
Abstract
Cryopreservation is a common methodology for long-term microalgae storage. Current cryopreservation methods are based on using diverse cryoprotectants and two-step cooling protocols, followed by sample storage at the temperature of liquid nitrogen (− 196 °C). However, the use of this methodology requires a continuous liquid N2 supply as well as facilities with dedicated equipment, which is not affordable for every laboratory. In our work, we report on the successful development of a simple and cost-effective method for the long-term cryogenic storage of Tetradesmus obliquus at temperatures (− 80 °C) used in commonly available deep freezers that are more readily accessible to laboratories. Two procedures were evaluated that were originally devised for other microalgae; this was followed by the optimization of critical parameters such as the sample’s microalgal concentration and the cryoprotectant reagent’s incubation time. Cell viability was monitored using the survival rates obtained by direct agar plating and the growth recovery times in liquid cultures. Viability-related variables were recorded following different storage times of up to 3 years. The main operational factors involved in the process (cell concentration, incubation time, and storage time) were statistically analyzed with regard to their influence on the survival rate. The statistical analysis showed interdependence (a two-factor interaction) between the cellular concentration and the cryoprotectant’s incubation time, on the one hand, and between the incubation time and the storage time on the other. Survival rates above 70% were obtained under optimized conditions after 3 months of storage, along with 20–35% viabilities after 3 years. These results open up the possibility of extending this method to other Scenedesmaceae, or even other microalgal species, and for its use in resource-limited laboratories.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Microalgae comprise a heterogeneous group of photosynthetic microorganisms with great biotechnological potential, about which there has been renewed interest over recent years (Gangl et al. 2015; Kiesenhofer and Fluch 2018). However, one important limitation to developing microalgae biotechnology for wider use is the current inconvenience of preservation methodologies. The maintenance of agar-based algal collections is well-established; however, it is labor intensive, costly, and subject to contamination issues and genetic drift (Fenwick and Day 1992). Consequently, there are efforts being made by researchers to develop suitable preservation protocols. Nowadays, the most common and successful method for microalgal preservation is cryopreservation. This refers to the maintenance of biological samples in a state of “suspended animation” at low (i.e., cryogenic) temperatures (Karlsson and Toner 1996). The aim of the process is to preserve organisms in a state that avoids changes to their morphological, physiological, biochemical, and genetic properties. Cryopreservation can be affected by very diverse factors, for example, the particular species or strain, the cell size and form, the growth phase and rate, the growth medium composition, osmolarity, and culture aeration (Taylor and Fletcher 1998). The basic cryopreservation mechanism is the use of chemical compounds to mitigate ice crystal formation during the freezing process, which is considered the fundamental cause of severe cell damage (Arav et al. 1996; Mazur et al. 1972). Currently, the most robust and reliable technique yielding high cell viability (Taylor and Fletcher 1998; Fleck et al. 2006) is the two-step cryopreservation methodology. This consists of a dehydration and cold acclimation step with subsequent rapid freezing in liquid nitrogen (− 196 °C) to minimize ice crystal formation, followed by long-term storage at the same or a lower temperature. However, this methodology requires a continuous liquid N2 supply as well as facilities with dedicated equipment for an otherwise limited sample storage, thus making the use of this methodology expensive at the laboratory scale.
Members of the genus Tetradesmus, among them T. obliquus (formerly Scenedesmus obliquus; Wynne and Hallan 2016), have become very popular in different fields of limnology, covering aquatic research and technology as well as water management. Part of this success is down to their ability to adapt to harsh environmental conditions, their rapid growth, the ease of cultivating and handling them (Lürling 2003), and their biochemical composition. Tetradesmus species are known as a common food source for herbivorous zooplankton, a biofuel production source because of their high lipid contents, and a great source of high-value nutritional substances such as carotenoids and compounds with antibacterial properties (Chacón-Lee and González-Mariño 2010; Guedes et al. 2011).
The usual methodologies for the cryopreservation of common Tetradesmus species, such as Tetradesmus obliquus, are based on two-step protocols (Morris 1978; Day et al. 1997; Osório et al. 2004). The recommended procedures described by the principal repositories for living cells, such as the American Type-Culture Collection (ATCC), use MetOH as the cryoprotectant and a two-step freezing procedure for the long-term storage of these species at the temperature of liquid N2 (http://www.lgcstandards-atcc.org).
The present work aims to establish a suitable cryogenization protocol for the long-term storage of the microalgal species T. obliquus at − 80 °C, the working temperature of standard lab freezers. Such a methodology (the only one available in the literature) has been developed by GeneArt® (a division of Thermo Fisher Scientific) for Chlamydomonas and Chlorella; hence, we used this as a starting point to find an optimal cryogenization protocol. We evaluated two procedures devised for those particular microalgal species, followed by the optimization of critical parameters to finally obtain suitable conditions for the cryopreservation of T. obliquus.
Materials and methods
Biological material
The microalgae T. obliquus (Turpin; Wynne) was purchased from the Culture Collection of Algae and Protozoa (CCAP 276/3B, Oban, UK). Microalgal cells were freed from bacterial contamination through serial dilution plating, and axenicity of the isolated microalgae was checked by plating in heterotrophic media before using them in our experiments.
Culture media and cryopreservation reagents
Tris-acetate-phosphate (TAP) culture medium was purchased as a ready-made solution from Gibco (Gibco™ TAP Growth Media, Thermo Fisher Scientific, Waltham, MA USA). The GeneArt® Cryopreservation Kit (Thermo Fisher Scientific, Waltham, MA USA) was used for the cryogenization protocols. The kit includes a so-called cryopreservation reagent A, which is added to the culture medium as an optional pre-conditioning step, and a cryopreservation reagent B that is used for final resuspension and cell storage. MetOH p.a. grade (Panreac, Castellar del Vallés, Spain) was used as the cryoprotectant. For growing T. obliquus on solid media, TAP-agar plates were prepared by adding microbiological agar (Panreac, Castellar del Vallés, Spain) to liquid TAP at a concentration of 13 g/L; this was autoclaved before plating.
Tetradesmus obliquus culture and maintenance
The liquid culture of T. obliquus was performed under myxotrophic conditions in TAP medium. Algae were cultured in Erlenmeyer flasks (50 or 100 mL) in growth cabinets at 26 °C under continuous light (100 μmol × m−2 × s−1 intensity) with agitation of 150 rpm. Starter cultures were initiated from Tetradesmus cells grown as spots on TAP-agar plates and gently resuspended in 0.5 mL of TAP, followed by inoculation in 25 mL of the same medium. Growth was monitored by measuring the optical density at 600 nm (OD600), after appropriate dilution, in a spectrophotometer. Prior to the cryogenization treatments, cellular concentrations of T. obliquus cultures were estimated using an equation obtained from regression analysis (Online Resource 1).
Cryopreservation methodology
The following cryogenization procedures (depicted in Online Resource 2) were used:
-
Standard procedure for Chlamydomonas reinhardtii (method A)
T. obliquus cells were grown up to the late-logarithmic phase (an OD600 of around 0.8–0.9) under standard culture conditions. The culture was then re-inoculated at a final OD600 of 0.1 in 25 mL of TAP medium containing 0.5 mL of cryopreservation reagent A (the cell pre-conditioning step) from the GeneArt® Cryopreservation Kit, and cultivation lasted for 3 days (an OD600 of about 0.4). The cells (ca. 2.5 × 107) were harvested from the appropriate volume of culture (as determined from the particular OD and applying the regression equation shown in Online Resource 1) by centrifugation at 2700 rpm (800×g) for 5 min in 15-mL falcon tubes, and the supernatant was carefully removed. Sedimented cells were slowly resuspended in 1 mL of cryopreservation reagent B from the GeneArt® Cryopreservation Kit to give a final cellular concentration of 2.5 × 107 c/mL; they were then incubated at room temperature for 30 min. Aliquots (240 μL) of the cell suspension were placed in cryovials (Sarstedt, ref. 72.693.005, La Roca del Vallés, Spain) and inserted in a Mr. Frosty® freezing container (Thermo Fisher Scientific, Waltham, MA USA) without the sponge insert or added isopropanol. The empty container slots were filled with empty vials to ensure a proper cooling profile (1 °C/min). The freezing container was put into a standard deep freezer (Heraeus, Hanau, Germany) at − 80 °C and after a minimum of 4 h, the vials were quickly moved to a final box container for extended storage at the same temperature (Online Resource 2).
-
Standard procedure for Chlorella vulgaris (method B)
This protocol was identical to that used in method A, except for the absence of the pre-conditioning step and the final cell concentration in cryopreservation reagent B, which in this case was 108 c/mL (Online Resource 2).
-
MetOH procedure (control)
This method was adapted from that described by the ATCC at http://www.lgcstandards-atcc.org for the cryogenization of Tetradesmus species in liquid N2. Briefly, T. obliquus cells were grown to late-logarithmic phase following the procedure explained above. Sedimented cells were carefully resuspended in 2 mL of TAP medium and gently mixed with 2 mL of freshly prepared TAP containing 10% MetOH to attain a final cellular concentration of 107 c/mL. After incubation at room temperature for 10 min, aliquots (500 μL) of cell suspension were placed in cryovials and processed for storage at − 80 °C as described in the previous methods (Online Resource 2).
-
Non-treated control cells
T. obliquus cells were grown and harvested as described above. The pelleted cells were carefully resuspended in 4 mL of TAP medium to attain a final cellular concentration of 107 c/mL. After incubation at room temperature for 10 min, aliquots (500 μL) of cell suspension were placed in cryovials and processed, as described in the previous methods for storage at − 80 °C (Online Resource 2).
Viability estimation procedure
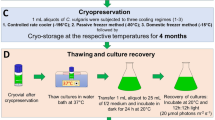
The performance of the cryogenization procedures was evaluated by monitoring the cell viability output. The workflow schedule is shown in Fig. 1.

The workflow used for the simultaneous determination of the parameters related to cell viability, RT and CC (cfu/mL), from samples subjected to cryogenic treatment and storage at − 80 °C (see the explanation in the “Materials and methods” section)
Thawing of samples
The same thawing procedure was used for the different freezing protocols employed. Cryovials containing the frozen cell samples were removed from − 80 °C storage and then immediately placed inside a dry ice container to minimize the damage caused by temperature fluctuations before thawing. Then, the vials were taken out of the container and immediately immersed into a 35 °C water bath. The cells were quickly thawed by gently swirling the vial until the sample thawing was complete (around 1–2 min). As a precaution, 90% ethanol was applied to the vial cap to avoid contamination before opening.
Culture inoculation
In order to make reliable comparisons between the different cryogenization methods, all the starting cultures were adjusted to an identical initial cell concentration (1.2 × 105 c/mL). Starting cultures were inoculated in 25 mL of TAP in 50-mL flasks. For samples from the MetOH procedure, a prior wash step with TAP was included to eliminate the cryoprotectant that might be toxic to the cells once they thawed. For the non-treated cells, a similar wash step was performed for comparative purposes.
Viability assessment
Cell viability after freezing and storage for a given time period was estimated using two “ad hoc” parameters. One was the recovery time (RT), taken as the time required by a microalgae culture to reach half of the exponential growth phase. The second was the survival rate (SR), which was estimated by counting the number of colony-forming units (cfu) growing on the agar plates inoculated with appropriately diluted samples taken from the thawed cryovials (Fig. 1).
Recovery time
TAP liquid cultures were inoculated as previously described, adjusting them to the same initial concentration (1.2 × 105 c/mL). They were cultured under standard conditions over a minimum period of 7 days. The OD600 was recorded from 0.5-mL samples every day, and the OD vs. time curve was obtained (Online Resource 3). The lag phase duration, and therefore the medium logarithmic phase attained, depended (among other factors) on the number of viable cells inoculated at the start of the culture. Consequently, the RT also depended on the number of viable cells, and so it could be used as an indicator of cell viability. The time required by the microalgal cultures to reach half of the exponential growth phase (RT) was graphically determined (Online Resource 3) from the replicate experiments, and the RT means were calculated, along with the standard deviations.
Survival rate
Determining the number of viable microalgal cells was achieved by plating seriated dilutions from the initial culture on TAP-agar medium. A 0.5 mL aliquot was separated from the inoculated TAP medium (see “Cryopreservation methodology”) and used to prepare 1/10 seriated dilutions. Ten microliter aliquots of each dilution (1:1, 1:10, 1:100, and 1:1000) were applied to TAP-agar plates in triplicate, and the plates were grown for a minimum of 10 days in growth cabinets under standard conditions. The number of viable cells was estimated from the cfu number and the actual cell concentrations (cfu/mL) in the vial samples; this was calculated taking into account the dilutions performed. The SR was calculated as the percentage ratio between the sample estimate and the control estimate.
Optimization of the cryogenization method
Cryogenization method A was chosen for the optimization of some of the variables involved in performing this methodology. In particular, the cryoprotectant incubation time (IT) and the cell concentration (CC) of the sample to be frozen were evaluated, together with the storage time (ST). Four different ITs (15 min, 30 min, 45 min, and 60 min), three different CCs (107 c/mL, 2 × 107 c/mL, and 4 × 107 c/mL) with reagent B (the cryoprotectant), and five different STs (1 day, 15 days, 1 month, 3 months, and 3 years) were assessed following a full factorial experimental design with three replicates per experimental combination.
In all the experiments, the samples were evaluated for cell viability under standardized conditions. Inoculations after sample thawing were always adjusted to a final concentration of 1.2 × 105 c/mL prior to cell viability evaluation, as described in the following section, and these were replicated in three identical Erlenmeyer flasks.
Results
Testing of cryopreservation methods for − 80 °C storage
Two parameters were used in the present study to measure the viability of the T. obliquus samples subjected to cryopreservation procedures after storage in a standard deep freezer (− 80 °C). One was the RT (see Online Resource 3). This parameter had the advantage of being simple, fast to obtain, and easy to reference. However, its sensitivity proved to be relatively low since, as will be shown, substantial differences in the number of viable cells are required to have a significant impact on the RT. As an alternative, a direct estimation of the number of viable cells was obtained by counting the number of cfu after appropriate dilutions from the thawed cell samples. This methodology was more laborious but more accurate than the previous one. Using this method, the SRs were estimated.
As a first step, the two standard protocols (Online Resource 2) described in the GeneArt commercial kit for C. reinhardtii and C. vulgaris cryopreservation (here designated as methods A and B, respectively) were applied to evaluate their performance with T. obliquus. The goal was to select a basic method to be used for further optimization. Two control procedures were applied for comparison (Online Resource 2); the non-treated cells were only subjected to direct freezing in the culture medium at a standard cellular density. Additionally, cells were treated with 5% MetOH as a cryoprotectant agent following the protocol recommended by the ATCC for storage of different Tetradesmus species under N2. A small aliquot of the starting culture was used to determine viable CC (cfu/mL) by plating the appropriate dilutions while the bulk of the culture was used for growth monitoring and RT determination (Fig. 1).
Cell viability over 3 months of storage for the two cryogenic treatments (methods A and B) was estimated by calculating the RT of samples experiencing different storage periods. These were compared to the RT obtained for the non-treated cells (NT) and those treated using the ATCC method established for liquid N2 storage based on the use of MetOH. As expected, the RT values for the unfrozen cells (t = 0) were quite similar (around 2 days) for all treatments (Fig. 2a). However, storing cells for just 1 day (t = 1 day) has a clear effect on the NT and MetOH samples, increasing their RT to around 4 days, while it had no appreciable impact on cells treated with the A or B methods (Fig. 2a). The comparison between the RT values at t = 0 day and t = 1 day can be considered as a measure of the impact on viability specifically as a result of the freezing/thawing process. This can be separated from the storage effect, which can be evaluated by studying cell viability over longer periods of time.

(a) RT for T. obliquus cell samples subjected to different treatments before cryogenic storage at − 80 °C. RT values were recorded after different storage times, from 0 day (t = 0) to 3 months (t = 3 months). (b) OD600 vs. time curves showing RT after storage for 3 months at − 80 °C of T. obliquus samples subjected to different cryogenic treatments
When storage was prolonged, a significant increase in RT values was obtained for MetOH-treated cells (Fig. 2). In this case, it took up to 9 days for the samples stored for 3 months to reach the medium log phase, indicating a drastic reduction in cell viability. A moderate rise in NT cells was observed after the same storage period (Fig. 2). A much lower RT increase was recorded for cells subjected to cryogenization method B, while storage over 3 months had virtually no effect on samples treated using method A (Fig. 2). Interestingly, although there was a similar RT increase in the NT and MetOH-treated cells after 1 day of freezing (from 2 to 4 days), the storage effect was greater in MetOH-treated cells (the RT increased from 4 days at t = 1 to 9 days at t = 3 months) than in non-treated cells (the RT increased from 4 to 5 days) (Fig. 2a). This suggests a cytotoxic effect from MetOH at − 80 °C (Best 2015), which was apparently avoided at the lower liquid-N2 temperature (− 196 °C) in the standard protocols.
The determinations of viable CCs and SR after 3 months of storage for samples subjected to the different treatments agreed with the RT values obtained. Figure 3a shows the viable CCs obtained by direct plating on TAP-agar medium (after appropriate serial dilution) of the thawed samples. As expected, a low viable CC (5.8 × 104 cfu/mL) corresponding to an SR of 0.6% (Fig. 3c) was obtained for the NT control cells, while much higher survival values (around 107 cfu/mL) were obtained for samples treated using methods A and B. The SR obtained for method A (ca. 75%) was clearly higher than for method B (ca. 45%), which agreed with the RT values (Fig. 2b). It seems clear that the two cryopreservation methods tested, particularly method A, worked properly by maintaining relatively high viability over a 3-month storage period (ca. 5.8 × 104 cfu/mL).

Viability of T. obliquus samples subjected to different cryogenization methods, after storage for 3 months at − 80 °C. Treatments were as follows: NT, non-treated cells; MetOH, cells subjected to the ATCC protocol designed for the storage of Tetradesmus species in N2; and methods A and B, developed by GeneArt for Chlamydomonas and Chlorella species, respectively. (a) Estimated number of cfu/mL after direct agar plating of the microalgal samples. (b) Same as before but leaving an O/N recovery of the microalgal samples before plating. (c) SR calculated from the same experimental as in a
Remarkably, no viable cells were recorded after plating the MetOH-treated samples, even when growth was recorded in RT experiments after 9 days (Fig. 2b). It is possible that the low number of remaining viable cells, perhaps damaged by the MetOH treatment (Okumura et al. 2001), was the main reason for the lack of growth in solid medium. However, it is also possible that the actual number of viable cells plated in TAP-agar medium was underestimated. To check the latter scenario, a parallel experiment was carried out by plating the cells after O/N recovery (14 h) in liquid TAP medium without shaking. As shown in Fig. 3b, viable cells increased for all treatments (except for the MetOH samples) after O/N recovery, with values ranging from 3.5 times that obtained by direct plating of NT cells to 7.8 times for method A cells. These values were much higher than expected for the generation time of T. obliquus cells, and so this effect was a clear indication that the recovery period allows the growth of a higher fraction of cells. The inclusion of an O/N recovery period after sample thawing would therefore be advisable, from a practical point of view, in order to obtain more viable cells. However, SR estimations using the direct plating of cells, which we also carried out in this work, seemed to be more accurate since it avoided an additional source of variability; namely, cell division during recovery.
In short, method A was more effective for T. obliquus cell cryopreservation, based on the RT and SR values, and it was therefore chosen for further optimization in the experiments described in the following section.
Optimization of experimental conditions for the cryopreservation of Tetradesmus obliquus
Cryogenization method A, which is recommended for the cryogenic storage of C. reinhardtii, was chosen for the optimization of certain variables considered to be causal factors, which might affect cryopreservation performance. In particular, the IT with the cryoprotectant agent, the CC of the sample to be stored, and the ST of the microalgal sample were evaluated in this part of the work. As in the previous section, the RT and the SR were the response variables studied.
In method A, following a pre-conditioning treatment, the Tetradesmus cells were resuspended in the cryoprotectant solution where they remained for a variable time (IT) to allow penetration of the cryoprotectant substance into the cell. This is an important factor since it determines the intracellular concentration of the cryoprotectant and its counteracting effect on ice crystal formation in the cells (Christner 2010; Morris 1978). On the other hand, the CC in the sample was also expected to play an important role, in terms of viability, for various reasons—it can affect cryoprotectant penetration and can also release growth inhibitors (generated from the cell wall) during storage at high cellular densities, which might negatively affect viability (Piasecki et al. 2009). These considerations make it difficult to predict the impact of CC, and so it should be empirically determined along with IT.
As detailed in the “Materials and methods” section, a full factorial design was followed with four pre-freezing ITs, three CCs, and five STs as the main effects on the viability parameters, RT and SR.
The RT values progressively increased with longer STs, although these variations were less than 1 day for all the treatments, even after 3 months of storage (Fig. 4). This indicated that cell viability was good enough to allow the rapid recovery of samples stored over this period. Figure 5 shows the SR over the ST for the Tetradesmus samples subjected to different factor values. In all cases, a progressive reduction in viability over time was observed. This oscillated between 30 and 70% after 3 months of storage. Overall, the best results were obtained when cells were incubated with cryoprotectant for 30 min. Accordingly, high SR values (52–70%) were obtained for all the CCs, as recorded after 3 months of storage. In addition, and in contrast to other incubation periods, at 30-min IT, there was no reduction in viability after 1 day of storage (comparing t = 0 and t = 1 day), which was indicative of the positive role of the cryoprotectant in minimizing the effect of the freezing/thawing cycle. The most effective preservation was obtained at 30 min, with a high CC (4 × 107 c/mL) and an SR of 70% (Fig. 5). Another successful factor combination was obtained with 45-min IT at a low CC (107 c/mL), which resulted in 60% SR (Fig. 5). Interestingly, again no freezing/thawing cycle effect (t = 0 vs. t = 1 day) appeared to happen under these conditions.

RT of T. obliquus cell samples subjected to cryogenization method A, after different STs, and under different IT and CC conditions

SR of T. obliquus cell samples subjected to cryogenization method A, after different STs, and under different IT and CC conditions
To further statistically analyze these trends, we performed a multifactorial analysis of variance (ANOVA) for the SR, using the data from up to 3 months of storage.Footnote 1 All the factors analyzed showed a high statistically significant (P < 0.001) effect on SR and there were some significant two-order interactions between them: SR-CC and IT-ST, which will be discussed later.
Measurements of the SR and RT were made on samples stored up to 3 years. Samples subjected to 30 and 45 min of cryoprotectant treatment showed an SR of around 20% for all the CCs. A relatively high SR (greater than 30%) was also obtained with 15 min of treatment at a high CC. Overall, these data allow us to conclude that viable samples were obtained with different treatments even after such a prolonged storage period at − 80 °C.
Regarding the RT, values of between 4 and 5 days (approximately) were obtained after 3 years of storage under the different cryogenic conditions. However, the differences observed between the RTs did not correlate with their respective SRs after such a long period. As mentioned before, a great variation in the number of viable cells was required to get an appreciable impact on the culture lag period and, consequently, on the time required to reach the exponential phase. Therefore, although the RT seemed to be adequate in detecting marked reductions in cell viability, it was not sensitive enough to detect differences within the range of our optimization experiments.
Discussion
The available methods for the efficient cryogenic storage of T. obliquus, and many other microalgal species, rely on a continuous supply of liquid nitrogen and the use of dedicated equipment and facilities. The development of cheaper and more convenient methodologies, allowing the long-term storage of samples in commonly used deep freezers at − 80 °C, would be a desirable alternative that would also avoid keeping microalgal collections in agar at the laboratory scale, a practice prone to genetic drift and random mutation.
Two different protocols have been developed by GeneArt for the cryogenic storage of two green algae at − 80 °C: C. reinhardtii (method A) and Chlorella vulgaris (method B). In our work, we have shown that method A, which includes a pre-conditioning step, performed better when applied to another green algae species, T. obliquus, resulting in higher SRs. This method was subsequently used as a starting point to optimize certain critical factors such as the CC of samples and the cryoprotectant IT.
Overall, the best results were obtained when Tetradesmus cells were incubated for 30 min in the cryoprotectant. Under these conditions, a non-significant reduction in viability was produced by the freezing/thawing cycle, irrespective of the CC. Moreover, SRs above 70% were obtained after 3 months of storage at − 80 °C for samples with a high CC (4 × 107 cfu/mL), and a viability of around 20% remained after 3 years. Another successful combination was obtained at a lower CC (107 cfu/mL) but at the longer IT of 45 min, showing no reduction in survival after the freezing/thawing cycle, and reaching viabilities of around 60% after 3 months, and above 35% after 3 years of storage.
Finding different optimal condition combinations was indicative of an interaction between the two factors, CC and IT, which was corroborated by ANOVA analysis. Increasing IT from 15 to 30 min had a positive effect on the SR (Figs. 5 and 6); this can be attributed to increased cryoprotectant penetration counteracting intracellular ice crystal formation. However, this effect was conditioned by the CC. While increasing IT resulted in a drastic reduction in cell viability at medium or high CC (Fig. 5), incubation for 45 min strongly improved SR at medium CC (Fig. 6). It has been reported that high CC reduces cryopreservation efficiency because of a low molecular weight toxic substance being enzymatically released from the cell wall upon cell death (Piasecki et al. 2009). It was therefore possible that prolonged cryoprotectant incubation combined with higher CC was generating relatively high levels of such a toxic compound, which was, in turn, affecting survival. Conversely, the production of such a compound would be limited at a lower CC, allowing further survival improvement resulting from longer cryoprotectant incubation; however, lower CC also elicited a reduction in cell viability that cannot be explained so simply. This indicated that SR variation related to the IT was dependent on the CC. The interaction plot for these two factors (Fig. 6) clearly demonstrates this. The SR for both low and high CCs shows a similar trend, with relatively parallel curves, where the maximum SR is attained after 30 min of cryoprotectant incubation. However, at intermediate CC, the interaction plot curve is clearly displaced, showing a maximum SR at 45 min of IT (Fig. 6). This interaction indicates that cell viability is a result of several factors interacting in a complex way. An important consequence of this viability-affecting interaction between factors is that one cannot predict the best value for either CC or IT in terms of cryopreservation efficiency, so these have to be empirically determined with combined experiments.

Interaction plot for the CC and IT pair-factors produced from a multifactorial ANOVA where the main effects were IT, CC, and ST, and the response variable was SR. Non-parallel curves are indicative of factor interaction. Statistical analysis was performed using Statgraphics Centurion XVII (v.17.2) software
The viability evaluation in our experiments was performed routinely by direct TAP-agar plating after diluting the thawed samples. This medium proved suitable for the purpose, unlike others such as the nutrient agar medium (EN ISO 16266), in which no colony growth was obtained even from unfrozen microalgal samples. This might be because the previous microalgae culture was grown in TAP, and therefore failed to adapt to a different medium. Nonetheless, it is possible that cryoprotectant reagent treatment somehow weakens microalgal cells thus contributing to their inability to adapt. Although, as mentioned above, SRs were obtained through direct plating for quantification purposes, we have also shown that overnight microalgae recovery in liquid TAP medium prior to plating dramatically increases the number of viable cells. Therefore, for practical purposes, a recovery step is advisable when plating microalgal cells from cryogenized samples, or when a large number of recovered cells are required.
There are very few works in the literature regarding the cryogenic storage of microalgae at − 80 °C. Benhra et al. (1994) assayed sucrose in combination with either polyvinylpyrrolidone or MetOH for the cryopreservation of Scenedesmus subspicatus using different cooling protocols. The viabilities obtained ranged from 0 to 60% but they were recorded after only 10 days of storage at − 80 °C. A recent work on C. vulgaris (Morschett et al. 2016) used L-proline, ethylene glycol, and DMSO as alternative cryoprotectants, together with a similar cooling methodology to ours. They reported SRs of around 63% in post-thaw experiments although, again, long-term storage was not assayed. The commercial methodology used in this work was initially optimized for C. reinhardtii and Chlorella vulgaris. Successful cryopreservation for these species after 2 years has been reported although the viability results were not published.
In our work, very good viabilities, of around 100%, were obtained in after-thaw (1-day storage) experiments under optimized conditions. However, contrary to what is found with the N2 cryogenization methodologies, a progressive decline in viability is observed at − 80 °C (Figs. 4 and 5). This is probably because, at this temperature, the cells do not reach the completely inactive metabolic state obtained at − 196 °C. Nevertheless, SRs of 20–35% were obtained in this work for T. obliquus after 3 years of storage at − 80 °C. These values are lower than those obtained with the N2 two-step cryogenic methodologies, which have reported viabilities well above 50% (Morris 1978; Day et al. 1997; Osório et al. 2004). However, our results can be regarded as encouraging considering the higher storage temperature used, even with the oscillations caused by opening the freezer in typical laboratory use. Moreover, potential genetic drift following recovery of the cryogenized samples (in the case of Tetradesmus species) is not an issue given that sexual reproduction has rarely been reported, and only under very specific and unusual laboratory conditions (Trainor 1996).
In summary, the successful development of a simple and cost-effective methodology has been reported for the cryogenic storage of T. obliquus at temperatures (− 80 °C) used in common deep freezers, opening up the possibility of extending the procedure to other Scenedesmaceae species.
Notes
Previous statistical analysis showed that the SR did not fulfill the normality requirement. This problem was conveniently resolved by applying the square root transformation to the SR data.
References
Arav A, Zeron Y, Leslie SB, Behboodi E, Anderson GB, Crowe JH (1996) Phase transition temperature and chilling sensitivity of bovine oocytes. Cryobiology 33:589–599. https://doi.org/10.1006/cryo.1996.0062
Benhra A, Ferard JF, Vasseur P (1994) Factorial design to optimize the viability of the alga Scenedesmus subspicatus after cryopreservation. Cryo-Letters 15:269–278
Best BP (2015) Cryoprotectant toxicity: facts, issues, and questions. Rejuvenation Res 18:422–436. https://doi.org/10.1089/rej.2014.1656
Chacón-Lee TL, González-Mariño GE (2010) Microalgae for “healthy” foods-possibilities and challenges. Compr Rev Food Sci Food Saf 9:655–675. https://doi.org/10.1111/j.1541-4337.2010.00132.x
Christner B (2010) Bioprospecting for microbial products that affect ice crystal formation and growth. Appl Microbiol Biotechnol 85:481–489. https://doi.org/10.1007/s00253-009-2291-2
Day JG, Watanabe MM, Morris GJ, Fleck RA, McLellan MR (1997) Long-term viability of preserved eukaryotic algae. J Appl Phycol 9:121–127. https://doi.org/10.1023/A:1007991507314
Fenwick C, Day JG (1992) Cryopreservation of Tetraselmis suecica cultured under different nutrients regimes. J Appl Phycol 4:105–109
Fleck RA, Pickup RW, Day JG, Benson EE (2006) Characterisation of cryoinjury in Euglena gracilis using flow-cytometry and cryomicroscopy. Cryobiology 52:261–268. https://doi.org/10.1016/j.cryobiol.2005.12.003
Gangl D, Zedler JAZ, Rajakumar PD, Martinez EMR, Riseley A, Włodarczyk A, Purton S, Sakuragi Y, Howe CJ, Jensen PE, Robinson C (2015) Biotechnological exploitation of microalgae. J Exp Bot 66:6975–6990
Guedes AC, Amaro HM, Malcata FX (2011) Microalgae as sources of high added-value compounds-a brief review of recent work. Biotechnol Prog 27:597–613
Karlsson JOM, Toner M (1996) Long-term storage of tissues by cryopreservation: critical issues. Biomaterials 17:243–256. https://doi.org/10.1016/0142-9612(96)85562-1
Kiesenhofer DP, Fluch S (2018) The promises of microalgae — still a long way to go. FEMS Microbiol Lett 365. https://doi.org/10.1093/femsle/fnx257
Lürling M (2003) Phenotypic plasticity in the green algae Desmodesmus and Scenedesmus with special reference to the induction of defensive morphology. Ann Limnol Int J Limnol 39:85–101. https://doi.org/10.1051/limn/2003014
Mazur P, Leibo SP, Chu EHY (1972) A two-factor hypothesis of freezing injury. Evidence from Chinese hamster tissue-culture cells. Exp Cell Res 71:345–355. https://doi.org/10.1016/0014-4827(72)90303-5
Morris GJ (1978) Cryopreservation of 250 strains of chlorococcales by the method of two-step cooling. Br Phycol J 13:15–24. https://doi.org/10.1080/00071617800650031
Morschett H, Reich S, Wiechert W, Oldiges M (2016) Simplified cryopreservation of the microalga Chlorella vulgaris integrating a novel concept for cell viability estimation. Eng Life Sci 16:36–44. https://doi.org/10.1002/elsc.201500056
Okumura Y, Koyama J, Takaku H, Satoh H (2001) Influence of organic solvents on the growth of marine microalgae. Arch Environ Contam Toxicol 41:123–128. https://doi.org/10.1007/s002440010229
Osório HC, Laranjeiro CN, Santos LMA, Santos MF (2004) First attempts to cryopreserve strains from the Coimbra Collection of Algae (ACOI) and the use of image analysis to assess viability. Nova Hedwigia 79:227–235. https://doi.org/10.1127/0029-5035/2004/0079-0227
Piasecki BP, Diller KR, Brand JJ (2009) Cryopreservation of Chlamydomonas reinhardtii: a cause of low viability at high cell density. Cryobiology 58:103–109. https://doi.org/10.1016/j.cryobiol.2008.11.001
Taylor R, Fletcher RL (1998) Cryopreservation of eukaryotic algae - a review of methodologies. J Appl Phycol 10:481–501
Trainor FR (1996) Reproduction in Scenedesmus. Korean J Phycol 11:183–201
Wynne MJ, Hallan JK (2016) Reinstatement of Tetradesmus G. M. Smith (Sphaeropleales, Chlorophyta). Feddes Repert 126:83–86. https://doi.org/10.1002/fedr.201500021
Funding
This study was funded by the Ministerio de Economía, Industria y Competitividad, Gobierno de España (grant number AGL2016-74866-C3-2-R).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
This article does not contain any studies with human participants performed by any of the authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 558 kb)
Rights and permissions
About this article

Cite this article
Garrido-Cardenas, J.A., Han, X., Alonso, D.L. et al. Evaluation and optimization of a methodology for the long-term cryogenic storage of Tetradesmus obliquus at − 80 °C. Appl Microbiol Biotechnol 103, 2381–2390 (2019). https://doi.org/10.1007/s00253-019-09650-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-019-09650-0