
Abstract
Ficellomycin is a peptide-like antibiotic which exhibits potent in vitro activity against Penicillium oxalicum and Staphylococcus aureus, even against strains resistant to most clinically used antibiotics. The gene cluster responsible for ficellomycin biosynthesis was cloned from Streptomyces ficellus and sequenced. It was found to contain 26 ORFs and is located within 30 kb of contiguous DNA. Targeted disruption of the encoding genes revealed that most were involved in the functional section of ficellomycin biosynthesis, such as peptide assembly, regulation, resistance, and biosynthesis of the precursor of ficellomycin 2-[4-guanidyl-1-azabicyclo[3.1.0]hexan-2-yl] glycine (2-GAHG). Within the 2-GAHG synthesis pathway, a sulfate adenylyltransferase appears to be involved in the synthesis of the pharmaceutically important 1-azabicyclo[3.1.0]hexane ring moiety, which has been reported to cause DNA cross-linking or impairment of semiconservative DNA replication.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The peptide-like antibiotic ficellomycin was originally isolated from the culture broth of Streptomyces ficellus (NRRL8067), together with two other natural products displaying antibiotic activities. One of them was designated as feldamycin and the other was later identified as Nojirimycin (Argoudelis AD et al. 1976). Ficellomycin displays high in vitro activity against Penicillium oxalicum and Staphylococcus aureus, even against strains resistant to penicillin, streptomycin, neomycin, macrolides, and lincosamides, like the methicillin-resistant S. aureus (MRSA). It is also moderately effective in vivo, where it exhibited a CD50 of ca. 7.6 mg kg−1 in mice infected with S. aureus (Foulke-Abel J et al. 2011). The structure of ficellomycin was elucidated in 1989, revealing it to be valyl-2-[4-guanidyl-1-azabicyclo[3.1.0]hexan-2-yl] glycine by NMR, MS, and derivatization studies, which also revealed the presence of a highly strained azabicyclo[3.1.0]hexane (aziridine) ring system (Kuo MS et al. 1989). It seems likely that this aziridine moiety is the structural feature responsible for the antibiotic activity of ficellomycin, since it is found in effective alkylating agents, and is a key determinant of the antitumor activity of structurally related natural products (Brulikova L et al. 2012; Ismail FM et al. 2009) (Fig. 1). Reusser and co-workers revealed that ficellomycin selectively impairs semiconservative DNA replication in DNA polymerase I—deficient Escherichia coli that had been rendered permeable to nucleotides by toluene treatment (Reusser F 1977). Specifically, this antibiotic causes the accumulation of 34S DNA species, likely as a result of DNA alkylation. Further investigation indicated that ficellomycin neither inhibits steps occurring during the early initiation of replication nor the polymerases, but instead causes the formation of deficient 34S DNA fragments. These fragments lack the ability to integrate into larger DNA pieces and eventually the complete bacterial chromosome during the maturation and assembly process of the bacterial chromosome (Reusser F 1977).

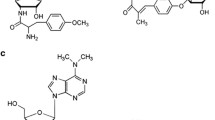
Chemical structures of four aziridine-containing natural products whose gene clusters have been identified. The characteristic aziridine moieties are indicated in bold lines
These unusual structural features and prominent biological activity, coupled with the unique mode of action, have attracted much attention in the chemical community. Up until recently, several attempts have been made to synthesize ficellomycin, but no total synthesis has been reported to date (Ismail FM et al. 2009). However, our interest was to decipher how this natural compound is biosynthesized and which enzymes are involved in the biosynthesis of the unusual building blocks of ficellomycin, such as the pharmaceutically important 1-azabicyclo[3.1.0]hexane ring and the guanidine group. We are also interested in harnessing the biosynthetic machinery of ficellomycin to generate new structural derivatives of this antibiotic through the use of metabolic engineering. In this paper, we describe the isolation, sequencing, analysis, and confirmation of the ficellomycin biosynthetic gene cluster from S. ficellus. Bioinformatic analysis of this gene cluster, combining recent years’ research progress, provided a detailed molecular blueprint for ficellomycin biosynthesis. Furthermore, targeted gene disruptions and tandem analysis of their fermentation products indicate that a sulfate adenylyltransferase may be involved in the synthesis of the pharmaceutically important 1-azabicyclo[3.1.0]hexane ring system, and an amidinotransferase may catalyze the formation of the guanidine group, which is also a key functional group in some medicinally important natural compounds (Staschke KA et al. 1995). Additionally, a number of novel genes were found in this cluster, whose functions are still unknown.
The aim of the present research was to determine the overall architecture of ficellomycin biosynthetic gene cluster in S. ficellus and highlight the occurrence of poorly understood, but potentially novel modes of enzyme-catalyzed aziridine ring biosynthesis. The availability of the ficellomycin gene cluster sequence and the proposed biosynthetic pathway will be of great assistance to genetic engineering for the production of this unusual antibiotic, and will hopefully inspire attempts to apply this knowledge for combinatorial biosynthesis. It should be possible to genetically engineer the ficellomycin producer to improve its productivity, or to generate a series of new compounds with higher antibacterial, antifungal, or antitumor activities.
Materials and methods
Bacterial strains, plasmids, fosmids, and reagents
The ficellomycin producing strain S. ficellus (NRRL8067) was purchased from the Agricultural Research Service Culture Collection (USA). E. coli strains DH5ɑ (Life Technologies Inc., USA) and EPI300 (Epicenter, USA) were routinely used as hosts for E. coli plasmids, fosmids, and E. coli-Streptomyces shuttle vectors. P. oxalicum (CICC2667) was purchased from the China General Microbiological Culture Collection Center (Beijing, China). E. coli ET12567 containing the nontransmissible plasmid pUZ8002, fosmid pOJ446, plasmid pKC1139, and pSET152 with the ermE* promoter were preserved by our own lab (Unpublished data); the pGEM-T easy cloning vector was from Promega (Madison, Wisconsin, USA). The antibiotics of kanamycin, chloramphenicol, ampicillin, apramycin, nalidixic acid, and thiostrepton were from Sigma (Sigma-Aldrich, Saint Louis, Missouri, USA). The DNA purification kit, restriction endonucleases, T4 DNA ligase, and LA-Taq DNA polymerase were purchased from Takara (Otsu, Japan). Media and other chemical reagents were purchased from Sinopharm Chemical Reagent Beijing Company Limited (Beijing, China).
DNA isolation, manipulation, and sequencing
Isolation of chromosomal DNA from S. ficellus and agarose gel electrophoresis were performed according to standard methods (Hopwood DA et al. 1985). The QIAprep Spin Miniprep Kit (Qiagen, USA) was used to isolate plasmids and fosmids from E. coli. Primer synthesis and DNA sequencing were performed at the Chinese National Human Genome Center (Beijing, China). PCR was performed using LA-Taq DNA polymerase (Takara). All the primers used in this study were listed in the supplemental data (Table S1).
Genomic library construction and screening
A genomic library of S. ficellus was constructed in the fosmid vector pOJ446 according to the standard protocol. Purified DNA was digested with BamHI to give 30 to 50-kb fragments that were subsequently ligated into the BamHI site of pOJ446 and packaged into lambda phage by using a Gigapack III XL packaging extract kit (Stratagene, Cedar Creek, TX, USA), and the packaged cosmid pool was used to infect E. coli EPI300, according to the manufacturer’s instructions. A total of 6000 cosmid-containing clones were isolated and individually frozen at − 80 °C in microtiter plate wells.
The cosmid library was screened by PCR amplification of the conserved region in the NRPS adenylation domain, which was considered to be involved in ficellomycin assembly. PCR primers were designed to amplify internal fragments of NRPS genes, corresponding to the region between the conserved motifs A3 and A7, previously identified in NRPS adenylation domains (Ayuso-Sacido A and Genilloud O 2005). Primer design took into consideration Streptomyces codon bias (Wright F and Bibb MJ 1992). The primers used for the screening procedure were as follows: A3F (5′-GCSTACSYSATSTACACSTCSGG-3′) and A7R (5′-SASGTCVCCSGTSCGGTAS-3′). Boiled cells from each cosmid-containing clone were used as a source of template DNA for PCR amplification. The resulting positive clones were subsequently sequenced to confirm their identity and the flanking regions.
Sequence analysis
Nucleotide sequence assembly was performed using the Vector NTI software (Invitrogen, USA). The open reading frames (ORFs) within the sequence were predicted using the online software FramePlot 3.0 beta (http://watson.nih.go.jp/~jun/cgi-bin/frameplot-3.0b.pl/). The corresponding deduced proteins were compared with other known proteins in the databases using available BLAST methods (http://www.ncbi.nlm.nih.gov/blast/). Amino acid sequence alignments were performed using CLUSTALW (http://www.genome.jp/tools-bin/clustalw). The NRPS-PKS architecture was analyzed using tools available at the NRPS-PKS online website (http://nrps.igs.umaryland.edu/nrps/), and the prediction of the conserved substrate-binding pocket of the A domain was performed using the online program NRPS predictor (http://nrps.informatik.uni-tuebingen.de/Controller?cmd=SubmitJob) (Rausch C et al. 2005; Agüero-Chapin G et al. 2016).
Construction of gene inactivation mutants and genetic complementation
All mutant strains used in this study were generated by homologous recombination according to a published method (Liu W and Shen B 2000), with minor modifications as follows: The plasmid pKC1139 was used to construct each of the inactivation recombinants, and each of the targeted genes was replaced with a thiostrepton resistance gene. For conjugation, the recombinant plasmids were first introduced into E. coli ET12567/pUZ8002, which was subsequently cultivated in LB medium (10 g/L tryptone, 10 g/L NaCl, 5 g/L yeast extract) supplemented with kanamycin (30 μg/mL), chloramphenicol (25 μg/mL), and apramycin (50 μg/mL), until reaching an optical density at 600 nm of 0.3 to 0.4. The cells were harvested, washed twice with LB medium, and used as the conjugational donor cells. S. ficellus spores (105 to 109) were washed and resuspended in 2 × YT medium (5 g/L tryptone, 5 g/L yeast extract, 2.5 g/L NaCl), and incubated at 50 °C for 10 min to activate germination. After additional incubation at 37 °C for 2 to 5 h, the spores were pelleted, resuspended in LB, and used as the conjugational recipient cells. The donor (10 mL) and recipient (10 mL) cells were mixed and spread evenly onto MS medium (20 g/L D-mannitol, 20 g/L soybean meal, and 20 g/L agar powder) supplemented with 10 mM MgCl2. The plates were incubated at 30 °C for 16 h, after which each plate was covered with 1 mL of sterile water supplemented with nalidixic acid (50 μg/mL) and apramycin (50 μg/mL). The plates were continuously incubated at 30 °C for a further 4 to 5 days until exconjugants appeared. Double-crossover mutants were selected based on their apramycin sensitivity—thiostrepton resistance phenotype—and were further confirmed by diagnostic PCR using the corresponding primers (Table S1, Fig. S1-5). The complementary strains were respectively generated by introducing each of the specific pSET152-derived plasmids (include the ermE* promoter and a downstream complementary gene) or the corresponding cosmids into the mutants by either conjugation or polyethylene glycol (PEG)-mediated protoplast transformation (Kieser T et al. 2000). The vectors used for gene disruption or complementation and various disruptant trains were listed in Table S2.
Fermentation of wild-type S. ficellus and its derived mutant strains
Both the wild-type and mutant S. ficellus strains were grown on GYM (4 g/L glucose, 10 g/L malt extract, 4 g/L yeast extract, 2 g/L calcium carbonate, 20 g/L agar powder) plates at 28 °C for sporulation. Seed cultures were prepared by inoculating 250-mL shake-flasks containing 50 mL of seed medium (25 g/L glucose and 25 g/L Pharmamedia, pH 7.2) with spores, and incubating at 28 °C and 250 rpm for 72 h on a rotary shaker. For fermentation, 10 mL of the seed culture was used to inoculate 500-mL shake-flasks containing 100 mL of fermentation medium (10 g/L glucose monohydrate, 5 g/L starch, 5 g/L Pharmamedia, and 2 g/L calcium carbonate, pH 7.2), which were incubated for 72 to 96 h at 28 °C and 250 rpm on a rotatory shaker. The fermentation supernatants were harvested by centrifugation (Eppendorf 5415C centrifuge, 4 °C, 10 min, 10,000 g) at various time-points (48, 60, 72, 84, and 96 h) and assayed for their antifungal activity against P. oxalicum. Peak titers were generally obtained after 72 h of incubation.
Ficellomycin bioassay
Ficellomycin production and purification was preliminarily measured using a microbiological disc-plate assay procedure with P. oxalicum as the assay organism. Aliquots comprising 200 μl of the fermentation supernatant were added to stainless steel cylinders on PDB agar plates (20 g/L glucose monohydrate, 20 g/L potato extract, 20 g/L agar powder) that were pre-seeded with a 48-h-grown P. oxalicum seed culture at a concentration of 1% (vol/vol). The plates were incubated at 30 °C for 24 h, and the biological activity was estimated by measuring the sizes of the inhibition zones.
LC-MS/MS analysis of the fermentation products
After finishing the fermentation, the entire culture broths (approximately 100 mL) of the wild-type and mutant strains were filtered through approximately 50 g of diatomaceous earth (Jinan Bona Biological Technology Co., Ltd., China). The filter cake was discarded and one milliliter of the clear filtrate was freeze-dried to give the crude fermentation products. The products were resuspended in 0.5 mL of ultrapure water and passed through a 0.2-μm syringe filter before LC-MS/MS analysis on an LCMS-IT-TOF instrument (Shimadzu, Japan) equipped with a Spherisorb NH2 (3.0 μm, 150 × 2.1 mm) normal-phase column (Waters, USA). The conditions for LC-MS analysis were as follows: Solvent A was water containing 0.1% formic acid, and solvent B was acetonitrile containing 0.1% formic acid. The program comprised a linear gradient comprising 80–40% solvent B from 0 to 20 min, followed by 40% solvent B from 21 to 30 min. The flow rate was 0.2 mL/min and the injection volume was 10 μL.
Nucleotide sequence accession number
The sequence of the ficellomycin biosynthetic gene cluster has been deposited in GenBank under the accession number KY454693.
Results
Cloning, sequencing, and analysis of the ficellomycin biosynthetic gene cluster
To isolate the ficellomycin biosynthetic gene cluster from S. ficellus, a cosmid library of the organism’s chromosomal DNA was constructed. The genomic library contained 6000 colonies. Restriction enzyme analysis of 20 randomly selected cosmids confirmed that the average size of the inserts was about 35 to 45 kb. Based on the structure of ficellomycin, a nonribosomal peptide synthetase (NRPS) was proposed to be involved in ficellomycin biosynthesis using valine as the starting unit. NRPSs have highly conserved core motifs located in the adenylation (A) and peptidyl carrier protein (PCP) domains, which permit the use of degenerate PCR primers to specifically screen for cosmids containing NRPS genes. Consequently, a pair of degenerate oligonucleotide primers (A3F and A7R) targeting regions encoding the A motifs were designed, and approximately 6000 clones of the S. ficellus genomic library were screened. Twenty-five cosmids containing at least a portion of the NRPS-encoding gene were identified by the PCR-based screening method. Analysis of the sequences revealed that some of these cosmids overlapped each other and that some NRPS-encoding genes were identical. Consequently, five independent putative NRPS-encoding genes were identified and designated as nrps1, nrps2, nrps3, nrps4, and nrps5. To verify the involvement of these five NRPSs in ficellomycin biosynthesis, they were individually inactivated by replacing their respective adenylation domains in the genome of S. ficellus with the thiostrepton resistance gene (tsr) cassette. (Fig. S1) Fermentation and LC-MS analysis of five mutants revealed that only the nrps1 disruption mutant (S. ficellus M1) failed to produce ficellomycin, suggesting that NRPS1 is the synthetase responsible for ficellomycin biosynthesis in S. ficellus. To further confirm this finding, a gene complementation experiment was conducted. The cosmid pFIC1, which contains the entire NRPS1 encoding gene together with its upstream region, was transferred into S. ficellus M1 by PEG-mediated protoplast transformation to obtain the recovered strain S. ficellus MR1. Fermentation analysis indicated that the strain S. ficellus MR1 recovered its ability to synthesize ficellomycin (Fig. 2 and Fig. 3a). The gene disruption and complementation experiments therefore confirmed that the NRPS1 gene was necessary for ficellomycin biosynthesis and was involved in ficellomycin assembly.

LC-MS analysis of different fermentation products (extracted ion chromatograms of m/z [M+H]+ 313.1970 corresponding to ficellomycin). a Ficellomycin standard. b Fermentation products of the wild-type S. ficellus strain. c Fermentation products of the nrps1 distruption strain (S. ficellus M1). d Fermentation products of S. ficellus MR1

MS spectrum of ficellomycin [M+H]+ or [M+Na]+. a Ion at m/z = 313.1970. b MS/MS spectrum of the parent ion at m/z = 313.1970
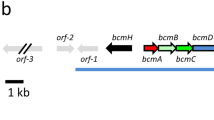
Probe-primers were designed according to the flanking regions of nrps1, and the genomic library of S. ficellus was screened by PCR. Sequence analysis revealed several overlapping fosmids that span a 55-kb region of DNA. To ensure full coverage of the entire ficellomycin gene cluster, further chromosome walking was conducted using a similar strategy. Finally, screening of the library yielded a contiguous DNA region spanning about 70 kb of the chromosome, which was mapped to the five overlapping fosmids pFIC1, pFIC2, pFIC3, pFIC4, and pFIC5 (Fig. 4a). DNA sequencing of the five cosmids yielded a 65.9-kb DNA sequence with 72.3% overall GC content. Bioinformatic analysis of this region revealed 55 open reading frames (ORFs) (Fig. 4b). The predicted functions of the ORFs were assigned by comparing their respective translated products with known proteins in public databases (Table 1). Twenty-six of these identified ORFs, from fic11 to fic36 (Fig. 4b and Table 1), were proposed to constitute the majority of the ficellomycin gene cluster based on the functions of their deduced products and the previously identified azinomycin B gene cluster.

Cloned DNA region on the chromosome of S. ficellus that harbors the ficellomycin biosynthetic gene cluster. a Overlapping fosmid inserts cloned from the S. ficellus genome that harbor the ficellomycin biosynthetic gene cluster. Solid black bars indicate regions whose sequence has been determined. B BamHI. b Genetic organization of the ficellomycin gene cluster. Proposed functions of individual ORFs are labeled and summarized in Table 1
Ficellomycin contains the pharmaceutically important 1-azabicyclo[3.1.0]hexane-2-ylidene core (AHYC) moiety, and genes that are most likely to direct AHYC biosynthesis in the azinomycin B pathway have been proposed (Zhao Q et al. 2008). Comparison of the two gene clusters revealed that thirteen ORFs in the ficellomycin cluster exhibited significant sequence identity with their respective counterparts in the azinomycin cluster (Table 1). Although the arrangement of these ORFs in the two gene clusters was different, similar strategies are likely used by ficellomycin and azinomycin producing strains to generate the AHYC.
Analysis of the ficellomycin nonribosomal peptide synthetase (NRPS)
Gene inactivation and complementation experiments have shown that NRPS1 is involved in ficellomycin biosynthesis. Sequence analysis revealed that it is a single-module NRPS with 1383 AA, which contains one condensation (C) domain, one adenylation (A) domain, one peptidyl carrier protein (PCP) domain, and one thioesterase (TE) domain. The predicted substrate of the A domain is glutamic acid. The organization of these domains in NRPS1 is shown in Fig. 5b, and the most immediately notable feature of NRPS1 is the lack of an initiation module, which selects the first amino acid (valine) and binds it covalently to the PCP1 domain. According to the results of library screening and sequencing, the initiation module of the ficellomycin assembly line is likely located at a different chromosomal locus. Recently, increasing numbers of biosynthetic pathways with genes located in different chromosomal loci were found, including the ansamitocin (Yu T-W et al. 2002), moenomycin (Ostash B et al. 2007), meilingmycin (He Y et al. 2010), and rhodochelin pathway (Bosello M et al. 2011). This phenomenon may be caused by chromosomal rearrangement in one Streptomyces species after the horizontal transfer of the original clusters from another.

Proposed biosynthetic pathway of ficellomycin. a Pathway for the crucial building block 2-[4-guanidyl-1-azabicyclo[3.1.0]hexan-2-yl] glycine. b A possible mode for NRPS-directed skeleton assembly of ficellomycin. The question mark (?) indicates the unidentified initiation module
The presence of a TE domain indicates that NRPS1 is the termination module, which specifically selects the last amino acid, activates the carboxylate with ATP to generate aminoacyl-AMP, and finally installs the aminoacyl group onto the thiolate of the adjacent PCP domain. For ficellomycin, the last amino acid should be 2-[4-guanidyl-1-azabicyclo[3.1.0]hexan-2-yl] glycine (2-GAHG, a nonproteinogenic amino acid). By contrast, the predicted substrate of the A domain of NRPS1 is glutamic acid, which has a very different structure. According to the NRPS1 substrate prediction, two hypotheses were proposed. Firstly, the nonproteinogenic amino acid 2-GAHG may originate from glutamic acid. This speculation is in agreement with previous isotope-labeled feeding studies of azinomycin B, the most closely related natural product to ficellomycin. According to these feeding experiments, the aziridino [1, 2a] pyrrolidinyl amino acid building block of azinomycin B originates from glutamic acid (Liu C et al. 2006). Consequently, the rather similar structure in ficellomycin might also be derived from the same substrate. Secondly, it may be that glutamic acid rather than 2-GAHG is specifically activated and covalently bound to the PCP domain of NRPS1. Only then would the installed or assembled glutamic acid moiety be further modified to form the unusual building block 2-GAHG (Fig. 5a). For verification of this assumption, the fermentation broth of the nrps1 disruption mutant (S. ficellus M1) was analyzed by LC-MS. The results indicated that the double-crossover mutation completely abolished the production of ficellomycin, but no free 2-GAHG intermediate or its corresponding derivatives were detected. These results partially supported our hypothesis.
Biosynthesis of the nonproteinogenic amino acid moiety in ficellomycin
Our results indicate that most of the proposed genes directing AHYC biosynthesis in the azinomycin B pathway appear to be present within the ficellomycin cluster (Table 1). For the AHYC moiety in azinomycin B, most steps of the biosynthetic pathway have been proposed (Zhao Q et al. 2008). Based on the similarity between the clusters and the molecules, we propose that the biosynthetic pathway of ficellomycin follows a similar pattern. As shown in Fig. 5a, a detailed 2-GAHG biosynthetic pathway was proposed. Fic29 exhibits high sequence similarity to LysX that catalyzes the N-acetylation of 2-aminoadipate in the bacterial lysine (Lys) biosynthetic pathway, suggesting that the first step toward AHYC starts with Glu via protection of the α-amino group. Fic20 and Fic21, which respectively share high sequence similarity to various N-acetylglutamate kinases and phosphate reductases in the ornithine (Orn) biosynthetic pathway, may activate the γ-carboxyl group of N-acetylglutamic acid by phosphorylation, and then reduce it to form N-acetylglutamyl-γ-semialdehyde. Fic23 and Fic24, which respectively share high homology with the N- and C-terminal subunits of transketolases, are responsible for two-carbon unit extension on the C5 position. Subsequently, Fic16, which resembles ornithine-oxo-acid transaminase—a member of the pyridoxal-phosphate-dependent aminotransferase family—is likely responsible for the transamination from ornithine onto the 6-keto group, fulfilling all atomic requirements for the formation of the azabicyclic skeleton to yield AHYC (Fig. 5a). Fic32 exhibits high sequence similarity to a family of acyl-CoA dehydrogenases, suggesting that an ɑ-β dehydrogenation reaction is required to initiate the closing of the first nitrogen-containing five-membered ring.
The genes fic17, fic18, fic19, and fic28 encode proteins resembling sulfate adenylyltransferase subunit 1, subunit 2, adenylsulfate kinase, and sulfotransferase, respectively. According to Bicker and Fischer’s research (Bicker U and Fischer W 1974), they are likely to be involved in aziridine formation. As shown in Fig. 6, in the presence of ATP and sulfate, a sulfate adenylyltransferase (Fic17, FiC18) catalyzes the formation of adenosine 5′-phosphosulfate (APS), and an adenylsulfate kinase (Fic19) subsequently generates 3′-phophoadenosine-5′-phosphosulfate (PAPS), which is a sulfate donor for a variety of acceptor groups. PAPS can then be enzymatically linked by a sulfotransferase (Fic28) to the hydroxyl group of compound 5. The sulfated hydroxyl group of compound 6 would then become a good leaving group, and an intramolecular nucleophilic attack by the vicinal secondary amine could form the aziridine.

The proposed aziridine ring formation mechanism
To validate the role of the sulfate adenylyltransferase in ficellomycin biosynthesis, we inactivated the crucial fic17 gene by replacing it with a thiostrepton resistance cassette to produce the mutant strain ΔSL17-1 (Fig. S5). However, the ΔSL17-1 strain still produced ficellomycin, even if the productivity was drastically decreased compared to the wild type (Figs. 7 and 8). To further confirm the function of the sulfate adenylyltransferase in ficellomycin biosynthesis, the ability of the wild-type fic17 gene to complement the disrupted fic17 gene was tested in the mutant strain ΔSL17-1. The pSET152 backbone was used to construct the expression vector pSF17, in which the expression of fic17 is under the control of the constitutive ermE* promoter. Both the pSF17 construct and the pSET152 vector as a control were separately introduced into the S. ficellus ΔSL17-1 mutant strain, and culture supernatants from each transformant were assayed for ficellomycin production. As expected, pSF17 restored ficellomycin production of S. ficellus ΔSL17-1 to the wild-type level, whereas a significant reduction of ficellomycin production was still detected in the control in which pSET152 was introduced into S. ficellus ΔSL17-1, when they were cultured under identical conditions (Figs. 6 and 7). The gene disruption and complementation experiments proved the involvement of sulfate adenylyltransferase in ficellomycin biosynthesis, but also suggested that some other sulfate adenylyltransferase gene or mechanism likely exist which enables low levels of aziridine biosynthesis.

Determination of ficellomycin production by assaying the antifungal activity against P. oxalicum. A1–A3 the wild-type strain of S. ficellus. B1–B3 the fic17 disruption strain S. ficellus ΔSL17-1. C1–C3 the S. ficellus ΔSL17-1 strain transformed with the complementation plasmid pSF17. D1–D3 the S. ficellus ΔSL17-1 strain transformed with the plasmid pSET152

LC-MS analysis of ficellomycin production by different strains of S. ficellus (extracted ion chromatograms of m/z [M+H]+ 313.1970 corresponding to ficellomycin). a The wild-type strain of S. ficellus. b The fic17 disruption strain S. ficellus ΔSL17-1. c S. ficellus ΔSL17-1 transformed with the complementation plasmid pSF17. d S. ficellus ΔSL17-1 transformed with plasmid pSET152
In addition, three unique genes, fic33, fic25, and fic36, were identified in the ficellomycin gene cluster. Gene disruption and complementation experiments validated their necessity for ficellomycin biosynthesis (Fig. 9). Fic33, which exhibits high sequence similarity to dehydrogenases, was proposed to be responsible for the hydroxylation of compound 2 to form compound 3. Fic25 and Fic36 respectively showed 75 and 68% identity to glutamine-scyllo-inositol transaminase and scyllo-inosamine-4-phosphate amidinotransferase. Given that an aminotransferase and an amidinotransferase have been reported to catalyze the transamination and transamidination that yields guanidine in the streptomycin biosynthesis pathway (Ohnuki T et al. 1985), we propose that Fic25 and Fic36 may be responsible for the transamination of compound 3 and transamidination of compound 4 to form the guanidine group of ficellomycin as shown in Fig. 5a. However, the timing of angucycline formation, which may occur prior to or after the formation of the aziridine ring, is also uncertain.

Bioassay of ficellomycin production by different strains of S. ficellus. A1 and A2 fermentation products of the nrps 1 disruption strain S. ficellus M1, A3 fermentation products of the wild-type strain S. ficellus, A4 fermentation products of the nrps 1 recovered strain, B1 and B2 fic25 disruption strain S. ficellus M2, B3 wild-type S. ficellus, B4 fic25 recovered strain, C1 and C2 fic36 disruption strain S. ficellus M3, C3 wild-type S. ficellus, C4 fic36 recovered strain, D1 and D2 fic33 disruption strain S. ficellus M5, D3 wild-type S. ficellus, D4 fic33 recovered strain
Genes encoding self-resistance and other functions
Fic11 resembles a family of proteins annotated as inner membrane ATP-binding-cassette (ABC) transporters. In bacteria, they transport molecules through the permeability barriers of the cell membrane, and their cargo molecules are extremely diverse, including small to medium-sized molecules such as sugars, amino acids, and lipids, as well as large, bulky compounds including peptides, proteins, and antibiotics (Braibant M et al. 2000). Fic12 resembles an arginine/ornithine binding protein, which is an additional protein partner required by ABC transporters, and is responsible for binding the substrates and delivering them to the transmembrane domains (TMDs) (Braibant M et al. 2000; Lewinson O and Livnat-Levanon N 2017). It is plausible that Fic11 and Fic12 may function as self-resistance proteins that actively export the synthesized ficellomycin out of the cells. Fic34 exhibited high sequence similarity to the LuxR family of DNA-binding proteins, such as RifZ in rifamycin biosynthesis, that are widespread in secondary metabolism and annotated as pathway-specific transcriptional regulators (Li C et al. 2017). Regulation of ficellomycin production and possibly the expression of self-resistance determinants are believed to involve Fic34.
Finally, two genes within the ficellomycin gene cluster encode proteins whose functions cannot be assigned to the proposed ficellomycin biosynthetic pathway. Fic15 resembles a carboxylase from Streptomyces sp. TAA204 and Fic22 resemble the lysine biosynthesis protein LysW. In addition, some genes, such as fic26 and fic27, could not be predicted based on sequence analysis alone. Interestingly, they both have corresponding homologs in the azinomycin cluster, respectively showing intermediate level of sequence identity to AziU3 and AziU2. However, their involvement in ficellomycin biosynthesis remains to be determined.
The probable boundaries of the ficellomycin gene cluster were assigned based on the deduced functions of the products of the genes on the cluster termini and flanking genes, as well as by comparison with the azinomycin gene cluster. There are no ORFs in the sequenced region upstream of fic11 with predicted functions necessary for ficellomycin formation. Likewise, the region located downstream of fic36 has no counterparts in the azinomycin biosynthetic gene cluster, or genes with a possible function in ficellomycin biosynthesis. Therefore, we propose that the bona fide boundaries of the ficellomycin gene cluster are defined by orf11 and orf36. Whether any of the genes in the flanking regions that encode products of unknown function are involved in the regulation of antibiotic production or resistance remains to be determined.
Discussion
Aziridine alkaloids are a group of rare natural products with many unique properties. They have mainly been isolated from either microorganisms or plants, but have also been detected in some marine species (Ismail FM et al. 2009). A wide spectrum of pharmacological activities is associated with this family of compounds. Reported activities for purified aziridine alkaloids include strong antitumor, antibacterial, and general antimicrobial activities (Ismail FM et al. 2009). Due to the impressive array of diverse biological activities exhibited by these small-ring-containing compounds, as well as their potential applications as therapeutic agents or mechanistic probes for the study of enzymatic catalysis, a thorough understanding of their biosynthesis mechanism is a crucial first step in maximizing their potential as probes or as specific tools for the rational design of drugs with optimal in vivo specificity. Nevertheless, our understanding of the biosynthetic mechanisms responsible for aziridine formation remains elusive. In fact, only a handful of aziridine-containing natural products are known, and the biosynthetic gene clusters for only three of these have been published: the mitomycin cluster from Streptomyces lavendulae (Mao Y et al. 1999), the azinomycin cluster from Streptomyces sahachiroi (Zhao Q et al. 2008), and the azicemicin cluster from Kibdelosporangium sp. MJ126-NF4 (Ogasawara Y, and Liu HW 2009). The functions of most genes related to mitomycin biosynthesis, except for the enzymes involved in the self-resistance mechanism (Mcr, Mrd, Mct) and the final O-methylation step (MitM), were not biochemically characterized (Mao Y et al. 1999). Notably, this lack of biochemical data includes the genes for aziridine ring formation (Mao Y et al. 1999). For azinomycin biosynthesis, a reaction sequence for the construction of the azabicyclic moiety starting from glutamic acid was proposed. A hydroxylation catalyzed by the P450 enzyme AziC9 and a deacylation catalyzed by AziC10 are proposed to be the final steps leading to AHYC, but how the aziridine ring is constructed remains uncertain (Zhao Q et al. 2008). In azicemicin, feeding experiments using deuterium-labeled amino acids revealed that aspartic acid is the precursor of the aziridine moiety (Ogasawara Y, and Liu HW 2009). Furthermore, a possible pathway for aziridine ring formation in azicemicin was proposed on the basis of the assigned functions of genes in its biosynthetic gene cluster. However, the identity of the enzyme that catalyzes the last cyclization step was not revealed by complementation studies (Ogasawara Y, and Liu HW 2009). Therefore, the details of the biochemically very interesting aziridine ring formation reaction remain obscure. Nevertheless, the identification of the ficellomycin gene cluster further enriches the gene repository with genes related to aziridine biosynthesis and provides some new clues for the elucidation of the detailed catalytic mechanism.
Comparatively speaking, ficellomycin and azinomycin are more closely structurally related to each other than to the other aziridine-containing compounds, and they both contain the azabicyclic moiety (AHYC) (Fig. 1). The comparison between the ficellomycin gene cluster and the azinomycin biosynthesis gene clusters highlighted the conserved similarities between the two clusters in terms of gene presence and function. Except for some ORFs whose functions have been tentatively assigned as related to the assembly of the azabicyclic backbone, there are still some functionally unassigned homologous genes, which also exist in both gene clusters. These include sulfate adenylyltransferase, adenylsulfate kinase, sulfotransferase, and some functionally unknown protein-encoding genes (fic17, fic18, fic19, fic26, fic27, and fic28). We hope that these sequences will provide new clues for deeper research into the detailed mechanism of aziridine ring biosynthesis. Interestingly, even though the exact mechanism remains elusive to this day, Bicker and Fischer reported the first enzymatic aziridine synthesis from beta amino-alcohols as early as 1974 (Bicker U and Fischer W 1974). In their work, beta amino-alcohols were incubated with homogenized rat liver extract, and in the presence of both ATP and sulfate, they were converted into the corresponding aziridine compounds. The authors proposed that sulfate adenylyltransferase, adenylsulfate kinase, and sulfotransferase were involved in the aziridine ring formation (Bicker U and Fischer W 1974). Even more interestingly, putative sulfate adenylyltransferase genes are present in all the three known aziridine-biosynthetic clusters, i.e., those of mitomycin, azinomycin, and ficellomycin (mmcU/mmcV, aziH1/aziH2, and fic17/18, respectively) (Thibodeaux CJ et al. 2012). The azinomycin and ficellomycin clusters both also encode putative APS kinases (AziH3 and Fic19, respectively) and sulfotransferases (AziC1 and Fic28, respectively). It is therefore reasonable to speculate that the three enzymes that are also part of the sulfur assimilation pathway are involved in aziridine biosynthesis (Fig. 6). To prove our speculation, fic17 disruption and complementation experiments were conducted. The results on one hand proved the involvement of sulfate adenylyltransferase in ficellomycin biosynthesis. However, they suggested that on the other hand, the gene fic17 was not necessary for ficellomycin biosynthesis, since the disruption mutant ΔSL17-1 did not completely lose the capacity for ficellomycin production. There appears to be two possible reasons for this. First, another sulfate adenylyltransferase gene may exist outside the ficellomycin gene cluster, which can partially compensate for the role of fic17 in ficellomycin synthesis. Secondly, another, minor mechanism for aziridine ring construction may exist, and in the absence of adenylyltransferase, an alternative biosynthetic mechanism may be employed. For example, the sequenced azicemicin cluster lacks all three of these genes, and its producer strain Kibdelosporangium sp. MJ 126-NF4 likely employs a distinct strategy to generate the aziridine unit (Ogasawara Y, and Liu HW 2009). To finally elucidate the detailed aziridine-formation mechanism, additional experiments, such as precursor-incorporation studies, isotope tracer experiments, genetic analysis, enzymological studies, etc., will be needed, while our understanding of the biosynthetic mechanisms responsible for aziridine ring formation is steadily improving.
This work represents perhaps the most comprehensive characterization of the gene cluster responsible for ficellomycin biosynthesis in S. ficellus since the identification of ficellomycin in 1977. Our results are somewhat consistent with Dr. Wen Liu’s research on AHYC biosynthesis in the azinomycin B pathway (Zhao Q et al. 2008) and provide clues for a detailed aziridine biosynthesis mechanism. Many, if not all of the genes responsible for the formation of the nonproteinogenic amino acid 2-GAHG are located within the ficellomycin gene cluster. These genes are of special interest because their functions are poorly understood, and some of them might be useful as probes for identifying related natural-product biosynthetic genes from other microorganisms and plants. The cloned genes presented here therefore provide a key starting point for future molecular-genetic and biochemical studies on ficellomycin biosynthesis and general natural-product assembly. One important advantage of having this information will likely be demonstrated through the genetic manipulation of the candidate regulator genes, which in turn will hopefully yield an evident increase in ficellomycin production. In addition, expression and disruption of selected genes should be useful for engineering the biosynthesis of clinically valuable ficellomycin analogs, as well as more complex hybrid natural-product systems.
References
Agüero-Chapin G, Pérez-Machado G, Sánchez-Rodríguez A, Santos MM, Antunes A (2016) Alignment-free methods for the detection and specificity prediction of adenylation domains. Methods Mol Biol 1401:253–272
Argoudelis AD, Reusser F, Whaley HA, Baczynskyj L, Mizsak SA, Wnuk RJ (1976) Antibiotics produced by Streptomyces ficellus. I Ficellomycin J Antibiot 10:1001–1006
Ayuso-Sacido A, Genilloud O (2005) New PCR primers for the screening of NRPS and PKS-I systems in actinomycetes: detection and distribution of these biosynthetic gene sequences in major taxonomic groups. Microb Ecol 1:10–24
Bicker U, Fischer W (1974) Enzymatic aziridine synthesis from beta amino-alcohols—a new example of endogenous carcinogen formation. Nature 455:344–345
Bosello M, Robbel L, Linne U, Xie X, Marahiel MA (2011) Biosynthesis of the siderophore rhodochelin requires the coordinated expression of three independent gene clusters in Rhodococcus jostii RHA1. J Am Chem Soc 12:4587–4595
Braibant M, Gilot P, Content J (2000) The ATP binding cassette (ABC) transport systems of Mycobacterium tuberculosis. FEMS Microbiol Rev 4:449–467
Brulikova L, Hlavac J, Hradil P (2012) DNA interstrand cross-linking agents and their chemotherapeutic potential. Curr Med Chem 3:364–385
Foulke-Abel J, Agbo H, Zhang H, Mori S, Watanabe CM (2011) Mode of action and biosynthesis of the azabicycle-containing natural products azinomycin and ficellomycin. Nat Prod Rep 4:693–704
He Y, Sun Y, Liu T, Zhou X, Bai L, Deng Z (2010) Cloning of separate meilingmycin biosynthesis gene clusters by use of acyltransferase-ketoreductase didomain PCR amplification. Appl Environ Microbiol 10:3283–3292
Hopwood DA, Bibb MJ, Chater KF, Kieser T, Bruton CJ, Kieser HM, Lydiate DJ, Smith CP, Schrempf H (1985) Genetic manipulation of Streptomyces: a laboratory manual. John Innes Foundation, Norwich
Ismail FM, Levitsky DO, Dembitsky VM (2009) Aziridine alkaloids as potential therapeutic agents. Eur J Med Chem 9:3373–3387
Kieser T, Bibb MJ, Butter M, Chater KF, Hopwood DA (2000) Practical Streptomyces genetics. John Innes Foundation, Norwich
Kuo MS, Yurek DA, Mizsak SA (1989) Structure elucidation of ficellomycin. J Antibiot 3:357–360
Lewinson O, Livnat-Levanon N (2017) Mechanism of action of ABC importers: conservation, divergence, and physiological adaptations. J Mol Biol 5:606–619
Li C, Liu X, Lei C, Yan H, Shao Z, Wang Y, Zhao G, Wang J, Ding X (2017) RifZ (AMED_0655) is a pathway-specific regulator for rifamycin biosynthesis in Amycolatopsis mediterranei. Appl Environ Microbiol 8:03201–03216
Liu C, Kelly GT, Watanabe CM (2006) In vitro biosynthesis of the antitumor agent azinomycin B. Org Lett 6:1065–1068
Liu W, Shen B (2000) Genes for production of the enediyne antitumor antibiotic C-1027 in Streptomyces globisporus are clustered with the cagA gene that encodes the C-1027 apoprotein. Antimicrob Agents Chemother 2:382–392
Mao Y, Varoglu M, Sherman DH (1999) Molecular characterization and analysis of the biosynthetic gene cluster for the antitumor antibiotic mitomycin C from Streptomyces lavendulae NRRL 2564. Chem Biol 4:251–263
Ogasawara Y, Liu HW (2009) Biosynthetic studies of aziridine formation in azicemicins. J Am Chem Soc 50:18066–18068
Ohnuki T, Imanaka T, Aiba S (1985) Isolation of streptomycin-nonproducing mutants deficient in biosynthesis of the streptidine moiety or linkage between streptidine 6-phosphate and dihydrostreptose. Antimicrob Agents Chemother 3:367–374
Ostash B, Saghatelian A, Walker S (2007) A streamlined metabolic pathway for the biosynthesis of moenomycin A. Chem Biol 14:257–267
Rausch C, Weber T, Kohlbacher O, Wohlleben W, Huson DH (2005) Specificity prediction of adenylation domains in nonribosomal peptide synthetases (NRPS) using transductive support vector machines (TSVMs). Nucleic Acids Res 18:5799–5808
Reusser F (1977) Ficellomycin and feldamycin; inhibitors of bacterial semiconservative DNA replication. Biochemistry 15:3406–3412
Staschke KA, Colacino JM, Baxter AJ, Air GM, Bansal A, Hornback WJ, Munroe JE, Laver WG (1995) Molecular basis for the resistance of influenza viruses to 4-guanidino-Neu5Ac2en. Virology 2:642–646
Thibodeaux CJ, Chang WC, Liu HW (2012) Enzymatic chemistry of cyclopropane, epoxide, and aziridine biosynthesis. Chem Rev 3:1681–1709
Wright F, Bibb MJ (1992) Codon usage in the G+C-rich Streptomyces genome. Gene 1:55–65
Yu T-W, Bai L, Clade D, Hoffmann D, Toelzer S, Trinh KQ, Xu J, Moss SJ, Leistner E, Floss HG (2002) The biosynthetic gene cluster of the maytansinoid antitumor agent ansamitocin from Actinosynnema pretiosum. Proc Natl Acad Sci USA 12:7968–7973
Zhao Q, He Q, Ding W, Tang M, Kang Q, Yu Y, Deng W, Zhang Q, Fang J, Tang G, Liu W (2008) Characterization of the azinomycin B biosynthetic gene cluster revealing a different iterative type I polyketide synthase for naphthoate biosynthesis. Chem Biol 7:693–705
Funding
This research was financially supported by the National Natural Science Foundation of China (Grant No. 81373309) and the National High-Tech Research and Development Plan of China (Grant No. 2013AA102803, Task No. 2013AA102803C).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary materials
ESM 1
(PDF 409 kb)
Rights and permissions
About this article

Cite this article
Liu, Y., Li, M., Mu, H. et al. Identification and characterization of the ficellomycin biosynthesis gene cluster from Streptomyces ficellus . Appl Microbiol Biotechnol 101, 7589–7602 (2017). https://doi.org/10.1007/s00253-017-8465-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-017-8465-4