performed a transcriptional analysis on the genes involved in l-ornithine and bacitracin biosynthesis in order to better understand how these modifications led to increased l-ornithine and bacitracin levels in the optimized bacitracin-producing strain. The following genes were monitored and included genes involved in central metabolism (pgi, pfkA, pyk, and gap), l-ornithine biosynthesis (rocG, gltA, ppnK1, argB, argC, argD, argJ), and bacitracin production ( bacA and bacT). A 4-fold increase in the expression level of ppnK1 was observed in the optimized strain DW2ΔproBΔproJΔargR::ppnk1, indicating that the ppnK1 gene was successfully integrated and overexpressed. The expression levels of the genes pfkA, pyk and gapA, and gapB encoding glycolytic enzymes were increased 2.5-, 2.8-, 1.2-, and 1.7-fold respectively compared with DW2 (Fig. 5b). The expression levels of argCJBD genes involved in l-ornithine biosynthesis in the optimal strain were 1.75-, 2.1-, 1.5-, and 1.4-fold higher than those in DW2 respectively, due to the disruption of the arginine repressor ArgR (Fig. 5b). Since glutamate is an important precursor for l-ornithine production, the genes involved in glutamate biosynthesis, rocG and gltA, were monitored (Fig. 6a). Only rocG gene showed 1.5-fold upregulation, while gltA gene showed no significant difference compared with the control (Fig. 6b). Surprisingly, the expression levels of bacitracin biosynthesis genes were upregulated 1.5-fold in the optimized strain. Therefore, the regulation levels of these two genes were also investigated in the strain DW2ΔargR (Fig. 6c). Similar to the optimized strain, the expression levels of these genes were also elevated, suggesting that upregulation of the bac operon resulted from argR disruption.

The expression levels of genes involved in l-ornithine and bacitracin biosynthesis. a Genes in l-ornithine biosynthesis pathway. b Genes in glycolysis. c Genes in bacitracin biosynthesis. The significant levels were indicated by *(P < 0.05), **(P < 0.01)
Discussion
A number of studies have aimed to improve bacitracin production via genetic modification. For instance, the entire 49-kb bacitracin biosynthesis operon (bacT and bacABC) from B. licheniformis ATCC 10716 was integrated into a B. subtilis strain for heterologous expression, and a high level of bacitracin production was achieved (Eppelmann et al. 2001). The deletion of global transcription regulator AbrB resulted in a ~ 4-fold increase in the expression levels of bacT and the bacABC operon. However, the bacitracin production did not improve with these modifications (Wang et al. 2017a). Therefore, factors other than the expression levels of bacitracin biosynthesis genes play decisive roles in bacitracin production, including the availability of the precursor amino acid building blocks. Zhu et al. deleted the gene encoding the leucine-responsive regulator Lrp, and overexpressed the gene encoding the branch chain amino acid (BCAA) importer BrnQ to increase the intracellular BCAA pool, which resulted in a 22.4% increase of bacitracin production (Zhu et al. 2018). Moreover, Cai et al. enhanced the expression of nitrogen regulators CodY, TnrA, and GlnR to improve the supply of BCAA, which lead to increased bacitracin production (Cai et al. 2019b). The same group also enhanced bacitracin production by blocking an antisense sRNA (aprA) to improve the supply of the precursor amino acids (Cai et al. 2019a). These studies demonstrated that the increased availability of precursor is beneficial to improve bacitracin production. Moreover, the addition of the bacitracin-precursor amino acid l-ornithine has been shown to have a positive effect on bacitracin production (Chen et al. 2013).
In this study, we improved bacitracin production in B. licheniformis via enhancement of de novo biosynthesis of l-ornithine. Based on our results, several genetic modifications, including the disruption of the branch pathway for proline biosynthesis and the arginine repressor AgrR, as well as overexpression of ATP-NAD kinase, led to significant improvement in bacitracin production. Thus, these strategies were combined to construct the optimized bacitracin-producing strain DW2ΔproBΔproJΔargR::ppnk1. NADPH acts as an essential component for the biosynthesis of many metabolites. For instance, overexpression of Zwf and PpnK to improve NADPH supply could effectively enhance l-isoleucine production in C. glutamicum (Shi et al. 2013; Shi et al. 2015). Lysine production could be improved by 60% through altering coenzyme specificity of GAPDH from NADH-dependence to NADPH-dependence to enlarge the intracellular NADPH pool in C. glutamicum (Bommareddy et al. 2014). Additionally, strengthening NADPH supply has been proven to enhance the production of l-ornithine (Jiang et al. 2013b) and bacitracin (Zhu et al. 2019). Compared with the control strain, the intracellular concentration of l-ornithine and bacitracin production increased by 25.9% and 31.9% respectively (Fig. 3) via episomal overexpression of ppnK1 gene, which was consistent with previous research. Considering NADPH plays a crucial role in the biosynthesis of many amino acids, the improvement of bacitracin production could also be due in part to the enhanced availability of other precursor amino acids. Compared with the wild type strain DW2, the intracellular level of NADPH was increased in DW2ΔproBΔproJΔargR::ppnk1, but the NADPH/NADP+ ratio was lower (Table 2). These results indicated that overexpression of ppnk1 effectively increased the pool of NADP+, but that NADP+ had not been effectively converted into NADPH. Therefore, strengthening the expression of NADPH-generating enzymes, such as isocitrate dehydrogenase ICDH, glucose-6-phosphate 1-dehydrogenase Zwf, or glutamate dehydrogenase RocG, in the cells could potentially increase the levels of l-ornithine and benefit bacitracin production.
The expression of the arg operon for l-ornithine biosynthesis is regulated by the arginine repressor ArgR. As expected, after the deletion of argR, the expression levels of argCJBD genes were about 1.5-fold higher than those of the control (Fig. 6a), which was consistent with previous studies on l-ornithine production in C. glutamicum (Kim et al. 2015). Interestingly, the transcription levels of the bacitracin biosynthesis operon also increased after the deletion of ArgR (Fig. 6c), suggesting that the expression of these genes was also repressed by ArgR. Based on previous research, the bac operon was directly repressed by the global transcription regulator AbrB (Wang et al. 2017a). Further research is necessary to identify whether or not ArgR directly represses the bac operon or indirectly through the regulation of AbrB or other transcriptional regulators. Overexpression of glycolytic genes gap, pfkA, and pyk was able to improve l-ornithine production in C. glutamicum (Jiang et al. 2013b; Zhang et al. 2018). The upregulation of these three genes was also observed in DW2ΔproBΔproJΔargR::ppnk1 (Fig. 6b), which could contribute to the improvement of the enhanced l-ornithine production in B. licheniformis. Lee and Cho reported that the availability of glutamate was a rate-limiting factor for l-ornithine production in E. coli (Lee and Cho 2006). Thus, the expression levels of rocG and gltA genes were also monitored in DW2ΔproBΔproJΔargR::ppnk1, and the results showed that the rocG was upregulated while gltA expression was similar to the wild type strain, which is consistent with the previous study that showed that RocG regulated glutamate biosynthesis, while GltA was responsible for glutamate degradation in B. licheniformis (Tian et al. 2017).
In conclusion, bacitracin production using B. licheniformis was successfully improved through enhanced availability of one precursor amino acid, l-ornithine. In optimized bacitracin-producing strain DW2ΔproBΔproJΔargR::ppnk1, the genes proB and proJ were deleted to block the branch pathway, the repressor gene argR was knocked out to enhance the expression of genes involved in l-ornithine biosynthesis and bacitracin biosynthesis operon, and another copy of the ppnK1 gene expressed under a strong promoter was integrated into the genome to increase the supply of the cofactor NADPH. After these modifications, the intracellular l-ornithine was increased by 71.4%, leading to a 16.5% increase in bacitracin production. This study provided a novel strategy for enhancing bacitracin production and developed a strain capable of industrial-level production of bacitracin.
References
Bommareddy RR, Chen Z, Rappert S, Zeng AP (2014) A de novo NADPH generation pathway for improving lysine production of Corynebacterium glutamicum by rational design of the coenzyme specificity of glyceraldehyde 3-phosphate dehydrogenase. Metab Eng 25:30–37. https://doi.org/10.1016/j.ymben.2014.06.005
Cai D, Zhang B, Rao Y, Li L, Zhu J, Li J, Ma X, Chen S (2019a) Improving the utilization rate of soybean meal for efficient production of bacitracin and heterologous proteins in the aprA-deficient strain of Bacillus licheniformis. Appl Microbiol Biotechnol 103:4789–4799. https://doi.org/10.1007/s00253-019-09804-0
Cai D, Zhu J, Zhu S, Lu Y, Zhang B, Lu K, Li J, Ma X, Chen S (2019b) Metabolic engineering of main transcription factors in carbon, nitrogen, and phosphorus metabolisms for enhanced production of bacitracin in Bacillus licheniformis. ACS Synth Biol 8(4):866–875. https://doi.org/10.1021/acssynbio.9b00005
Chen X, Xie F, Zeng X, Li D, Chen S, Li J, Wang Z (2013) Supplementations of ornithine and KNO3 enhanced bacitracin production by Bacillus licheniformis LC-11. Ann Microbiol 64(2):509–514
Drablos F, Nicholson DG, Ronning M (1999) EXAFS study of zinc coordination in bacitracin A. Biochim Biophys Acta 1431(2):433–442
Elander RP (2003) Industrial production of beta-lactam antibiotics. Appl Microbiol and Biotechnol 61(5-6):385–392. https://doi.org/10.1007/s00253-003-1274-y
Eppelmann K, Doekel S, Marahiel MA (2001) Engineered biosynthesis of the peptide antibiotic bacitracin in the surrogate host Bacillus subtilis. J Biol Chem 276(37):34824–34831. https://doi.org/10.1074/jbc.M104456200
Haavik HI, Froyshov O (1975) Function of peptide antibiotics in producer organisms. Nature 254(5495):79–82
Hwang GH, Cho JY (2014) Enhancement of L-ornithine production by disruption of three genes encoding putative oxidoreductases in Corynebacterium glutamicum. J Ind Microbiol Biotechnol 41(3):573–578. https://doi.org/10.1007/s10295-013-1398-8
Jiang LY, Chen SG, Zhang YY, Liu JZ (2013a) Metabolic evolution of Corynebacterium glutamicum for increased production of L-ornithine. BMC Biotechnol 13:47. https://doi.org/10.1186/1472-6750-13-47
Jiang LY, Zhang YY, Li Z, Liu JZ (2013b) Metabolic engineering of Corynebacterium glutamicum for increasing the production of L-ornithine by increasing NADPH availability. J Ind Microbiol Biotechnol 40(10):1143–1151. https://doi.org/10.1007/s10295-013-1306-2
Johnson BA, Anker H, Meleney FL (1945) Bacitracin: a new antibiotic produced by a member of the B. Subtilis group. Science 102(2650):376–377. https://doi.org/10.1126/science.102.2650.376
Kim SY, Lee J, Lee SY (2015) Metabolic engineering of Corynebacterium glutamicum for the production of L-ornithine. Biotechnol Bioeng 112(2):416–421. https://doi.org/10.1002/bit.25440
Konz D, Klens A, Schorgendorfer K, Marahiel MA (1997) The bacitracin biosynthesis operon of Bacillus licheniformis ATCC 10716: molecular characterization of three multi-modular peptide synthetases. Chem Biol 4(12):927–937
Lee YJ, Cho JY (2006) Genetic manipulation of a primary metabolic pathway for L-ornithine production in Escherichia coli. Biotechno Lett 28(22):1849–1856. https://doi.org/10.1007/s10529-006-9163-y
Liu Z, Yu W, Nomura CT, Li J, Chen S, Yang Y, Wang Q (2018) Increased flux through the TCA cycle enhances bacitracin production by Bacillus licheniformis DW2. Appl Microbiol Biotechnol 102:6935–6946. https://doi.org/10.1007/s00253-018-9133-z
Morris M (1994) Primary structural confirmation of components of the bacitracin complex. Biol Mass Spectrom 23(2):61–70. https://doi.org/10.1002/bms.1200230204
Qi G, Kang Y, Li L, Xiao A, Zhang S, Wen Z, Xu D, Chen S (2014) Deletion of meso-2,3-butanediol dehydrogenase gene budC for enhanced D-2,3-butanediol production in Bacillus licheniformis. Biotechnol Biofuels 7(1):16. https://doi.org/10.1186/1754-6834-7-16
Shalak MVI (1971) Bacitracin-a new preparation in poultry farming. Tr Beloruses Set’ShokhozAkad 90:42
Shi F, Li K, Huan X, Wang X (2013) Expression of NAD(H) kinase and glucose-6-phosphate dehydrogenase improve NADPH supply and L-isoleucine biosynthesis in Corynebacterium glutamicum ssp. lactofermentum. Appl Biochem Biotechnol 171(2):504–521. https://doi.org/10.1007/s12010-013-0389-6
Shi F, Li K, Li Y (2015) Comparative proteome analysis of global effect of POS5 and zwf-ppnK overexpression in L-isoleucine producing Corynebacterium glutamicum ssp. lactofermentum. Biotechno Lett 37(5):1063–1071. https://doi.org/10.1007/s10529-015-1768-6
Tian G, Wang Q, Wei X, Ma X, Chen S (2017) Glutamate dehydrogenase (RocG) in Bacillus licheniformis WX-02: enzymatic properties and specific functions in glutamic acid synthesis for poly-gamma-glutamic acid production. Enzyme Microb Technol 99:9–15. https://doi.org/10.1016/j.enzmictec.2017.01.002
Wang D, Wang Q, Qiu Y, Nomura CT, Li J, Chen S (2017a) Untangling the transcription regulatory network of the bacitracin synthase operon in Bacillus licheniformis DW2. Res Microbiol 168(6):515–523. https://doi.org/10.1016/j.resmic.2017.02.010
Wang Q, Zheng H, Wan X, Huang H, Li J, Nomura CT, Wang C, Chen S (2017b) Optimization of inexpensive agricultural by-products as raw materials for bacitracin production in Bacillus licheniformis DW2. Appl Biochem Biotechnol 183(4):1146–1157. https://doi.org/10.1007/s12010-017-2489-1
Wang Q, Zheng H, Wan X, Huang H, Li J, Nomura CT, Wang C, Chen S (2017c) Optimization of inexpensive agricultural by-products as raw materials for bacitracin production in Bacillus licheniformis DW2. Appl Biochem Biotechnol 183:1146–1157. https://doi.org/10.1007/s12010-017-2489-1
Zhang B, Yu M, Wei WP, Ye BC (2018) Optimization of L-ornithine production in recombinant Corynebacterium glutamicum S9114 by cg3035 overexpression and manipulating the central metabolic pathway. Microb Cell Fact 17(1):91. https://doi.org/10.1186/s12934-018-0940-9
Zhang B, Yu M, Zhou Y, Li Y, Ye BC (2017) Systematic pathway engineering of Corynebacterium glutamicum S9114 for L-ornithine production. Microb Cell Fact 16(1):158. https://doi.org/10.1186/s12934-017-0776-8
Zhu J, Cai D, Xu H, Liu Z, Zhang B, Wu F, Li J, Chen S (2018) Enhancement of precursor amino acid supplies for improving bacitracin production by activation of branched chain amino acid transporter BrnQ and deletion of its regulator gene lrp in Bacillus licheniformis. Synth Syst Biotechnol 3(4):236–243. https://doi.org/10.1016/j.synbio.2018.10.009
Zhu S, Cai D, Liu Z, Zhang B, Li J, Chen S, Ma X (2019) Enhancement of bacitracin production by NADPH generation via overexpressing glucose-6-phosphate dehydrogenase Zwf in Bacillus licheniformis. Appl Biochem Biotechnol 187(4):1502–1514. https://doi.org/10.1007/s12010-018-2894-0
Zuo S, Xiao J, Zhang Y, Zhang X, Nomura CT, Chen S, Wang Q (2018) Rational design and medium optimization for shikimate production in recombinant Bacillus licheniformis strains. Process Biochem 66:19–27
Funding
This work was supported by the National Natural Science Foundation of China (Grand No: 31500074)
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical approval
This paper does not contain any studies with human participants or animals performed by any of the authors
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 155 kb)
Rights and permissions
About this article

Cite this article
Yu, W., Li, D., Jia, S. et al. Systematic metabolic pathway modification to boost l-ornithine supply for bacitracin production in Bacillus licheniformis DW2. Appl Microbiol Biotechnol 103, 8383–8392 (2019). https://doi.org/10.1007/s00253-019-10107-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-019-10107-7