
Abstract
Biological nitrogen fixation (BNF) through the enzyme nitrogenase is performed by a unique class of organisms known as diazotrophs. One interesting facet of BNF is that it produces molecular hydrogen (H2) as a requisite by-product. In the absence of N2 substrate, or under conditions that limit access of N2 to the enzyme through modifications of amino acids near the active site, nitrogenase activity can be redirected toward a role as a dedicated hydrogenase. In free-living diazotrophs, nitrogenases are tightly regulated to minimize BNF to meet only the growth requirements of the cell, and are often accompanied by uptake hydrogenases that oxidize the H2 by-product to recover the electrons from this product. The wild-type strain of Azotobacter vinelandii performs all of the tasks described above to minimize losses of H2 while also growing as an obligate aerobe. Individual alterations to A. vinelandii have been demonstrated that disrupt key aspects of the N2 reduction cycle, thereby diverting resources and energy toward the production of H2. In this work, we have combined three approaches to override the primary regulation of BNF and redirect metabolism to drive biological H2 production by nitrogenase in A. vinelandii. The resulting H2-producing strain was further utilized as a surrogate to study secondary, post-transcriptional regulation of BNF by several key nitrogen-containing metabolites. The improvement in yields of H2 that were achieved through various combinations of these three approaches was compared and is presented along with the insights into inhibition of BNF by several nitrogen compounds that are common in various waste streams. The findings indicate that both ammonium and nitrite hinder BNF through this secondary inhibition, but urea and nitrate do not. These results provide essential details to inform future biosynthetic approaches to yield nitrogen products that do not inadvertently inhibit BNF.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hydrogen gas (H2) could augment fossil fuel usage due to its carbon neutral combustion and ability to generate electricity in fuel cell applications. Currently, H2 is primarily produced from non-renewable sources such as fossil fuel reserves of natural gas (Rittmann and Herwig 2012). Reducing fossil fuel usage and mitigating climate change make the presently used thermochemical processes less desirable than biological ones in which H2 is generated from waste streams that feed H2-producing organisms or utilize photosynthetic species (Bandyopadhyay et al. 2010; Masukawa et al. 2010; Weyman et al. 2010). Biologically generated H2 (biohydrogen) provides an eco-friendly and renewable method with which to take advantage of these waste streams, but the processes yielding biohydrogen suffer from low productivity and high complexity which have thus far prevented them from being broadly adopted by industry (Ghirardi et al. 2005; Noar et al. 2015). Therefore, efforts to provide novel biohydrogen production routes are actively being pursued by laboratories across the globe (Bandyopadhyay et al. 2010; Fixen et al. 2016; Masukawa et al. 2010; Rey et al. 2007; Schütz et al. 2004; Weyman et al. 2010).
There are currently two primary routes to biohydrogen production. The first is light-dependent and relies on a photosynthetic species such as an alga or cyanobacterium (Bandyopadhyay et al. 2010; Bothe et al. 2010; Ghirardi et al. 2005; Masukawa et al. 2010; Tamagnini et al. 2002; Weyman et al. 2010). The second is light-independent (Elsharnouby et al. 2013; Rittmann and Herwig 2012; Sinha and Pandey 2011), and occurs in many anaerobic environments, including in most marshland sediments. Both of these routes use either a nitrogenase or hydrogenase to generate molecular H2. The general biological nitrogen fixation (BNF) reaction catalyzed by the complex metalloenzyme nitrogenase produces H2 as a by-product when dinitrogen (N2) in the atmosphere is reduced into ammonia (NH3) as shown in Eq. 1 (Burgess and Lowe 1996):
Azotobacter vinelandii is a free-living, nitrogen-fixing bacterium (diazotroph). It is both a strict aerobe and a model organism for studies focused on BNF (Setubal et al. 2009). A. vinelandii naturally evolves very low levels of H2 during BNF, as any H2 produced is generally recycled by the uptake hydrogenase (Fig. 1). Hydrogenase catalyzes the reversible oxidation of H2 gas to recapture the electrons consumed by nitrogenase to reduce the energetic costs of BNF through Eq. 2:

Simplified schematic showing select metabolic routes to hydrogen (H2) and ammonia (NH3) production in A. vinelandii. The diagram depicts strain AZBB275 which has a variant form of the Mo-dependent nitrogenase which is limited in its ability to fix dinitrogen into NH3 (dashed line) but not limited in its proton reduction (larger arrow). This strain is also unable to consume H2 as both hydrogenases have been knocked out (red x); only the membrane-bound hydrogenase is shown as it is believed to be the primary H2 consumer associated with nitrogenase
Many of the current approaches to produce biohydrogen only use hydrogenases (Ghirardi et al. 2005; Radakovits et al. 2010; Sinha and Pandey 2011; Tamagnini et al. 2002). However, this approach is limited by the reversible nature of the hydrogenase reaction (Eq. 2). The H2 by-product of nitrogenase is the result of a unidirectional reaction (Eq. 1), which cannot be driven backwards to any great extent (Guth and Burris 1983; Shaw et al. 2014). Therefore, in the absence of a hydrogenase, any H2 produced by A. vinelandii could become a terminal product, assuming there are no other biological processes that would utilize this H2 within the cell.
A. vinelandii is genetically tractable, and can be modified to increase its H2 production. However, the full potential of the biohydrogen production scheme has not been adequately explored in A. vinelandii, although individual approaches that increase nitrogenase-driven H2 production have been independently reported by employing both in vivo and in vitro approaches (Barahona et al. 2016; Barney et al. 2004; Noar et al. 2015). One such approach was to knock out the uptake hydrogenase genes in A. vinelandii, which results in H2 evolution as the organism can no longer oxidize the H2 by-product of BNF (Barney et al. 2017; Noar et al. 2015). Another is to make certain amino acid substitutions near the active site of the FeMo-cofactor in the NifD subunit (α-subunit) of nitrogenase, resulting in a variant form of the enzyme which is deficient in its ability to reduce N2, but unaffected in its ability to reduce protons to H2 (Barney et al. 2004; Masukawa et al. 2010). One specific amino acid substitution that shifts the selectivity of the nitrogenase active site is the modification to the nifD gene at the position of valine 70 (nifDV70I) of the Mo-dependent nitrogenase (Barney et al. 2004).
The final approach to increase the levels of H2 in A. vinelandii is by deregulating nitrogenase. This approach was first reported in 1992 and recently reconstructed in our laboratory (Bali et al. 1992; Barney et al. 2015; Brewin et al. 1999). The resulting strain yields considerable amounts of NH4+ into the spent medium, and maintains elevated levels of nitrogenase expression during both the exponential and stationary phases of growth, whereas wild-type A. vinelandii dramatically decreases nitrogenase expression during stationary phase growth (Barney et al. 2017). This indicated that a deregulated strain could continue to drive the activity of nitrogenase, even when substantial amounts of NH4+ have accumulated in the medium and cell division had reached stasis.
Our goals in this study were twofold. First, we sought to optimize the internal pathways of A. vinelandii through genome engineering to improve H2 production by the molybdenum-based nitrogenase. Second, we aimed to demonstrate the utility of our optimized strain to serve as a tool for monitoring whole-cell nitrogenase activity in response to nitrogenous compounds found in waste streams or as potential metabolites in the cell, including NH3, the natural product of the nitrogenase enzyme. Our results revealed a unique secondary inhibition profile for various nitrogenous metabolites that appears to be novel and acting outside of primary regulation. Once we uncovered this secondary inhibition of BNF, we further investigated this by eliminating additional intracellular pathways to identify which specific chemical species were inhibiting BNF, and which might be benign if left unaltered. These results provide strong evidence that NH4+ and NO2− downregulate BNF, while NO3− and urea have little effect on this secondary inhibition. A summary of these results and their implications on BNF and potential biosynthetic approaches to increase yields of BNF toward novel nitrogen compounds is presented.
Methods
Reagents, bacteria, and media
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO) or Fisher Scientific (Pittsburgh, PA). A. vinelandii DJ (ATCC BAA-1303), the wild-type strain used, was originally obtained from Dennis Dean (Virginia Tech) and is both deficient in alginate production and highly transformable (Setubal et al. 2009). A. vinelandii strains (Table 1) were grown aerobically at 28 °C using Burk’s (B) medium. B medium contained per liter of deionized water: sucrose (20 g), MgSO4•7H2O (0.2 g), CaCl2•2H2O (90 mg), Na2MoO4•H2O (0.25 mg), FeSO4•7H2O (5 mg), KH2PO4 (0.2 g), and K2HPO4 (0.8 g), adjusted to pH 7.5. For H2 assays, strains were grown in sterile filtered B medium supplemented with 20 mM MOPS buffer. To test H2 production at varied pH, strains were grown in B medium supplemented with a multiple buffer system containing 10 mM MES, 10 mM MOPS, and 10 mM TAPS adjusted to each specified starting pH value. Kanamycin was excluded from the assays, but included on plates used to maintain specific strains. Strains containing the nifDV70I amino acid substitution were grown on B medium agar plates supplemented with ammonium (20 mM), urea (10 mM), or nitrate (10 mM). Ammonium was provided as ammonium sulfate and nitrate was added as sodium nitrate.
Construction of A. vinelandii strains
All plasmids were constructed and maintained within Escherichia coli JM109 which was obtained from New England Biolabs (Ipswich, MA). The construction of strains AZBB163 and DJ1373 and methods for A. vinelandii genome editing have been described previously (Barney et al. 2004, 2015; Eberhart et al. 2016). A complete list of the strains, plasmids, and primers used are listed in Supplementary Tables S1, S2, and S3.
Protein quantification
To quantify the total amount of cellular protein, isolated cells were pelleted at > 12,000 g by centrifugation, and then resuspended in 1 mL of water and sonicated for 50 s (Misonix LX-2000, Qsonica, Newtown, CT) inside of a 1.5-mL tube. Cell lysate was then added to 1.0 mL of Coomassie Plus (Bradford) assay reagent (Pierce, Rockford, IL), mixed by inverting the cuvette several times, and incubated at room temperature for 10 min. Samples were read at a wavelength of 595 nm (Varian Inc., Palo Alto, CA). Samples were compared to a standard curve prepared using bovine serum albumin (BSA) as a standard (Pierce, Rockford, IL). Alternatively, total protein was also measured using the Biuret assay (Chromý et al. 1974). While this did require a larger quantity of cells based on the working range of the assay, protein could be liberated and measured through Biuret without the need for sonication. Samples were compared to a standard curve prepared using BSA as a standard, which was also measured based on the absorbance at 280 nm using the common extinction coefficient for BSA (ε = 43,824 M−1 cm−1).
Hydrogen measurements
Hydrogen gas (H2) was measured by transferring a specific amount of cells scraped from a two-day old plate to 3 mL of medium in a 20-mL serum vial such that the starting OD600 was 1.5. The serum vials were then capped with stoppers, sealed, and preincubated for 2 hours at 28 °C, 180 rpm and a 45° angle. This preincubation was incorporated to assure that cells had adapted to the liquid culture conditions prior to initiating any measurements. After the 2 hours, the culture head space was flushed with fresh air, and the serum bottles were resealed. At this point, 250 μL was drawn from the headspace and analyzed. The sample was then allowed to incubate for another 2 hours under the same conditions. Sampling was done during the second incubation period at regular intervals depending on the assay. Gas composition of the headspace was determined using a gas chromatograph equipped with a thermal conductivity detector (GC-8A, Shimadzu Scientific, Kyoto, Japan) with argon as the carrier gas and a molecular sieve 5A column, similar to what has been described previously (Barney et al. 2005; Dos Santos et al. 2007). H2 consumption was quantified by the same method, but a starting OD600 of 0.375 was used and 250 μL of H2 at atmospheric pressure was added after thoroughly venting the culture. This lower OD600 was selected for accurate measurement of H2 consumption, as control experiments revealed that gas transport and solubility could limit activity at higher concentrations of cells.
Results
Nitrogenase-driven H2 production
Three modifications were incorporated into the A. vinelandii genome to increase the production of H2 by this model bacterium: a modification to nifL (nifL::KanR) that deregulates nitrogenase (Bali et al. 1992; Barney et al. 2015; Brewin et al. 1999), incorporation of a mutation near the active site of nitrogenase (nifDV70I) that results in a preference of the enzyme for H+ reduction over N2 reduction (Barney et al. 2004), and deletion of the uptake hydrogenase complex (Δhyd) that recycles the H2 by-product of the nitrogenase reaction (Barney et al. 2017; Noar and Bruno-Bárcena 2016). Various combinations of these three modifications were constructed and are shown in Fig. 2 in order of increasing H2 production (blue bars). The strain AZBB275 incorporating all three modifications (nifL::KanR nifDV70I Δhyd) had the highest H2 production (5.6 μmol h−1 (mg protein)−1), a 28-fold increase from the wild-type strain DJ (0.2 μmol h−1 (mg protein)−1). The largest effect by a single-component modification was seen for the hydrogenase deletion strain, AZBB261 (Δhyd). The variant form of the molybdenum-dependent nitrogenase containing the nifDV70I amino acid substitution (DJ1373) resulted in an increase in H2 production to around 0.7 μmol h−1 (mg protein)−1, indicating that this mutant alone results in sufficient H2 production to overwhelm the capability of the uptake hydrogenase to recycle the H2 under these conditions.

Hydrogen produced by wild-type and mutant strains of A. vinelandii in the absence (blue bars) or presence (gray bars) of 5 mM ammonium (NH4+). Relevant genetic features of each strain are listed on the right. Strains with nifDV70I result in a nif− phenotype. Strains designated with Δhyd are lacking both the soluble (Avin_04360–Avin_04410) and membrane-bound (Avin_50500–Avin_50590) hydrogenases (N = 3, mean ± SD)
Since the various manipulations made here were expected to have a detrimental effect on the growth rates of some of these strains, we tested several key constructs to determine the doubling time of each strain. As shown in Table 2, the wild-type strain yielded doubling times of ~ 2.3 h when supplemented with urea, and ~ 3.3 h when grown diazotrophically, which correlates well with previous reports (Curatti et al. 2005; Hill et al. 1999; Martin et al. 1989). AZBB163 (nifL::KanR), which is deregulated for BNF, yielded a slight increase in doubling time with urea, and a slightly larger increase under diazotrophic conditions, in addition to the lower maximal OD obtained at stationary phase that has been reported previously (Barney et al. 2017). Strain AZBB269 (nifL::KanR Δhyd), which contains the deletion of the hydrogenases in combination with the deregulated BNF, showed an increase over AZBB163 under both sets of growth conditions. Finally, AZBB275, which grows similar to AZBB269 in the presence of urea, showed a dramatic loss in growth rate under diazotrophic conditions, indicating that BNF is no longer occurring at sufficient rates to allow the cells to grow under these conditions.
Nitrogenase inhibition by NH4 +
The effect of including elevated levels of ammonium (NH4+) was also probed in this study. The inclusion of 5 mM NH4+ in the growth medium reduced the levels of H2 produced by all strains (Fig. 2, gray bars). This concentration of NH4+ was chosen as it resulted in strong inhibition in the hydrogenase deletion strain (AZBB261, Δhyd), though it should be noted that these levels of NH4+ exceed anything that would accumulate in any of the strains except AZBB269 containing the modification to the nifL region (nifL::KanR), and even AZBB269 does not accumulate these levels of NH4+ during the short timeframe of the assays employed here. In the final strain, AZBB275 (nifL::KanR nifDV70I Δhyd), there was a fivefold reduction when compared to growth in nitrogen-free medium. Both of the strains (AZBB269 and AZBB275) containing the nifL::KanR modification retained the ability to produce H2 at ~ 1 μmol h−1 (mg protein)−1. Near complete repression of nitrogenase activity by 5 mM NH4+ was seen in all strains lacking the nifL modification (Fig. 2, all but AZBB269 and AZBB275). Strain AZBB275, which contains the nifDV70I amino acid substitution, is essentially unable to fix nitrogen under the these assay conditions or even under typical growth conditions (Table 2), which would limit any background inhibition by NH4+ in this optimized strain. One caveat of the nifDV70I modification is that it makes these strains dependent on an external source of nitrogen to attain growth rates similar to the other strains (Table 2).
Continuous production of H2
To determine if the rate of H2 production changed during the 2-h assay, levels of H2 and oxygen (O2) were measured in 15-min increments. Linear H2 production was observed throughout the 2-h duration of the assay (Fig. 3), indicating that rising levels of H2 did not inhibit further production at these concentrations. This was further correlated with a linear decrease in O2 reaching a final concentration indicating 33% consumption of O2. This was in agreement with a preliminary experiment indicating that the cells required about 2 h following introduction into the liquid culture to achieve maximal exponential growth and hydrogen production levels. This 2-h preincubation time was employed throughout all of these experiments.

H2 production and O2 consumption by A. vinelandii strain AZBB275 (nifL::KanR nifDV70I Δhyd) over time. a Hydrogen and associated b oxygen levels reported as micromole per milligram starting protein (N = 3, linear least squares regression)
Production versus consumption rates of H2
Strains AZBB275 (nifL::KanR nifDV70I Δhyd) and AZBB163 (nifL::KanR) were further tested for the production or consumption of H2 by A. vinelandii to compare and contrast these two competing reactions (Fig. 4). A volume equivalent to 9 μmol of H2 (slightly greater than the amount produced during 2 hours by AZBB275 in this experiment) was added to three of the four cultures. Strain AZBB275 showed linear H2 production up to 16 μmol of H2 when initially started with this spiked amount of H2. Supplementing AZBB275 with exogenous NH4+ (20 mM) reduced the rate of production, but not entirely, similar to what was found in Fig. 2. Strain AZBB163, which still contained both of the hydrogenase complexes, consumed H2 at a rate greater than its production capabilities, resulting in a continuous decrease in the H2 that was spiked into this sample. Rates of H2 production by AZBB275 were similar in the samples with or without the initial quantity of spiked H2, further confirming that the H2 produced was not limiting further production of H2 through any noticeable inhibition.

H2 produced and consumed by A. vinelandii strains AZBB275 (nifL::KanR nifDV70I Δhyd) and AZBB163 (nifL::KanR) when cultured under conditions with or without exogenously provided ammonium and exogenously provided H2. AZBB163 was introduced at an OD600 of 0.375 while all AZBB275 samples were introduced at an OD600 of 1.5. Samples to which 250 μL H2 was added are noted (+ H2). (N = 3, linear least squares regression)
Additional nitrogenous compounds as inhibitors of nitrogenase
NH4+ added to the medium inhibited H2 production by AZBB275 (Figs. 2 and 4). To determine the potential of other nitrogenous compounds to inhibit H2 production, AZBB275 (nifL::KanR nifDV70I Δhyd) was screened with urea, nitrite (NO2−), and nitrate (NO3−). AZBB275 was suitable for this analysis because the nifDV70I amino acid substitution results in a very limited ability to fix N2 (Table 2), so that NH4+ levels accumulating within the cell would be negligible. Nitrogenase activity was repressed by all three of these N compounds in addition to NH4+ (Fig. 5): ~ 80% with 10 mM NH4+, ~ 70% with 3 mM NO2−, ~ 40% with 10 mM NO3−, and ~ 70% with 10 mM urea. As the rates of inhibition by NH4+ and NO2− were greater than what was found for urea and NO3−, we speculated that it might not be urea or NO3−, but instead the products of further enzymatic activities within the cell that were resulting in their inhibition. Repression by urea was removed in strain AZBB312 (nifL::KanR nifDV70I Δhyd ΔureABC), which is based on strain AZBB275, but also contains a further disruption of the gene coding for the enzyme urease, which could hydrolyze urea into carbon dioxide and NH4+ (Fig. 1). Strain AZBB330 (nifL::KanR nifDV70I Δhyd ΔnasAB), which is also based on AZBB275, contains disruptions to the genes for both nitrate reductase and nitrite reductase (Fig. 1), eliminating further reduction of NO3− to NO2− or NO2− to NH4+, rendering the NO3− a terminal compound within the cell. Inhibition by NO2− was unchanged while NO3− inhibition was removed in the ΔnasAB strain AZBB330. The inability to further reduce NO2− to NH4+ was also confirmed for AZBB330. Concentration curves for NH4+ and NO2− (Fig. 6) in strains AZBB275 and AZBB330 showed a concentration-dependent sensitivity to both compounds. Growth rates of AZBB312 and AZBB330 were also measured, except that ammonium sulfate (10 mM) was substituted in place of urea for the nitrogen source as a control. AZBB312 grew with a doubling time of ~ 2.9 h with NH4+, but the doubling time dropped to ~ 43 h when grown on urea, indicating that urea is no longer a suitable substrate for this strain in the absence of the urease enzyme, as has been reported previously (Barney et al. 2015). AZBB330 grew with a doubling time of ~ 3.1 h with NH4+, but the doubling time dropped to ~ 109 h when grown on NO3−, indicating that nitrate was no longer a suitable substrate for this strain in the absence of the nitrate and nitrite reductase genes nasAB (Supplementary Table S4).

Effect of different nitrogen compounds on the amount of H2 produced by A. vinelandii AZBB275 (nifL::KanR nifDV70I Δhyd) and the two mutant strains AZBB312 (AZBB275 + ΔureABC) and AZBB330 (AZBB275 + ΔnasAB). Strains were incubated with medium that was not supplemented with any nitrogenous compounds, 10 mM NH4+, 3 mM nitrite (NO2−), 10 mM nitrate (NO3−), or 10 mM urea (CH4N2O). (N ≥ 3, mean)

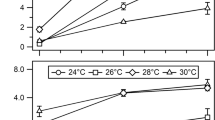
Effect of the concentration of NH4+ and NO2− on H2 production in A. vinelandii strains AZBB275 (nifL::KanR nifDV70I Δhyd) and AZBB330 (AZBB275 + ΔnasAB). a AZBB275 with NH4+, b AZBB275 with NO2−, and c AZBB330 with NO2−. (N = 2, running average)
Full cell assay susceptibility to pH effects
The effect of pH on H2 production as measured by in vitro assays using isolated nitrogenase (activity measured in an argon atmosphere) was studied previously (Pham and Burgess 1993). However, the strains constructed here allowed for in vivo measurement of the effect of the pH of the growth medium and the ability of the cells to produce nitrogenase-driven H2. H2 production increased with increasing pH over the range of 5.5–9.0. All other experiments described in this study were done in medium adjusted to pH 7.5, which was selected because it is close to the starting pH of typical B medium. Therefore, the H2 values presented in Fig. 7 were normalized to the mean value obtained for the pH 7.5 samples and converted to percentages. A. vinelandii struggled to grow at pH levels lower than 6 and showed minimal H2 production at low pH. Maximum H2 production (135%) plateaued at pH values of 8.5 and 9, indicating that mild alkaline conditions favor nitrogenase activity in vivo.

Effect of pH on H2 production and OD600 in A. vinelandii strain AZBB275 (nifL::KanR nifDV70I Δhyd). a H2 produced and associated b OD600 measurements obtained at the end of the 2-h assay. B medium was adjusted to pH 7.5 in all other experiments so all values in this figure were normalized to the mean of the pH 7.5 samples. (N = 3, LOESS regression and mean)
Discussion
H2 is produced by a range of bacteria, often through the action of reversible hydrogenases, and generally as a result of anaerobic processes (Elsharnouby et al. 2013; Rittmann and Herwig 2012; Sinha and Pandey 2011). A. vinelandii is unique from many bacteria, as it is an obligate aerobic nitrogen-fixing bacterium (diazotroph), and as such, it should be able to yield substantial H2 through the mechanism of nitrogenase under aerobic conditions (Noar and Bruno-Bárcena 2016; Setubal et al. 2009).
The initial goal of this study was to determine the potential to produce hydrogen gas (H2) via nitrogenase when the path to H2 was optimized in A. vinelandii using a combination of three different strategies. The majority of these strains were constructed from a parent strain devoid of both the vanadium-dependent and iron-only nitrogenases (ΔvnfHDGK and ΔanfHDGK, respectively (Eberhart et al. 2016)) to assure that the results measured were solely attributed to the molybdenum-dependent nitrogenase of A. vinelandii, although these other two nitrogenase systems would not be expected to be highly expressed under the growth conditions employed in this study, which included molybdenum in the medium (Barney et al. 2017). We anticipated that by incorporating a genomic modification which deregulates BNF, along with the inability to recycle H2 and the inclusion of a modification to the Mo-based nitrogenase that drives substrate preference away from N2 and toward proton reduction, we could construct a strain that approaches the full potential of H2 production by A. vinelandii. In addition, since this modification that results in a substrate preference toward proton reduction would limit BNF, the strain should produce low levels of intracellular NH4+, which could potentially inhibit the activity of the nitrogenase. We constructed and measured each modification in isolated strains both individually, and in combination (Fig. 2). Prior to these experiments, it was uncertain whether the effects of these combined modifications in A. vinelandii would complement one another to increase H2 yield, or result in a similar level of H2 production to what was seen for the individual modifications (Bali et al. 1992; Barney et al. 2004, 2017; Noar et al. 2015). By constructing and analyzing independent modification of all three approaches alone and in combination, we were able to assess the independent contributions of each (Fig. 2). In addition to targeting the gene generally associated with H2 uptake in A. vinelandii, we also deleted the second hydrogenase gene cluster in our Δhyd strains, which is not generally associated with H2 uptake (Barney et al. 2017; Noar and Bruno-Bárcena 2016), to further assure that no alternative pathways to consume H2 were present in the target strain. Our final strain AZBB275 contains modifications resulting in the deletion of 28 genes, and modifications to two additional genes to generate the central strain in these studies.
Rey et al. (2007) pursued a similar approach to redirect metabolism in Rhodopseudomonas palustris toward biological H2 production, but required anaerobic conditions to accomplish this task. Fixen et al. (2016) built upon the work of Rey et al. to achieve elevated H2 production and low levels of carbon dioxide and carbon monoxide reduction to shorter hydrocarbons by nitrogenase in R. palustris. Our modified A. vinelandii strain produced H2 at rates greater than those obtained by Fixen et al., who reported ~ 2 μmol h−1 (mg protein)−1 (based on Fig. 1b within reference (Fixen et al. 2016)). Because Fixen et al. used the Bradford assay with BSA as a standard, we employed the same strategy here for general protein quantifications. Prior studies have also reported H2 production in A. vinelandii based on disruptions of hydrogenase (Barney et al. 2017; Linkerhägner and Oelze 1995; Noar et al. 2015; Noar and Bruno-Bárcena 2016), which would be comparable to what was obtained here for our hydrogenase disruption strain AZBB261 (Δhyd). Our maximum yields of H2 were approaching 7 μmol h−1 (mg protein)−1 (Figs. 2 and 7) in our strain containing all three modifications (nifL::KanR nifDV70I Δhyd). The maximum production of H2 using purified molybdenum-dependent nitrogenase through in vitro studies is approximately 2200 nmol min−1 (mg MoFe protein)−1 (~ 132 μmol h−1 (mg protein)−1) (Barney et al. 2004), thought in vitro studies generally use Biuret assays for protein quantitation (Christiansen et al. 1998). Since we wanted to compare our values to prior reports that used different methods to quantify protein, we did a comparison of protein determinations with both methods. Our results comparing the quantitation of full cell proteins from A. vinelandii cells using both Bradford and Biuret assays indicate that Biuret assays yield protein quantities that are 50% higher than the values obtained by the Bradford assay, which is in line with the inconsistencies between different assays that have been reported previously (Stoscheck 1990). Taking this factor into account as well as the H2 yields obtained here and in prior reports with in vitro assays using the Biuret assay for protein quantification (Barney et al. 2015, 2009), we calculated that at least ~ 3.5% of the protein in AZBB275 is active MoFe enzyme in the cells. Nitrogenase is a two-component enzyme composed of the catalytic enzyme MoFe and the obligate reductase Fe protein. This high level of MoFe enzyme by convention indicates a similarly high level of Fe protein (Barney et al. 2009).
Masukawa et al. (2010) pursued a strategy in cyanobacteria including a deletion to the uptake hydrogenase and modifications near the active site of the molybdenum nitrogenase, and yielded results as high as ~ 24 μmol h−1 (mg chlorophyll a)−1. Weyman et al. (2010) and Schütz et al. (2004) reported yields of 100–140 nmol h−1 (μg chlorophyll a)−1 based on gene disruption of the uptake hydrogenase and modification near the active site of the MoFe protein of nitrogenase, respectively. Since each of these analyses is based on chlorophyll content, and not protein, it is difficult to make direct comparisons to our results. Bandyopadhyay et al. (2010) demonstrated yields of 3.5 μmol h−1 (mg protein)−1 in Cyanothece sp. ATCC 51142 grown under anaerobic conditions, though the levels under aerobic conditions were less than half of those rates. Bandyopadhyay et al. utilized the bicinchoninic acid assay for their protein determinations. An accurate comparison of each of these strains would require the analysis to be performed and reported using the same benchmark, as either total protein (including consideration of the protein assays employed) or total cell mass, and would need to also consider levels of nitrogenase enzyme present and the source of energy to drive the process (heterotrophic versus photoautotrophic growth).
Our next goal in this work was to use strain AZBB275 as a surrogate to assay intracellular nitrogenase activity in response to physiological nitrogenous compounds. Since H2 is a by-product of the BNF reaction, it could also act as a proxy of nitrogenase activity if the reactions that recycled H2 were removed. Generally, enzymatic characterization of nitrogenase is accomplished with either in vitro assays using purified enzyme or in vivo assays where live cells are subjected to the alternative substrate, acetylene. In vitro assays using pure enzymes are the optimal method of measuring nitrogenase activity, as this removes any effects of the regulatory system which modulates nitrogenase activity and expression. Alternatively, biotechnological applications aimed at exploiting nitrogenase as it exists within an organism would require robust whole-cell assays. In vivo assays using acetylene are hindered by low nitrogenase levels and activities, and require that acetylene, a substrate for the enzyme and also an inhibitor of BNF, always be present. This prevents testing of environmental conditions where no inhibitor is desired or in studies aimed to isolate the effect of a single compound on the internal functions of the cell.
The intent of this analysis was to determine if H2 production, and by extension ammonium (NH4+) production (Fixen et al. 2016), could be increased over current levels (Linkerhägner and Oelze 1995; Noar et al. 2015; Noar and Bruno-Bárcena 2016). It is well established that the presence of various nitrogenous compounds within the growth medium will repress BNF in A. vinelandii (Curatti et al. 2005; Klugkist and Haaker 1984; Little et al. 2006), and that the specific disruption to the nifL gene, resulting in nitrogenase deregulation, is able to produce copious quantities of NH4+ (Bali et al. 1992; Barney et al. 2015, 2017; Brewin et al. 1999), which might further inhibit BNF. As our strains contain different combinations of these modifications, including the nifL disruption that dramatically derepresses nitrogenase expression, it was of interest to determine if the presence of NH4+ or different nitrogenous compounds in the medium would continue to inhibit nitrogenase in this deregulated strain.
Repression of H2 production by high levels (5 mM) of NH4+ in AZBB275 (nifL::KanR nifDV70I Δhyd) (Fig. 2) demonstrates that nitrogen fixation has yet to be fully deregulated, even in the high NH4+-producing strain AZBB163 that is proving to be a powerful tool to better understand global nitrogen regulation and response within the cell (Barney et al. 2015, 2017). As AZBB163 can produce above 25 mM NH4+ (Barney et al. 2017; Brewin et al. 1999), and our results indicate that nitrogen fixation is inhibited by levels of NH4+ as low as 3 mM (Fig. 6), it is clear that secondary inhibition by the product is still limiting the potential yields that could be obtained by this strain. This indicates that there is a potential to improve yields further if the secondary repression mechanism in A. vinelandii could be identified and subsequently eliminated.
In addition to the inhibition of BNF by NH4+, we also found inhibition by urea, NO2−, and NO3−, though the urea inhibition could be eliminated if urease was disrupted and NO3− inhibition could be eliminated if nitrate reductase was disrupted, indicating that the primary inhibitory compounds were NH4+ and NO2− (Fig. 5). Therefore, NH4+ inhibition might be overcome if fixed nitrogen could be directed toward terminal products of urea or NO3−. The nitrogenous compound NO3− is reported to repress BNF in A. vinelandii, Cyanothece sp. ATCC 51142, and Klebsiella pneumoniae (Bandyopadhyay et al. 2010; Gutierrez et al. 1997; Hom et al. 1980; Strandberg and Wilson 1968), but our results demonstrate that this inhibition in A. vinelandii is likely the result of NO2−, and not NO3−, which agrees with previous findings for Gloeothece (Nägeli) sp. ATCC 27152 (Cheng et al. 1999). Urea inhibition of BNF has been reported in Anabaena cylindrica and in Herbaspirillum seropedicae. This inhibition was attributed to NH4+ in A. cylindrica, but urea inhibition of BNF appeared to be independent of NH4+ in H. seropedicae (Gusso et al. 2008; Mackerras and Smith 1986). Further optimization could be achieved by combining the disruption or deletion of nitrate reductase and urease to remove the inhibitive effects of NO3− and urea that may be present in nutrient-rich waste streams. This also indicated that higher direct biological production of nitrogen compounds for biofertilizer applications could be achieved if products could be directed toward urea or nitrate in strains lacking urease or nitrate reductase, respectively.
Deregulation of BNF in these strains is accomplished by disrupting the nifL gene, which results in the constitutive expression of NifA in the absence of the repressor NifL (Bali et al. 1992; Barney et al. 2015, 2017; Brewin et al. 1999). As such, the data suggests that the diminished activity of nitrogenase observed in the presence of NH4+ is likely due to a post-translational modification that has yet to be adequately detailed or described in A. vinelandii. Other organisms use post-translational regulation through the actions of DraT and DraG (Liang et al. 1991; Rey et al. 2007). These two proteins perform ADP-ribosylation of NifH in response to NH4+ (Liang et al. 1991). While comparative genomics has not revealed any homologs to DraT and DraG in A. vinelandii, a recent study proposed NifO as being similar in function to DraG when they adjusted the algorithm to better match different motifs (Rubel et al. 2016). Based on the conclusions drawn from our study, identification of the gene(s) responsible for encoding potential secondary regulatory protein(s) could result in further improvements in nitrogenase activity, and further the potential for aerobic hydrogen or NH4+ production by A. vinelandii.
Being able to remove nitrogenase repression by NH4+ might allow for the use of this strain for the aerobic production of H2 in N-rich environments. If nitrogenase activity were insensitive to NH4+, the productivity of this strain and range of applications would increase. Environments where external NH4+ was readily available would allow the strain to continue producing H2 while not limiting its ability to use NH4+ for growth.
The pH dependency of H2 production in active cultures was a curious phenomenon. Hydrogenases are well known to be pH dependent. However, strain AZBB275 (nifL::KanR nifDV70I Δhyd) no longer has either hydrogenase cluster. Because our strain lacks all known hydrogenase systems and both of the alternative nitrogenases (vanadium and iron only), it is assumed that all the H2 produced in AZBB275 is a direct result of the molybdenum-dependent nitrogenase. Nitrogenase is also known to be pH sensitive with an optimum pH of 7.5 when tested in vitro (Pham and Burgess 1993). It is uncertain if the pH effect seen with AZBB275 is related to an effect on the nitrogenase within the cell, or if it has altered the overall metabolism to favor the production of H2. Interestingly, our results indicated that the production of H2 favors low availability of protons in the culture medium (Fig. 7).
Nitrogenase-driven H2 production has some advantages over fermentative and photosynthetic processes. A. vinelandii, as an aerobic, heterotrophic organism with no light requirement, could potentially be modified to withstand fixed N sources. This makes it an interesting candidate for producing H2 from various waste streams, which are often both nutrient rich and opaque. This study presents an alternative approach to biohydrogen production which may be better suited to mixed culture approaches (Elsharnouby et al. 2013; Rittmann and Herwig 2012; Sinha and Pandey 2011) or versus cyanobacteria (Masukawa et al. 2010; Tamagnini et al. 2002). Creating mixed aerobic cultures could also have application when coupled together with sugar-releasing algae to yield a two-step process that produces both H2 and NH3 from sunlight (Arriola et al. 2017; Barney et al. 2015).
References
Arriola MB, Velmurugan N, Zhang Y, Plunkett MH, Hondzo H, Barney BM (2017) Genome sequences of Chlorella sorokiniana UTEX 1602 and Micractinium conductrix SAG 241.80: implications to maltose excretion by a green alga. Plant J 93(3):566–586
Bali A, Blanco G, Hill S, Kennedy C (1992) Excretion of ammonium by a nifL mutant of Azotobacter vinelandii fixing nitrogen. Appl Environ Microbiol 58(5):1711–1718
Bandyopadhyay A, Stockel J, Min HT, Sherman LA, Pakrasi HB (2010) High rates of photobiological H2 production by a cyanobacterium under aerobic conditions. Nat Commun 1:7
Barahona E, Jiménez-Vicente E, Rubio LM (2016) Hydrogen overproducing nitrogenases obtained by random mutagenesis and high-throughput screening. Sci Rep 6:9
Barney BM, Igarashi RY, Dos Santos PC, Dean DR, Seefeldt LC (2004) Substrate interaction at an iron-sulfur face of the FeMo-cofactor during nitrogenase catalysis. J Biol Chem 279(51):53621–53624
Barney BM, Laryukhin M, Igarashi RY, Lee HI, Dos Santos PC, Yang TC, Hoffman BM, Dean DR, Seefeldt LC (2005) Trapping a hydrazine reduction intermediate on the nitrogenase active site. Biochemistry 44(22):8030–8037
Barney BM, Lukoyanov D, Igarashi RY, Laryukhin M, Yang TC, Dean DR, Hoffman BM, Seefeldt LC (2009) Trapping an intermediate of dinitrogen (N2) reduction on nitrogenase. Biochemistry 48(38):9094–9102
Barney BM, Eberhart LJ, Ohlert JM, Knutson CM, Plunkett MH (2015) Gene deletions resulting in increased nitrogen release by Azotobacter vinelandii: application of a novel nitrogen biosensor. Appl Environ Microbiol 81(13):4316–4328
Barney BM, Plunkett MH, Natarajan V, Mus F, Knutson CM, Peters JW (2017) Transcriptional analysis of an ammonium-excreting strain of Azotobacter vinelandii deregulated for nitrogen fixation. Appl Environ Microbiol 83(20):e01534–e01517
Bothe H, Schmitz O, Yates MG, Newton WE (2010) Nitrogen fixation and hydrogen metabolism in cyanobacteria. Microbiol Mol Biol Rev 74(4):529–551
Brewin B, Woodley P, Drummond M (1999) The basis of ammonium release in nifL mutants of Azotobacter vinelandii. J Bacteriol 181(23):7356–7362
Burgess BK, Lowe DJ (1996) Mechanism of molybdenum nitrogenase. Chem Rev 96(7):2983–3011
Cheng J, Hipkin CR, Gallon JR (1999) Effects of inorganic nitrogen compounds on the activity and synthesis of nitrogenase in Gloeothece (Nägeli) sp. ATCC 27152. New Phytol 141(1):61–70
Christiansen J, Goodwin PJ, Lanzilotta WN, Seefeldt LC, Dean DR (1998) Catalytic and biophysical properties of a nitrogenase apo-MoFe protein produced by a nifB-deletion mutant of Azotobacter vinelandii. Biochemistry 37(36):12611–12623
Chromý V, Fischer J, Kulhánek V (1974) Re-evaluation of EDTA-chelated biuret reagent. Clin Chem 20(10):1362–1363
Curatti L, Brown CS, Ludden PW, Rubio LM (2005) Genes required for rapid expression of nitrogenase activity in Azotobacter vinelandii. Proc Natl Acad Sci U S A 102(18):6291–6296
Dos Santos PC, Mayer SM, Barney BM, Seefeldt LC, Dean DR (2007) Alkyne substrate interaction within the nitrogenase MoFe protein. J Inorg Biochem 101(11–12):1642–1648
Eberhart LJ, Knutson CM, Barney BM (2016) A methodology for markerless genetic modifications in Azotobacter vinelandii. J Appl Microbiol 120(6):1595–1604
Elsharnouby O, Hafez H, Nakhla G, El Naggar MH (2013) A critical literature review on biohydrogen production by pure cultures. Int J Hydrog Energy 38(12):4945–4966
Fixen KR, Zheng YN, Harris DF, Shaw S, Yang ZY, Dean DR, Seefeldt LC, Harwood CS (2016) Light-driven carbon dioxide reduction to methane by nitrogenase in a photosynthetic bacterium. Proc Natl Acad Sci U S A 113(36):10163–10167
Ghirardi ML, King PW, Posewitz MC, Maness PC, Fedorov A, Kim K, Cohen J, Schulten K, Seibert M (2005) Approaches to developing biological H2-photoproducing organisms and processes. Biochem Soc Trans 33:70–72
Gusso CL, de Souza EM, Rigo LU, Pedrosa FD, Yates MG, Rego FGD, Klassen G (2008) Effect of an ntrC mutation on amino acid or urea utilization and on nitrogenase switch-off in Herbaspirillum seropedicae. Can J Microbiol 54(3):235–239
Guth JH, Burris RH (1983) Inhibition of nitrogenase-catalyzed NH3 formation by H2. Biochemistry 22(22):5111–5122
Gutierrez JC, Santero E, Tortolero M (1997) Ammonium repression of the nitrite-nitrate (nasAB) assimilatory operon of Azotobacter vinelandii is enhanced in mutants expressing the nifO gene at high levels. Mol Gen Genet 255(2):172–179
Hill S, He LH, Kennedy C (1999) Physiological characterisation of an Azotobacter vinelandii nifU-deletion mutant and its spontaneous Nif+ revertants that over-produce cytochrome bd. FEMS Microbiol Lett 175(2):185–191
Hom SSM, Hennecke H, Shanmugam KT (1980) Regulation of nitrogenase biosynthesis in Klebsiella pneumoniae: effect of nitrate. J Gen Microbiol 117(MAR):169–179
Klugkist J, Haaker H (1984) Inhibition of nitrogenase activity by ammonium chloride in Azotobacter vinelandii. J Bacteriol 157(1):148–151
Liang JH, Nielsen GM, Lies DP, Burris RH, Roberts GP, Ludden PW (1991) Mutations in the draT and draG genes of Rhodospirillum rubrum result in loss of regulation of nitrogenase by reversible ADP-ribosylation. J Bacteriol 173(21):6903–6909
Linkerhägner K, Oelze J (1995) Hydrogenase does not confer significant benefits to Azotobacter vinelandii growing diazotrophically under conditions of glucose limitation. J Bacteriol 177(20):6018–6020
Little R, Martinez-Argudo I, Dixon R (2006) Role of the central region of NifL in conformational switches that regulate nitrogen fixation. Biochem Soc Trans 34:162–164
Mackerras AH, Smith GD (1986) Evidence for direct repression of nitrogenase by ammonia in the cyanobacterium Anabaena cylindrica. Biochem Biophys Res Commun 134(2):835–844
Martin AE, Burgess BK, Iismaa SE, Smartt CT, Jacobson MR, Dean DR (1989) Construction and characterization of an Azotobacter vinelandii strain with mutations in the genes encoding flavodoxin and ferredoxin I. J Bacteriol 171(6):3162–3167
Masukawa H, Inoue K, Sakurai H, Wolk CP, Hausinger RP (2010) Site-directed mutagenesis of the Anabaena sp. strain PCC 7120 nitrogenase active site to increase photobiological hydrogen production. Appl Environ Microbiol 76(20):6741–6750
Noar JD, Bruno-Bárcena JM (2016) Protons and pleomorphs: aerobic hydrogen production in Azotobacters. World J Microbiol Biotechnol 32(2):8
Noar J, Loveless T, Navarro-Herrero JL, Olson JW, Bruno-Bárcena JM (2015) Aerobic hydrogen production via nitrogenase in Azotobacter vinelandii CA6. Appl Environ Microbiol 81(13):4507–4516
Pham DN, Burgess BK (1993) Nitrogenase reactivity: effects of pH on substrate reduction and CO inhibition. Biochemistry 32(49):13725–13731
Radakovits R, Jinkerson RE, Darzins A, Posewitz MC (2010) Genetic engineering of algae for enhanced biofuel production. Eukaryot Cell 9(4):486–501
Rey FE, Heiniger EK, Harwood CS (2007) Redirection of metabolism for biological hydrogen production. Appl Environ Microbiol 73(5):1665–1671
Rittmann S, Herwig C (2012) A comprehensive and quantitative review of dark fermentative biohydrogen production. Microb Cell Factories 11:18
Rubel ET, Raittz RT, Coimbra NA, Gehlen MA, Pedrosa FO (2016) ProClaT, a new bioinformatics tool for in silico protein reclassification: case study of DraB, a protein coded from the draTGB operon in Azospirillum brasilense. BMC Bioinformatics 17(Suppl 18):455
Schütz K, Happe T, Troshina O, Lindblad P, Leitão E, Oliveira P, Tamagnini P (2004) Cyanobacterial H2 production - a comparative analysis. Planta 218(3):350–359
Setubal JC, dos Santos P, Goldman BS, Ertesvåg H, Espin G, Rubio LM, Valla S, Almeida NF, Balasubramanian D, Cromes L, Curatti L, Du Z, Godsy E, Goodner B, Hellner-Burris K, Hernandez JA, Houmiel K, Imperial J, Kennedy C, Larson TJ, Latreille P, Ligon LS, Lu J, Maerk M, Miller NM, Norton S, O’Carroll IP, Paulsen I, Raulfs EC, Roemer R, Rosser J, Segura D, Slater S, Stricklin SL, Studholme DJ, Sun J, Viana CJ, Wallin E, Wang B, Wheeler C, Zhu H, Dean DR, Dixon R, Wood D (2009) Genome sequence of Azotobacter vinelandii, an obligate aerobe specialized to support diverse anaerobic metabolic processes. J Bacteriol 191(14):4534–4545
Shaw S, Lukoyanov D, Danyal K, Dean DR, Hoffman BM, Seefeldt LC (2014) Nitrite and hydroxylamine as nitrogenase substrates: mechanistic implications for the pathway of N2 reduction. J Am Chem Soc 136(36):12776–12783
Sinha P, Pandey A (2011) An evaluative report and challenges for fermentative biohydrogen production. Int J Hydrog Energy 36(13):7460–7478
Stoscheck CM (1990) Quantitation of protein. Methods Enzymol 182:50–68
Strandberg GW, Wilson PW (1968) Formation of the nitrogen-fixing enzyme system in Azotobacter vinelandii. Can J Microbiol 14(1):25–31
Tamagnini P, Axelsson R, Lindberg P, Oxelfelt F, Wünschiers R, Lindblad P (2002) Hydrogenases and hydrogen metabolism of cyanobacteria. Microbiol Mol Biol Rev 66(1):1–20
Weyman PD, Pratte B, Thiel T (2010) Hydrogen production in nitrogenase mutants in Anabaena variabilis. FEMS Microbiol Lett 304(1):55–61
Acknowledgements
We thank Yaniv Brandvain for suggestions related to statistical analysis of our data.
Funding
This work was supported by grants from the MnDRIVE transdisciplinary research initiative through the University of Minnesota based on funding from the state of Minnesota to B.M.B. and the National Institute of Food and Agriculture (Project Numbers MIN-12-070 and MIN-12-081).
Author information
Authors and Affiliations
Contributions
B.M.B., C.M.K., and M.H.P conceived the study and designed experiments. B.M.B. and M.H.P. designed plasmids and constructed strains. C.M.K, M.H.P., and R.A.L. developed methods and collected data. All authors contributed to writing the manuscript and discussion.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical statement
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
ESM 1
(PDF 345 kb)
Rights and permissions
About this article

Cite this article
Knutson, C.M., Plunkett, M.H., Liming, R.A. et al. Efforts toward optimization of aerobic biohydrogen reveal details of secondary regulation of biological nitrogen fixation by nitrogenous compounds in Azotobacter vinelandii. Appl Microbiol Biotechnol 102, 10315–10325 (2018). https://doi.org/10.1007/s00253-018-9363-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-018-9363-0