
Abstract
Isoprene is a useful phytochemical with high commercial values in many industrial applications including synthetic rubber, elastomers, isoprenoid medicines, and fossil fuel. Currently, isoprene is on large scale produced from petrochemical sources. An efficient biological process for isoprene production utilizing renewable feedstocks would be an important direction of research due to the fossil raw material depletion and air pollution. In this study, we introduced the mevalonate (MVA) pathway genes/acetoacetyl-coenzyme A thiolase (mvaE) and MVA synthase (mvaS) from Enterococcus faecalis (E. faecalis); MVA kinase (mvk) derived from Methanosarcina mazei (M. mazei); and phosphomevalonate kinase (pmk), diphosphomevalonate decarboxylase (mvaD), and isopentenyl diphosphate isomerase (idi) from Streptococcus pneumoniae (S. pneumoniae) to accelerate dimethylallyl diphosphate (DMAPP) accumulation in Escherichia coli (E. coli). Together with a codon-optimized isoprene synthase (ispS) from Populus alba (P. alba), E. coli strain succeeded in formation of isoprene. We then manipulated the heterologous MVA pathway for high-level production of isoprene, by controlling the gene expression levels of the MVA pathway genes. We engineered four E. coli strains which showed different gene expression levels and different isoprene productivities, and we also characterized them with quantitative real-time PCR and metabolite analysis. To further improve the isoprene titers and release the toxicity to cells, we developed the extraction fermentation by adding dodecane in cultures. Finally, strain BL2T7P1TrcP harboring balanced gene expression system produced 587 ± 47 mg/L isoprene, with a 5.2-fold titer improvement in comparison with strain BL7CT7P. This work indicated that a balanced metabolic flux played a significant role to improve the isoprene production via MVA pathway.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Isoprene (2-methyl-1, 3-butadiene) can be utilized as a platform chemical for production of high-value bio-based chemicals, such as the polymer building block of synthetic rubber, elastomers, and isoprenoid medicines. It is also a fossil fuel alternative since the inherent fuel properties of isoprene promote its chemical conversion to a transportation fuel additive to C10 and C15 hydrocarbon fuels (Xue and Ahring 2011). Currently, isoprene used in industrial applications is petroleum derived, or chemically synthesized. In nature, a large amount of isoprene is synthesized by green plants, fungi, bacteria, and green algae (Sharkey and Yeh 2001; Kuzma et al. 1995; Sanchez et al. 1991). Recently, growing research efforts have focused on bio-production of isoprene using microbial cells, especially E. coli, an important model organism in biological engineering and industrial microbiology.
Two metabolic pathways are known to synthesize isoprene: the 2C-methyl-d-erythritol 4-phosphate (MEP) pathway and the MVA pathway. MEP pathway starts with the condensation of pyruvate and d-glyceraldehyde 3-phosphate (G3P) to yield 1-deoxy-d-xylulose 5-phosphate (DXP), followed by intramolecular rearrangement and reduction. The MVA pathway consists of the condensation of three molecules of initial substrate acetyl-coenzyme A to MVA and other phosphorylations (Eisenreich et al. 2004; Rohmer 1999). In the previous study, the isoprene production was quite low (< 20 mg/L) when the native MEP pathway was employed in E. coli (Liu et al. 2014; Liu et al. 2015). In contrast, isoprene titer was 52 mg/L with a MVA pathway derived from Saccharomyces cerevisiae (S. cerevisiae) (Yang et al. 2012), which could even get a 3.5-fold increase after optimization of the MVA gene sources (Yang et al. 2016).
We thus assumed that the optimal selection of genes should be one of the factors that might improve isoprene production when applying the MVA pathway. It has been proven that the lower MVA pathway genes (mvk, pmk, mvaDi, and Idi) responsible for supplying isopentenyl diphosphate (IPP)/dimethylallyl diphosphate (DMAPP) from S. pneumoniae could accumulate higher CoQ10 (Zahiri et al. 2006), lycopene (Yoon et al. 2006), and β-carotene (Yoon et al. 2009) than other homologs from Staphylococcus aureus (S. aureus), Streptococcus pyogenes (S. pyogenes), S. pneumoniae, E. faecalis, or S. cerevisiae. The top portion genes (mvaE and mvaS) of MVA pathway from E. faecalis were found to have advantages in supplying MVA from acetyl-CoA (Yoon et al. 2009). Primak et al. reported that the mvk protein, which was usually feedback regulated by DMAPP and diphosphomevalonate (MVAPP), played an important regulatory role in biosynthesis of the MVA pathway-derived compounds. Interestingly, the mvk gene from M. mazei showed the ability to bypass this inhibition (Primak et al. 2011). In addition, gene ispS from P. alba exhibited the highest activity when compared with those from P. nigra and P. tremula (Yang et al. 2012).On the other hand, it has been reported that biosynthesis of terpenoid compounds through MVA pathway easily suffered from the metabolic flux imbalance, leading to accumulation of toxic metabolites, such as 3-hydroxy-3-methyl glutaryl coenzyme A (HMG-CoA) and IPP/DMAPP (Alonso-Gutierrez et al. 2015; Pitera et al. 2007). Similarly, keeping balance of the carbon flux through the MVA pathway thus may be crucial in high-level production of isoprene.
In this study, we firstly constructed a heterologous MVA pathway in E. coli by introducing the above specific set of genes as they had advantages in terms of high activities or productivity of other terpenoids. Then, based on the analysis between the initial functional strain BL7CT7P of the MVA pathway and the control strain, we designed another three constructs to achieve different gene expression levels by altering the gene expression cassettes, plasmid copy numbers, and promoters. We performed the manipulation of the gene expression levels of the MVA pathway for high-level production of isoprene. Finally, to further increase isoprene production, we developed the extraction fermentation and optimized the induction concentration and time using M9 medium with 10 g/L glucose.
Materials and methods
Strains, medium, and reagent
E. coli TOP10and E. coli BL21 (DE3) were used for cloning procedures and gene expression, respectively. LB medium (yeast extract 5 g/L, tryptone 10 g/L, NaCl 10 g/L) was used for gene cloning and the seed culture. M9 medium described previously (Liu et al. 2015) was prepared for isoprene production. Ampicillin (100 mg/L), Kanamycin (50 mg/L), and chloramphenicol (30 mg/L) were supplemented into the medium as needed. The restriction enzymes were purchased from NEB and Takara; T4 ligase and Gibson Assembly® Master Mix kit were purchased from NEB; DNA polymerase Phusion was purchased from Thermo Fisher Scientific. PUCE-ispS containing the ispS coding region from P. alba (GenBank No. AB198180) which has been used before (Liu et al. 2014; Liu et al. 2015) was synthetized by Beijing Ying Mao Sheng Ye Biotechnology Company. The genome DNA of S. pneumoniae CMCC 31001 was purchased from the National Center for Medical Culture Collections. Strain E. faecalis CGMCC 1.15424 was purchased from the China General Microbiological Culture Collection Center. The gene mvk from M. mazei was cloned by PCR amplification with the metagenomics of anaerobic active sludge which was rich in methane-producing bacteria M. mazei as the template (Dı’az et al. 2006).
Construction plasmids for the MVA pathway
The illustrated maps of the constructed plasmids were shown in Fig. 1b. The chemically synthesized ispS gene without the sequence for chloroplast transit peptide was cloned into pETDuet-1 using NcoΙ and XhoΙ, yielding plasmid pET-ispS. Gene encoding mvaE (GenBank no. AAG02439.1) and mvaS (GenBank no. AAG02438.1) amplified from E. faecalis CGMCC 1.15424 genome were respectively cloned into one MCS using BglII and XhoΙ, and another MCS using BamHΙ and SalΙ, yielding plasmid pRSF-mvaE-mvaS. Gene idi from S. pneumoniae CMCC 31001 genome (GenBank no. ABJ54064.1) was cloned into the second MCS location of plasmid pRSFduet-1 using NdeΙ and XhoΙ. The gene cassettes T7-idi and T7-ispS were transferred to pRSF-mvaE-mvaS using their respective enzyme site XbaΙ and SphΙ to yield plasmid pRSF4T7P, keeping one gene for one PT7 promoter. For plasmid pACYC3T7P, mvk gene from M. mazei genome (GenBank no. AAM31458.1) and pmk gene from S. pneumoniae CMCC 31001 genome (GenBank no. ABJ55414.1) were respectively digested with two enzymes EcoRΙ and NotΙ, and single enzyme FseΙ, and then cloned on the two MCSs of pACYCDuet-1 to obtain T7-mvk-T7-pmk. Gene mvaD (GenBank no. WP_000373458.1) was cloned into pACYCDuet-1 using BamHΙ and NotΙ to construct plasmid pACYC-mvk. We then constructed plasmid pACYC3T7P by transferring T7-mvk-T7-pmk to the plasmid pACYC-mvk using NheΙ. For plasmid pRSF2T7P, we initially used a commercial Gibson Assembly® Master Mix kit (New England Biolabs) to assemble the PCR fragments of vector pRSF-idi, gene mvaE and mvaS. Sequentially, we inserted ispS gene behind the second PT7 promoter between the sites of BamHΙ and NcoΙ to get plasmid pRSF2T7P. The PCR fragments of vector pTrc99A, mvk, pmk, and mvaD were assembled using the Gibson Assembly® Master Mix kit to get plasmid pTrcMPD. Plasmid Ap7C-ESMPDI was constructed by assembling the PCR fragments of vector pBbA7C, mvaE, mvaS, mvk-pmk-mvaD, and idi.

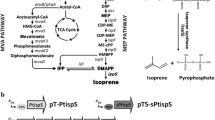
The MVA pathway and the MEP pathway for isoprene production in E. coli. a Illustration of the two pathways leading to the formation of isoprene. Genes overexpressed are indicated in red. b Construction of the isoprene production strains overexpressing the genes of the MVA pathway with two plasmids
All the primers of the genes, and all the plasmids and strains used in this study are listed in supplementary Table S1 and Table 1, respectively.
The extraction fermentation
Due to the high vapor pressure of isoprene, the 20-mL sealed vials of headspace sample injector were used to culture strains for their advantages of detection with gas chromatography mass-spectrometry (GC-MS). Colonies of strain BL7CT7P, BL5CT7P, BL2T7P1TrcP, and BL2T7P on plates were respectively picked up into 4-mL LB medium and cultured at 37 °C for overnight. The seed cultures were diluted 1:100 into 1 mL M9 medium in the 20-mL sealed vials and shaken at 37 °C at200 r.p.m. Until OD600 of the culture reached to 0.6, 0.8 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) and 20% dodecane (v/v) as overlay for trapping isoprene were supplemented into the cultures. Then, they were incubated at 30 °C to accumulate isoprene for 44 h with shaking at 200 r.p.m.
Detection isoprene by GC-MS
Isoprene production detection was operated by suctioning 1 mL of gas sample from the headspace of the sealed cultures with the headspace auto-injector. The gas was analyzed as described earlier (Julsing et al. 2007), using a GC-MS (Thermo Fisher, Trace ISQ America) equipped with an electron impact (EI) detector and a TG-WAXMS column (30 m × 0.25 mm, 0.25 μm film thickness). The conditions were set as follows: inlet at 200 °C, 1.1 mL min−1 constant flow of the nitrogen carrier gas, transfer line at 300 °C, ion source at 250 °C, scan m/z 50–300. The following oven temperature programs were carried out: the initial temperature was 40 °C for 1 min, then increased to 200 °C at 15 °C min−1 rate, and finally held for 1 min. The injector was maintained at 220 °C. The samples were incubated at 60 °C for 10 min, shaken for 10 s, and stirred 10 s before injection with the headspace needle of 70 °C. The sample drawn was 1 mL and split in the column with a ratio of 1/50. The product was quantified by direct comparison with standard isoprene (Sigma) dissolved in dodecane overlaying on water. The peak area was converted to isoprene concentration (mg isoprene /L broth) by comparing with a standard curve plotted with a set of known concentrations of isoprene.
Quantification of acetic acid and glucose
At the cultivated time points 4 h, 14 h, 24 h, 36 h, and 48 h, the cell culture aliquots were collected during fermentation for OD600 detection and sample preparation of detecting glucose and acetic acid. The concentrations of glucose and acetic acid in the culture supernatant were measured by HPLC using a refractive index detector (RID) equipped with an Aminex HPX-87H 300 × 7.8 column (25 cm, 0.4 cm i.d., Bio-Rad, USA) on an UltiMate 3000 HPLC system (Dionex, USA). Every 1.5 mL culture was sampled; of that, 200 μL was used for OD600 detection. About 1.3 mL culture supernatant was filtered through a spin-cartridge with a 0.45-μm nylon membrane, and 5 μL of the filtrate was eluted through the column at 50 °C and a flow rate of 0.6 mL/min with 5 mM H2SO4 as eluent for 25 min. Glucose and acetate were quantified by external standard calibration with authentic standards.
RNA isolation and real-time quantitative PCR (qPCR) analysis
An RNAprep Pure Cell/Bacteria Kit (TianGen) was used to isolate RNA from the IPTG-induced bacterial cells of BL7CT7P, BL5CT7P, BL2T7P1TrcP, and BL2T7 after being cultured for14 h. The total RNA concentrations were measured using a ScanDrop (Analytik Jena), and cDNA was synthesized with the FastQuant RT Super Mix (TianGen) using reverse transcriptase and the abovementioned commensurable RNA as template. To quantify the relative mRNA levels of gene mvaE, mvaS, mvk, pmk, mvaD, idi, and ispS, we designed primers for every gene to perform qPCR. The reverse transcription-synthesized cDNA was used as the template to quantify the relative copies of mRNA. The housekeeping glyceraldehyde-3-phosphate dehydrogenase (gapdh) gene was used as an internal reference to eliminate differences between samples. All the primers used in this qPCR are listed in Table S1. The qPCR reactions were performed with 2 μL of cDNA product in a 20-μL reaction system using Talent qPCR premix (Sybr Green) kit (TianGen) in the real-time Analytik Jena fluorescence PCR instrument. The PCR parameters were 1 cycle of 95 °C for 3 min, followed by 40 cycles of 95 °C for 5 s, 50 °C for 10 s, and 72 °C for 15 s. Every sample was measured in triplicate, and the results reported herein were the averages of the replicates.
Sample preparation and quantification of metabolites
At 14 h, 24 h, and 36 h time points, cell cultures were sampled and centrifuged in order to determine the intracellular levels of MVA, MVAP, MVAPP, and IPP/DMAPP. All the collected cell pellets after centrifugation were lysed and quenched with 250 μL methanol on ice, then 250 μL of water was added into the methanol lysate and mixed thoroughly. The supernatant was collected into a 3000 Da MW/CO centrifuge filter tube after centrifuging the methanol/water lysate for 3 min and a further centrifugation for 90 min at 13,000×g, 4 °C. Then, 0.5 mL water was added into the resulting filtrate, and frozen with liquid nitrogen. Finally, we lyophilized them for 1–2 days until to dry, and reconstituted in 90 μL 60% acetonitrile.
The extracted cellular metabolites were determined using liquid chromatography-mass spectrometry (LC-MS). The HPLC system consisted of a surveyor quaternary gradient pump, a vacuum degasser, a column temperature controller, and an auto-sampler (Shimadzu, LC-20AD). A HSS T3 column (1.7 μm, 100 × 2.1 mm) with the temperature maintained at 40 °C was used. The intermediates of the MVA pathway were separated by a linear gradient between solution A (0.05%NH3‧H2O, 2 mM ammonium acetate) and solution B (acetonitrile). The gradient was as follows: 0–2 min, 100% A; 2–3 min, 100% B; 3–6 min, 100% A; equilibration with 100% A. The total analysis time, including the equilibration, was 6 min for each analysis (Liu et al. 2015; Henneman et al. 2008). LC-MS analysis was performed on an AB Sciex 5500 Q-Trap triple quadrupole MS system. The operation parameters were as follows: declustering potential is − 100.0 V; entrance potential is − 10 V; ionization temperature is 580 °C. Curtain gas was set at 30 psi, collision gas at medium, and IonSpray voltage set to − 4500 V.
Results
Functional construction of a MVA pathway for isoprene production
Here, we employed the genes of mvaE and mvaS from E. faecalis, mvk from M. mazei, pmk, mvaD, and idi from S. pneumoniae CMCC 31001, and ispS from P. alba to construct a heterologous MVA pathway in E. coli, allowing biosynthesis of the universal precursors IPP/DMAPP of isoprenoid compounds from acetyl-CoA to enhance isoprene production(Fig. 1a). These seven genes were inserted into two plasmids pRSFDuet-1 and pACYCDuet-1 that could be used in combination (Fig. 1b). We first put each gene under control of an individual PT7 promoter in strain BL7CT7P in order to maximize the gene expression levels.
To test whether this arrangement was able to result inefficient isoprene production, we performed the isoprene production using strain BL7CT7P and the control strain harboring empty plasmids. We added 0.8 mM IPTG into the cultures when OD600 reached to 0.6. We monitored isoprene titer, cell growth, and the intracellular metabolite levels of the MVA pathway in BL7CT7P, and then compared them with those of the control strain. As shown in Table 2, 55.4 mg/L of isoprene was generated by BL7CT7P, but OD600 of the engineered cells was much lower than the control strain. The metabolites of MVA pathway in BL7CT7P were accumulated, especially the toxic IPP/DMAPP were 139-fold higher compared to the control. We suspected that the high-level expression of the MVA pathway genes probably resulted in a heavy burden to E. coli and caused an imbalance of the metabolic flux to isoprene in this functional strain BL7CT7P.
Strategies for controlling the expression levels of genes involved in MVA pathway
In previous studies on terpenoid production (Alonso-Gutierrez et al. 2015; Kevin et al. 2014; Redding-Johanson et al. 2011), it has been found that the expression levels of the pathway genes had a great influence on the production of terpenes. Therefore, we hypothesized that manipulation of the gene expression levels might be able to reduce the burden to cells, balance the carbon flux, and also increase the isoprene titer. Therefore, we subsequently tried different plasmid copies, gene expression cassettes, and promoters for expression of the MVA pathway genes in E. coli BL21 (DE3). However, ispS gene is always controlled by an independent PT7 promoter, leaving the carbon flux as much as possible to the target product of isoprene.
Briefly, we first decreased the gene expression cassettes on the high copy number (˃ 100) plasmid pRSF4T7P by co-expressing mvaE, mvaS, and idi within a single polycistron under control of a shared PT7 promoter, resulting in strain BL5CT7P. Then, we further decreased the gene expression cassettes by co-expressing mvk, pmk, and mvaD in a polycistron which was driven by aPTrc promoter. On the other hand, the medium copy number (~ 40) plasmid of pTrcMPD was used; otherwise the expression levels of mvk, pmk, and mvaD genes might be too low under the control of PTrc promoter, generating strain BL2T7P1TrcP. Next, we reduced the gene expression cassettes to two and integrated the six DMAPP-forming genes under the control of a shared PT7 promoter onto plasmid Ap7C-ESMPDI, which has a comparable low copy number (~ 12) of p15A origin of replication, obtaining strain BL2T7P (Fig. 1b).
Characterization of the manipulations using extraction fermentation
High concentration of isoprene is toxic to E. coli cells during fermentation (Whited et al. 2010), and also makes the GC-MS peak irregular and the standard curve nonlinear which leads to inaccurate quantification. However, according to our experience, dilution of the samples will lead isoprene compound running off and inaccurate detection because of its strong evaporation. We previously performed the isoprene fermentation in a sealed flask and detected it with solid-phase micro-extraction (SPME)-GC or GC-MS connected with headspace injector (Liu et al. 2014; Liu et al. 2015), leading to the isoprene concentrations both in culture and in the limited headspace comparably high. To address these problems, we introduced an organic layer between culture and headspace to employ the extraction fermentation by adding 20% dodecane during fermentation. We compared the two fermentation methods using strain BL2T7P and BL2T7P1TrcP, and as a result, the extraction fermentation had significant positive impacts on cell growth and isoprene production. As shown in Fig. 2a, b, the isoprene concentrations with dodecane overlay were 2-fold and 40% higher, and OD600 also presented 16 and 69% improvement over the strains without dodecane overlay respectively for strain BL2T7P and BL2T7P1TrcP. It indicated that the organic layer with stronger solubility trapped isoprene and reduced its toxicity to cells. We used the standard isoprene to test that the hold-up efficiency of isoprene in dodecane layer is 60.96% (Supplementary Material Method). This result indicates that most of the produced isoprene was trapped into the dodecane layer, reducing its toxicity to cells.

The effect of the extraction fermentation on isoprene production (a) and cell growth (b). Strain BL2T7P and BL2T7P1TrcP were cultured without dodecane (black bars) and with dodecane (gray bars) for isoprene production. The initial OD600 was 0.05 after inoculation. IPTG (0.8 mM) and 20% dodecane (v/v) as overlay for trapping isoprene were supplemented into the cultures when OD600 reached to 0.6. Then, they were incubated at 30 °C to accumulate isoprene for 44 h with shaking at 200 r.p.m. Data represent averages from three replicates with error bars
We then applied the extraction fermentation to analyze the isoprene production by these engineered strains. Comparison of isoprene titers among the four strains demonstrated that strain BL7CT7P with one promoter for each gene had a relatively low OD600 and a low isoprene titer of 117.4 ± 17.2 mg/L. But strain BL5CT7P with five gene expression cassettes produced higher isoprene than strain BL7CT7P, with the titer of 338.1 ± 101.5 mg/L at 48 h. It was indicated that co-expressing genes within a polycistron operon instead of monocistron was beneficial to increase isoprene production. Interestingly, a significant improvement of isoprene production was achieved by strain BL2T7P1TrcP with three gene expression cassettes, generating an isoprene titer of 563.9 ± 34.7 mg/L, which was nearly 5-fold higher than that of strain BL7CT7P. These results supported our hypothesis that regulation of gene expression levels might enhance isoprene production. As shown in Fig. 3a, strain BL2T7P produced almost the lowest isoprene with a titer of 105.4 ± 43.2 mg/L at 48 h, which was similar with that generated by strain BL7CT7P. The difference was that strain BL2T7P produced isoprene faster, grew faster, and had a higher cell density than strain BL7CT7P(Fig. 3b), which was probably due to the reduced cellular burden or the lower levels of toxic intermediates. Interestingly, we observed in Fig. 3c, d that both the glucose consumption and the acetate accumulation showed completely negative correlation with isoprene production in all four strains. It demonstrated that strain BL2T7P1TrcP consumed the least glucose and produced the least acetate (the final titer was 1.1 ± 0.2 mg/L), showing a 5.41-fold productivity compared to strain BL2T7P. In contrast, strain BL2T7P consumed more glucose and produced acetate of 2.8 ± 0.3 mg/L.

Comparison of isoprene production from E. coli strains overexpressing the MVA pathway. Isoprene production (a) and cell densities (b) at time point 24 h (black bars) and 48 h (gray bars). Time courses of glucose consumption (c) and acetate formation (d) from isoprene production strain BL7CT7P, BL5CT7P, BL2T7P1TrcP, and BL2T7P. All strains were cultured in sealed shake flasks with M9 medium containing 10 g/L glucose, and 0.8 mM IPTG and 20% dodecane were added when OD600 reached to 1.4. The data points and error bars represent means standard deviations of values from three independent biological replicates
Evaluation of different manipulations for isoprene production
In order to evaluate the four different manipulations, we investigated the gene transcription and metabolite production of the four engineered strains. We first carried out qPCR for all the MVA pathway genes in strain BL7CT7P, BL5CT7P, BL2T7P1TrcP, and BL2T7P. The relative mRNA levels of mvaE, mvaS, mvk, pmk, mvaD, idi, and ispS were normalized to gapdh. As exhibited in Fig. 4, the total relative mRNA levels were significantly different between the four samples. Generally, the mRNA levels of gene mvaE, mvk, and ispS were lower comparing to other genes. We observed that strain BL5CT7P exhibited lower levels of mvaS, mvk, pmk, and mvaD transcription, but higher levels of mvaE and idi, and similar levels of ispS transcription comparing to strain BL7CT7P. In contrast, the ratios of the cDNA levels derived from BL2T7P1TrcP far exceeded those from the other three strains. It indicated that the imbalanced expression of genes may lead to low transcriptional levels. Obviously, strain BL2T7P transcribed the lowest mRNA levels for all the genes among the four strains.

The relative mRNA levels from RT-PCR. The cDNA of gene mvaE, mvaS, mvk, pmk, mvaD, idi, and ispS was generated from strain BL7CT7P, BL5CT7P, BL2T7P1TrcP, and BL2T7P. Strains were cultured in M9 medium for 14 h and induced by 0.8 mM IPTG at 30 °C when OD600 was 1.4. The expression level of each gene was normalized with the value for the housekeeping gene gapdh. Results are the average of three technical replicates, with error bars showing the standard deviation from the means
Besides mRNA levels, metabolites were also analyzed. We isolated the intermediates and detected their relative concentrations at 14 h, 24 h, and 36 h time points. As illustrated in Fig. 5a, we found that BL7CT7P accumulated MVA, MVAP, and MVAPP over time, so the levels of IPP/DMAPP were significantly high at early growth phase and low at later phase. These results indicated that the metabolite flux was not constant, probably resulting from the over-expression of some genes and the insufficient expression of idi, ispS or their low activities. In contrast, strain BL5CT7P had much lower accumulations of intermediates (Fig. 5b),which was consistent with the mRNA levels that most genes were transcribed lower than that of strain BL7CT7P. Interestingly, BL2T7P1TrcP generated lower MVA and MVAPP, similar amount of MVAP and IPP/DMAPP (Fig. 5c), and higher isoprene titer compared with BL7CT7P (Fig. 3a).These results indicated that BL2T7P1TrcP showed a more balanced flux towards isoprene. Strain BL2T7P showed the lowest MVA, MVAP, and IPP/DMAPP levels among the four strains (Fig. 5d), and the shortage of the precursors IPP/DMAPP might be caused by the low gene expression.

Metabolite analysis of the MVA pathway in strain BL7CT7P (a), BL5CT7P (b), BL2T7P1TrcP (c), and BL2T7P (d). The metabolite levels of MVA, MVAP, MVAPP, and DMAPP/IPP were detected by LC-MS at time points 14 h (black bars), 24 h (gray bars), and 36 h (dark gray bars). The samples were collected from the same cultures with RT-PCR. Data represents averages of the peak area per OD600 from three replicates with error bars shown
Optimization of the extracted fermentation
In general, strains with different gene expression cassettes, promoters, and plasmid copy numbers might require different amount of inducer; thus, IPTG concentrations probably had different influences on the four strains. We then optimized the IPTG induction time and concentrations based on the extracted fermentation to further improve isoprene production. We induced the cultures with 0.8 mM IPTG at three different OD600: low ~ 0.6 (cultured 3 h), medium ~ 1.4 (cultured 4 h), and high ~ 2.2 (cultured 5.5 h), respectively. Then, we detected the isoprene production and cell densities after being induced for 36 h at 30 °C. As shown in Fig. 6a, we observed significant different isoprene production among the different induction times. Comparing with induced time at OD600 of 0.6, isoprene titers were increased 2.2-, 1.5-, 1.5-, and 2.8-fold respectively in BL7CT7P, BL5CT7P, BL2T7P1TrcP, and BL2T7P when they were induced at OD600 of 1.4. However, when the cells were induced at 2.2, they then respectively decreased 74.7%, 80.6%, 97.1%, and 57.0% comparing to those when induced at 1.4. The final OD600 were higher when the cells were induced at higher OD600. Therefore, cultured 4 h (OD600 of 1.4) is the optimal inducing time.

Isoprene production (a) and cell growth (b) when strains were induced at different OD600. Cultures were induced by 0.8 mM IPTG after respectively being cultivated for 3 h (black bars), 4 h (light gray bars), and 5.5 h (gray bars). Isoprene titers were measured after 36 h accumulation, and the induced OD600 and final OD600 were also monitored, respectively. Results are the average of three biological replicates with error bars representing the standard deviation from the means
Sequentially, we also investigated the effect of different IPTG concentrations. We used a series of IPTG concentrations in the range of 0.1 mM, 0.4 mM, 0.8 mM, and 1.2 mM to induce the four strains when OD600 was 1.4. As shown in Fig. 7, we observed that the isoprene titers generally increased with the IPTG concentration rising, but the extent was different among the four strains. When the IPTG concentration was increased to 1.2 from 0.1 mM, the isoprene titer was improved 3.3- and 2.7-fold in strain BL5CT7P and BL2T7P1TrcP, while only 29 and 71% in strain BL7CT7P and BL2T7P, respectively. In general, the titers of isoprene increased sharply when IPTG concentrations were increased to 0.8 from 0.1 mM, then slow down when IPTG concentrations were further increased to 1.2 mM. Finally, we concluded that 0.8 mM IPTG was deemed the optimal IPTG concentration for isoprene production considering isoprene titers, cell growth, and the cost of fermentation, with the isoprene titer increased to 587 ± 47 mg/L in strain BL2T7P1TrcP.

Isoprene production (a) and cell growth (b) when the cultures were induced with various IPTG concentrations. When OD600 were at about 1.4, strain BL7CT7P, BL5CT7P, BL2T7P1TrcP, and BL2T7P were induced with IPTG concentrations of 0.1 mM (black bars), 0.4 mM (light gray bars), 0.8 mM (white bars), and 1.2 mM (gray bars), respectively. Results are the average of three biological replicates with error bars representing the standard deviation from the means
Discussion
In conclusion, we introduced the superior genes for constructing MVA pathway and manipulated the 7-gene expression levels for greatly enhancing isoprene production in E. coli. Through altering the gene expression cassettes, plasmid copy numbers, and promoters, we obtained four engineered strains: BL7CT7P, BL5CT7P, BL2T7P1TrcP, and BL2T7P, in which BL2T7P1TrcP showed a more balanced system and highest isoprene production. We successfully regulated the gene expression levels which were characterized by mRNA levels and the metabolite levels monitored with LC-MS. To achieve higher isoprene production, we also developed the extraction fermentation with dodecane as overlay, and optimized IPTG concentration at 0.8 mM and induction time at OD600 of ~ 1.4. In this culturing condition, strain BL2T7P1TrcP produced the highest titer of ~ 587 ± 47 mg /L isoprene in M9 medium with 10 g/L glucose. Strains BL7CT7P and BL5CT7P exhibited a lower concentration of isoprene and slower cell growth, revealing that more promoters gave the cells more burden. On the contrary, strain BL2T7P produced the lowest isoprene but had the best cell growth, which we attribute to the fewer gene expression cassettes and lower gene expression levels. The manipulation here of the gene expression levels demonstrates the importance of metabolic engineering for the improvement of bio-product yield in a multi-gene pathway. In strain BL7CT7P, every gene was driven by a strong T7 promoter which could generate serious burdens to cells. In reverse, the significant effect of reducing burdens on isoprene production might be benefited from the serious burdens. However, strong T7 promoter often leads to inclusion body formation and thus enzymes lost their activities. Therefore, we will try to use weaker promoters for every gene to relieve burdens on cells to further increase isoprene production in later studies.
In our previous studies, we have engineered the MEP pathway for isoprene biosynthesis by overexpressing the key genes (Liu et al. 2014) and joint antisense RNA strategies for isoprene production (Liu et al. 2015) in E. coli BL21 (DE3). The optimization of the MEP pathway is worth pursuing, because it provides an increased mass yield of product (30.2% on glucose) compared to that by the MVA pathway of 25.2% (Whited et al. 2010). However, since the MEP pathway is under control of native E. coli regulation network with a lot of unknown factors, such as the metabolic nodes, we only produced isoprene at ~ 20 mg/L. The optimization of the carbon flux through the MEP pathway sufficient for commercial production of isoprene or isoprenoids is challenging based on the present research. MVA pathway, as a heterologous pathway, is easy to optimize and its carbon flux is easy to control. Therefore, the MVA pathway has been widely used in isoprenoid production including isoprene (Zurbriggen et al. 2012; Kim et al. 2016;Ye et al. 2016), isopentenol (Kevin et al. 2014), monoterpene (Alonso-Gutierrez et al. 2015), and sesquiterpene (Redding-Johanson et al. 2011). From these previous studies, we know that the expression levels of pathway genes had a huge effect on terpene production and that imbalanced expression led to metabolite accumulation.
In this work, we not only explored the superior genes derived from the selected organisms to construct MVA pathway but also manipulated gene expression levels for balanced expression of the pathway genes to improve isoprene production in E. coli. The strategies of polycistronic operons, promoter engineering, and plasmid copy numbers are efficient tools for regulation of gene expression levels. For their characterization, the relative mRNA levels and the intermediate detection facilitated a better understanding of the four engineered strains with different gene expression profiles. The variation in the levels of gene expression will likely impact the levels of protein synthesis. The protein levels of the MVA pathway enzymes will be useful for analysis of the different metabolite levels and expression levels in the four engineered strains and exploration of the bottlenecks in the pathway. Therefore, the targeted proteomics measurement will be focused in the next step to determine how much enzyme was actually made in each engineered strain.
We also developed the extraction fermentation, which is a feasible two-phase culture system to reduce toxicity to cells and address the evaporation of isoprene by trapping it with dodecane as overlay. Isoprene production increased nearly 2-fold by optimizing the extraction fermentation, IPTG concentrations, and the induction time. For these strains, the traditional induction at low OD600 (~ 0.6)was not optimal, but induction at a higher OD600 (~ 1.4) could allow the cells growing before expressing proteins and lead to higher isoprene production. These results were consistent with the study of terpene compound limonene production, in which the strain showed better limonene production when induced at the medium OD600 of 1.2 than that at OD600 of 0.2 and 2.5 (Alonso-Gutierrez et al. 2015). We suspected that the reasons for the poor OD600 and isoprene production when induced at low OD600 were from the burden of expressing the multiple heterologous enzymes, consumption of cellular resources and metabolites, and the synthesis of the metabolites being toxic to cells. Through manipulating the gene expression levels of MVA pathway and optimizing fermentation conditions, strain BL2T7P1TrcP with the highest isoprene productivity achieved a titer of 587 ± 47 mg/L in the sealed flasks. Nearly 30-fold enhancement of isoprene was achieved by controlling the gene expression levels of the MVA pathway compared to the titers of engineering MEP pathway. In the study of MVA pathway, we obtained a considerably higher titer of isoprene compared to the previous studies (Yang et al. 2012; Yang et al. 2016). These results supported that the heterologous MVA pathway in E. coli requires a balanced metabolic flux for high isoprene production in a multi-gene pathway system. However, the titer of isoprene is still not enough for the economic consideration. Although strain BL2T7P1TrcP rendered the highest isoprene productivities per OD600 among the four strains, as shown in Figs. 2 and 3, the poor cell growth might constrain its isoprene production. In the further studies, the bio-based fermentation process to scale-up isoprene production should be developed for increasing isoprene production. In addition, we should pursue more biotechnologies and engineering strategies from some aspects: (1) searching high activity enzymes, (2) building efficient pathways, (3) optimizing influx and efflux of key metabolites, (4) improving fermentation-based process and recovery technologies, (5) reducing feedback inhibition by isoprene product or intermediates on further synthesis, (6) using cheap feedstocks, and so on. The development of bio-isoprene needs industrial biotechnologies for the potential to enable isoprene production from renewable raw materials to deliver commercialization.
References
Alonso-Gutierrez J, Kim EM, Batth TS, Cho N, Hu Q, Chan LJ, Petzold CJ, Hillson NJ, Adams PD, Keasling JD, Martin GH, Lee TS (2015) Principal component analysis of proteomics (PCAP) as a tool to direct metabolic engineering. Metab Eng 28:123–133
Dı’az EE, Stams JMA, Amils R, Sanz LJ (2006) Phenotypic properties and microbial diversity of methanogenic granules from a full-scale upflow anaerobic sludge bed reactor treating brewery wastewater. Appl Environ Microbiol 72(7):4942–4949
Eisenreich W, Bacher A, Arigoni D, Rohdich F (2004) Biosynthesis of isoprenoids via the non-mevalonate pathway. Cell Mol Life Sci 61:1401–1426
Henneman L, van Cruchten AG, Denis SW, Amolins MW, Placzek AT, Gibbs RA, Kulik W, Waterham HR (2008) Detection of nonsterol isoprenoids by HPLC-MS/MS. Anal Biochem 383:18–24
Julsing MK, Rijpkema M, Woerdenbag HJ, Quax WJ, Kayser O (2007) Functional analysis of genes involved in the biosynthesis of isoprene in Bacillus subtilis. Appl Microbiol Biotechnol 75:1377–1384
Kevin W, George AC, Jain A, Batth TS, Baidoo EEK, Wang G, Adams PD, Petzold CJ, Keasling JD, Lee TS (2014) Correlation analysis of targeted proteins and metabolites to assess and engineer microbial isopentenol production. Biotechnol Bioeng 111:11
Kim J, Wang C, Jang H-J, Cha M-S, Park J-E, Jo S-Y, Choi E-S, Kim S-W (2016) Isoprene production by Escherichia coli through the exogenous mevalonate pathway with reduced formation of fermentation byproducts. Microb Cell Factories 15(1):214
Kuzma J, Nemecek-Marshall M, Pollock WH, Fall R (1995) Bacteria produce the volatile hydrocarbon isoprene. Curr Microbiol 30:97–103
Liu C-L, Fan L-H, Liu L, Tan T-W (2014) Combinational biosynthesis of isoprene by engineering the MEP pathway in Escherichia coli. Process Biochem 49:2078–2085
Liu C-L, Lv Q, Tan T-W (2015) Joint antisense RNA strategies for regulating isoprene production in Escherichia coli. RSC Adv 5:74892–74898
Pitera DJ, Paddon CJ, Newman JD, Keasling JD (2007) Balancing a heterologous mevalonate pathway for improved isoprenoid production in Escherichia coli. Metab Eng 9:193–207
Primak YA, Du M, Miller MC, Wells DH, Nielsen AT, Weyler W, Beck ZQ (2011) Characterization of a feedback-resistant mevalonate kinase from the archaeon Methanosarcina mazei. Appl Environ Microbiol 77:7772–7778
Redding-Johanson AM, Batth TS, Chan R, Krupa R, Szmidt HL, Adams PD, Keasling JD, Lee TS, Mukhopadhyay A, Petzold CJ (2011) Targeted proteomics for metabolic pathway optimization: application to terpene production. Metab Eng 13:194–203
Rohmer M (1999) A mevalonate-independent route to isopentenyl diphosphate. Compr Nat Products Chem 45–67
Sanchez JJ, Berenguer JA, Calderon V, Herce MD (1991) Spoilage of a bakery product by isoprene-producing molds. Rev Agroquim Technol Aliment 31:4
Sharkey TD, Yeh SS (2001) Isoprene emission from plants. Annu Rev Plant Physiol Plant Mol Biol 52:407–436
Whited MG, Feher JF, Benko AD, Cervin AM, Chotani KG, McAuliffe CJ, LaDuca JR, Ben-Shoshan AE, Sanford JK (2010) Development of a gas-phase bioprocess for isoprene-monomer production using metabolic pathway engineering. Ind Biotechnol 12:152–163
Xue J, Ahring BK (2011) Enhancing isoprene production by genetic modification of the 1-deoxy-d-xylulose-5-phosphate pathway in Bacillus subtilis. Appl Environ Microbiol 77:2399–2405
Yang J, Zhao G, Sun Y, Zheng Y, Jiang X, Liu W, Xian M (2012) Bio-isoprene production using exogenous MVA pathway and isoprene synthase in Escherichia coli. Bioresour Technol 104:642–647
Yang C, Gao X, Jiang Y, Sun B, Gao F, Yang S (2016) Synergy between methylerythritol phosphate pathway and mevalonate pathway for isoprene production in Escherichia coli. Metab Eng 37:79–91
Ye L, Lv X, Yu H (2016) Engineering microbes for isoprene production. Metab Eng 38:125–138
Yoon SH, Lee YM, Kim JE, Lee SH, Lee JH, Kim JY, Jung KH, Shin YC, Keasling JD, Kim SW (2006) Enhanced lycopene production in Escherichia coli engineered to synthesize isopentenyl diphosphate and dimethylallyl diphosphate from mevalonate. Biotechnol Bioeng 94:1025–1032
Yoon SH, Lee SH, Das A, Ryu HK, Jang HJ, Kim JY, Oh DK, Keasling JD, Kim SW (2009) Combinatorial expression of bacterial whole mevalonate pathway for the production of beta-carotene in E. coli. J Biotechnol 140:218–226
Zahiri HS, Yoon SH, Keasling JD, Lee SH, Won Kim S, Yoon SC, Shin YC (2006) Coenzyme Q10 production in recombinant Escherichia coli strains engineered with a heterologous decaprenyl diphosphate synthase gene and foreign mevalonate pathway. Metab Eng 8:406–416
Zurbriggen A, Kirst H, Melis A (2012) Isoprene production via the mevalonic acid pathway in Escherichia coli (Bacteria). BioEnergy Res 5(4):814–828
Acknowledgments
We thank Xiaozhou Luo (Lawrence Berkeley National Lab, Joint Bio-Energy Institute) for helping to revise this manuscript.
Funding
This work was supported by the State key laboratory of organic–inorganic composites, the National Basic Research Program of China (973 program) (2013CB733600), the National Nature Science Foundation of China (21390202, 21476017).
Author information
Authors and Affiliations
Contributions
C.-L. Liu and T.-W. Tan designed the study. C.-L. Liu and H.-R. Bi performed the experiments. C.-L. Liu, B. Hu, and L.-H. Fan are involved in the manuscript writing and editing. All the authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Ethical statement
This article does not contain any studies with human participants performed by any of the authors.
Electronic supplementary material
ESM 1
(PDF 443 kb)
Rights and permissions
About this article

Cite this article
Liu, CL., Bi, HR., Bai, Z. et al. Engineering and manipulation of a mevalonate pathway in Escherichia coli for isoprene production. Appl Microbiol Biotechnol 103, 239–250 (2019). https://doi.org/10.1007/s00253-018-9472-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-018-9472-9