
Abstract
The human intestine hosts various complex microbial communities that are closely associated with multiple health and disease processes. Determining the composition and function of these microbial communities is critical to unveil disease mechanisms and promote human health. Recently, meta-omic strategies have been developed that use high-throughput techniques to provide a wealth of information, thus accelerating the study of gut microbes. Metaproteomics is a newly emerged analytical approach that aims to identify proteins on a large scale in complex environmental microbial communities (e.g., the gut microbiota). This review introduces the recent analytical strategies and applications of metaproteomics, with a focus on advances in gut microbiota research, including a discussion of the limitations and challenges of these approaches.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Trillions of microbes, such as bacteria, fungi, and viruses, inhabit the human gut and could be considered as a virtual organ (Baothman et al. 2016; Ley et al. 2006; O’Hara and Shanahan 2006). In particular, diverse bacterial species represent the largest community in the gut microbiota. However, people recently realized and appreciated that gut microbes live symbiotically with us. The diverse and complex microbial communities play an essential role in human digestive function. For example, indigestible nutrients are digested or degraded by bacteria in the gut. In addition, depending on various factors (such as diet, mood, and age), the composition of gut microbiota is constantly changing, which might affect human health (Kasparovska et al. 2016; Mangiola et al. 2016; Odamaki et al. 2016). Nutrition is a very vital factor to affect the gut microbiota and human healthy (Graf et al. 2015; Sonnenburg and Backhed 2016). Malnutrition could cause disease by altering the gut microbial composition and delaying their normal development (Kane et al. 2015). Plant-based diets have been recommended to reduce the risk of colon cancer, type 2 diabetes, and heart diseases (Martin et al. 2013; Westergaard et al. 2014; Satija et al. 2016). The proportion of Bacteroidetes decreased and Firmicutes increased in heavy meat eaters compared to vegetarians (Zhang and Yang 2016), and an animal-based diet could result in significantly lower levels of short-chain fatty acids (SCFAs) which have anti-inflammatory effect (David et al. 2014; Iraporda et al. 2015). Recent finding indicated that dysbiosis of intestinal flora might be associated with, or cause, inflammation (Magrone and Jirillo 2013) and metabolic disease (Karlsson et al. 2013). For example, several beneficial members of the gut microbiota, such as Faecalibacterium prausnitzii, were significantly reduced in Crohn’s disease compared with healthy controls (Erickson et al. 2012). To further characterize the structure and function of the gut microbiota, the Human Microbiome Project (HMP) and Metagenomics of the Human Intestinal Tract (MetaHIT) projects were initiated (Human Microbiome Jumpstart Reference Strains et al. 2010; Human Microbiome Project 2012). Certain meta-omics approaches have been used to analyze large-scale gene or protein expressions and metabolite compositions (Gao et al. 2016; Levi Mortera et al. 2016; Xie et al. 2016; Zhao et al. 2016).
Metagenomic analysis has been used widely in gut microbial studies, employing different experimental methods. Generally, 16S ribosomal RNA subunit genes are highly conserved in bacterial species, providing useful information for taxonomic characterization (Barker et al. 2013; Costello et al. 2009; David et al. 2014). Although genome shotgun sequencing increases the available information for taxonomical characterization of the gut microbiota, it provides little information concerning the detailed and invaluable function of the gut microbiota. Moreover, these genomic-based approaches only predict potential functions, and do not reveal the extent and locations of protein expression. In recent years, metatranscriptomic analysis based on RNA sequencing has provided information of the functional characteristics and dynamic range of microbial communities (Wang et al. 2009). Typically, the metatranscriptome is studied via isolation of total RNA, construction of cDNA libraries and identification of the sequences. However, messenger RNA (mRNA) easily degraded in prokaryotes is unstable (Redon et al. 2005). Furthermore, mRNA levels do not represent the biological function significantly. The hypothesis that signaling molecules produced by the gut microbiota could lead to a variety of diseases is yet to be confirmed (Zhu et al. 2016). For example, short-chain fatty acids, such as acetate and propionate, produced by gut bacteria could be recognized as signal transduction molecules to regulate host energy metabolism (Donohoe et al. 2011; Zhu et al. 2016). Metabolomic analysis aims to characterize metabolite variations or the metabotype under a variety of conditions using nuclear magnetic resonance (NMR) spectroscopy or mass spectrometry (MS) approaches. Metabolomics provides an image of the entire metabolite composition of the microbial communities. Nevertheless, the details of the molecular mechanisms of gut microbiota remain ambiguous, because ultimately, proteins mediate the gut microbial function. Genes are transcribed into RNA, which are then translated into protein. Meanwhile, proteins can catalyze the synthesis of certain metabolites that regulate the physiological process of an organism or mediate its biological function directly. Therefore, metagenomics, metatranscriptomics, metaproteomics, and metabolomics are closely linked, and metaproteomics might play a critical part (Fig. 1). This paper reviews recent researches on metaproteomics of gut microbiota and highlights their methodological strategies and medical or biological applications. We also stress the importance of metaproteomics in understanding the potential biological function of gut microbiota and pay attention to their limitations. We conclude that metaproteomics is a promising tool for studying gut microbiota, and with the combination of other omics may get much greater gains in the gut microbial research.

Overview of the relationships among various meta-omics
Advantages of metaproteomics in gut microbiota research
Metaproteomics has been defined as the examination of a complete protein composition of environmental microbiota at a specific time (Wilmes and Bond 2004). Moreover, metaproteomic analysis provides valid information for environmental microorganisms, such as microbial activity, signal transduction, and metabolic pathways (Verberkmoes et al. 2009). In addition, it can also be used to investigate environment-microbe interactions (Mayne et al. 2016). Microbial metaproteomics analysis has become an efficient tool to investigate various processes in different environments, such as soil (Wang et al. 2011), marine (Lopez et al. 2002), and feces (Tanca et al. 2015). Adult feces contain about 75% water and 25% solid material (Rose et al. 2015). The major organic material of the solid matter is gut microbial cell (Stephen and Cummings 1980). In this respect, Tang et al. (2014) used a metaproteomic approach to explore the composition of microorganisms in chicken feces and revealed the adaptation process of the chicken gut microbiota.
Metaproteomic analyses of gut microbiota provide a unique perspective to understand microbial life processes. It could reveal the whole biological processes from the qualitative analysis of the activity of the microbes to the quantitative analysis of protein expressions and dynamic changes. Some proteins could serve as biomarkers for disease diagnosis, prognosis, and therapy. Protein expression in the gut microbiota might be altered in patients; therefore, metaproteomic analysis of gut microbiota might reveal the molecular mechanisms of diseases (Xu et al. 2016). It also could provide key information on complex protein networks and signal transduction pathways in certain diseases (Carrasco-Navarro et al. 2016). Therefore, to satisfy the demands of discovering the gut microbiota related biomarkers and addressing the relationship of functional redundancy and diversity of gut microbiota, it is necessary to use metaproteomics to gain the quantitative information of all proteins in the gut microbiota.
Metaproteomics methodology for gut microbiota research
Mass spectrometry is more suitable for analyzing complex samples and revealing their biological function (Schneider et al. 2012; Verberkmoes et al. 2009; Aires and Butel 2011). The availability of MS technology (especially LC-MS/MS) offers an opportunity to gain unprecedented insight into environmental microbes and makes the identification of proteins in gut microbial communities possible (Leary et al. 2012; Del Chierico et al. 2014). An optimized workflow could maximize protein identification, allowing us to analyze the metaproteome of gut microbiota successfully. Shotgun metaproteomic analysis has been applied widely to measure the total proteins in the gut microbiota (Tanca et al. 2015; Verberkmoes et al. 2009). As shown in Fig. 2, the critical steps for an efficient metaproteomic analysis include sample collection, protein extraction, protein isolation, MS analysis, and database searching.

Metaproteomic work flow used in the characterization of gut microbiota proteins
Sample collection
First, researchers need to collect fecal samples or samples from different compartments of the gut (e.g., the ileum, cecum, and the small and large intestine), where the microbial composition and diversity can be different. Collecting abundant high-quality samples is required for reliable results. Collecting samples from the gut poses challenges and limitations for analyzing the metaproteome of gut microbiota. For convenient sampling and saving costs, most investigators choose to isolate the gut microbiota from feces of human, which contain approximately 1011 bacteria per gram (Tlaskalová-Hogenová et al. 2011).
Centrifugation and filtering are two strategies to separate gut bacteria from other material in fecal samples. Apajalahti et al. (1998) and Kolmeder et al. (2012) used differential centrifugation to purify microorganisms. Briefly, feces are suspended by vortexing in sodium phosphate buffer containing Tween-80 or glass beads. The supernatants are collected, and insoluble particles are removed by low-speed centrifugation. This process is repeated several times to gather the cells. Microorganisms in the supernatants are then collected by high-speed centrifugation. The pellet is washed to remove the soluble compounds, such as viscous polysaccharides and mucins. Tanca et al. (2015) analyzed the data for the human gut microbiota proteome from feces with or without differential centrifugation and found that the former method is superior to the later for harvesting bacteria and proteins.
Recently, indirect double filtering, a non-centrifugation method, was developed to harvest gut bacteria (Xiong et al. 2015). Typically, bacterial cells and human cells or undigested food particles can be separated based on their different sizes. Bacteria are significantly smaller than human cells; therefore, large particles and human cells can be removed using a 20-μm filter, while microbial cells can be collected through a 0.22-μm filter. Using this approach, a higher number of microorganisms, especially low-abundance microbes, are enriched from complex fecal samples.
Protein extraction
Different chemical and physical methods may be used to prepare gut microbial proteins. SDS or Triton X-100 and guanidine are used as general detergents and denaturants to extract proteins, and the steps of protein extraction are well established. Commonly, a cell pellet is resuspended in 6 M urea, 2% SDS or 1% Triton X-100 solution to lyse cells for several minutes, and the proteins are recovered by high-speed centrifugation (Chen et al. 2016; Dhabaria et al. 2015; Peach et al. 2015). The similarities of the basic structures of urea and peptide bonds suggest that urea could reduce the efficiency of trypsin. In addition, detergent could interfere with protein quantification using BCA or Bradford assays. Moreover, the use of detergents has a detrimental effect on MS. Thus, chemical reagents that have negative influences on protein digestion or quantification should be diluted or removed. Commonly, a commercial C18 spin column is used to deplete undesirable substances of the sample (e.g., salt and detergents) before being analyzed by mass spectroscopy. To avoid the effects of chemical reagents, sonication is used frequently (Prauchner et al. 2013). Normally, microbial cells are sonicated three times, and the protein contents are collected. In practice, the efficiency of extraction varies by species because some proteins bound to DNA could be removed by centrifugation.
Protein isolation
Protein isolation is a critical step in metaproteomic studies. To improve the coverage of the proteome, proteins should be fractionated before MS analysis. Several protein separation strategies may be used to reduce the complexity of samples. Methods of protein fractionation have been developed to study the metaproteome of the gut microbiota, such as separation according to their physical properties, including molecular weight, charge, or hydrophobicity.
Gel-based methods, including one-dimensional (1D) or two-dimensional (2D) electrophoresis, can separate proteins based on their size or isoelectric point (Brunelle and Green 2014; O’Farrell 1975). In a 1D electrophoretic step, sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) is used commonly to generate protein fingerprints of gut microbiota. For example, Kolmeder et al. (2012) used a 1D-MS method to study human intestinal microbiota. In that study, proteins were separated into three fractions (proteins larger than 80 kDa, smaller than 35 kDa, and between 35 and 80 kDa) using SDS-PAGE according to their molecular weights. For downstream MS analysis, proteins bands in the gel are visualized via Silver or Coomassie blue staining, excised, and digested into peptides. Using this method, false positive results of MS may be avoided. Meanwhile, highly abundant proteins could be removed and low abundant proteins could be enriched, according to the magnitude of the staining. However, 1D electrophoresis fails to identify individual proteins because of its low resolution of separation.
Alternatively, 2D electrophoresis is used to separate proteins based on their molecular weight and charge (O’Farrell 1975). Briefly, proteins are separated based on their isoelectric point in the first dimension, and further separated by SDS-PAGE. Individual proteins or their isoforms can be visualized and characterized using computer-assisted image analysis of 2D-PAGE gels. Klaassens et al. (2007) identified 55 microbial proteins in infant feces using 2D electrophoresis-MS. However, there are several caveats to this method, such as laborious manual procedures with high costs and limited application. In addition, low abundance proteins might be lost during the procedure and are thus not identified.
Gel-free approaches rely on the development of chromatography techniques to separate proteins. High-pressure liquid chromatography (HPLC) is faster and more convenient than electrophoresis for separating peptide mixtures. In a typical metaproteomic experiment, a complex protein sample is digested into peptides followed by separation and identification by LC-MS. Reversed-phase liquid chromatography (RPLC) is the most frequent choice for proteomic study and a C18 column is generally used to separate peptides. RPLC separation is performed with a gradient elution program comprising a low proportion of methanol or acetonitrile and a high proportion of water as the mobile phase, which can be easily volatilized.
Technological and methodological improvements have advanced metaproteomic studies of the gut microbiota. A promising LC method, multidimensional liquid chromatography (MDLC), has been developed to allow the study of complex samples using several separation dimensions. MDLC has higher capacity and efficiency than traditional 1D liquid chromatography. Typically, size-exclusion chromatography (Cheruthazhekatt et al. 2013), strong cation exchange (SCX) chromatography (Mawuenyega et al. 2003; Zhou et al. 2015), and strong anion exchange (SAX) techniques (Ficarro et al. 2011) are always used in the first dimension, while traditional reverse phase chromatography is applied in the last dimension. These techniques have different selectivities.
To date, the combination of SCX chromatography and RP chromatography has been used widely to fractionate complex peptide mixtures. In a SCX-RP system, the acidified peptide mixture is loaded onto an SCX column, and fractions eluted from the first column are absorbed into an RP column (Betancourt et al. 2013; Quan et al. 2015; Slebos et al. 2008). Peptides are separated efficiently and easily by MDLC-MS; therefore, it has been used widely to identify the metaproteome of the gut microbiota. Brooks et al. (2015) applied a split-phase fused silica column, containing both SCX and RP materials, for 2D-LC. The peptide mixture was fractionated using an ammonium acetate solution in a gradient elution, followed by reverse phase chromatography. About 1149-2636 microbial proteins per sample were identified by the method of nano-2D-LC-MS/MS. Erickson et al. (2012) used a similar method to study the human host-microbiota signatures of Crohn’s disease.
Generally, in a gel-based method, proteins are digested into peptides after 1D or 2D-PAGE separation. For non-gel-based methods, proteins are digested before separation. A universal digestion procedure, named filter-aided sample preparation (FASP) to filtering out small molecular substances, has yet to be fully established (Wisniewski et al. 2009). In a FASP protocol, reduction, alkylation, and digestion are performed using a 10-kDa filter. Compared with the existing methods, FASP is a relatively time-consuming, but more efficient, process (Lipecka et al. 2016).
MS analysis
MS is an essential technology of metaproteomic research. Currently, ion sources of electrospray ionization (ESI) and matrix-assisted laser desorption ionization (MALDI) are ubiquitous in the study of proteomics (Jansen et al. 2005; Soltwisch et al. 2009; Whitehouse et al. 1985). ESI-MS/MS and MALDItandem time-of-flight (TOF/TOF) as the powerful tools for proteomics research could always be used to improve the accuracy of the protein identification by enhancing the separation efficiency, detection sensitivity, and the detection of flux significantly (Dumpala et al. 2009; Trufelli et al. 2011; Noordin and Othman 2013). ESI, combining ionization technology and LC, is a great choice for the analysis of complex samples and the identification of low abundance proteins (Kawashima et al. 2013). ESI generates ions of higher charge states and reduced mass-to-charge ratios, which can improve the compatibility and efficiency of the MS analyzer (Ho et al. 2003). Unlike ESI, MALDI generates +1 charged ions based on the short wavelength laser-induced desorption from the matrix, resulting in very large mass-to-charge ratios of peptides or proteins (Caprioli et al. 1997).
Mass analyzers, such as TOF, quadrupoles, and ion traps, are employed routinely in proteomics research. Scan speed, resolution, mass accuracy, and acquisition range are the primary parameters that determine the performance of mass analyzers. TOF analyzers determine the mass-to-charge ratios of the protein by detecting the ion flight time. Generally, a TOF analyzer runs in tandem with a quadrupole or additional TOF mass filters. Ion traps can capture ions according to the mass-to-charge ratios using magnetic and electric fields (Douglas et al. 2005). Although ion traps have low sensitivity for quantification, they are gaining ground because of their smaller size and lower cost (Ho et al. 2003).
In recent years, more and more new mass spectrometry technologies have been applied in proteomics research, such as LTQ-Orbitrap and Q Exactive systems. These technologies provide very high resolution and analytical ability, which will promote the development of proteomics significantly.
Database searching
Gut microbiota research is calling out for comprehensive reference metaproteome databases containing non-redundant sequences of gut microorganisms. The NCBInr database, with its high sequence coverage, is used frequently for protein characterization. However, removing redundant proteins is challenging. It is very time-consuming to get protein information because of the sheer size of the NCBInr database. To solve this problem, a combined database including different known gut microbial metagenome sequence databases are usually applied (Erickson et al. 2012; Kolmeder et al. 2012). However, they do not represent a comprehensive microbial map of the gut microbiota, because some genomes of uncultivable gut microbiota have not been sequenced. Thus, an ideal database with all potentially expressed proteins in the gut microbiota sample is urgently required.
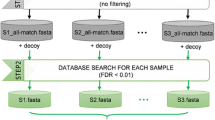
To solve this problem, Zhang et al. (2016c) proposed a high-performance and universal workflow for gut metaproteome identification and quantification, named MetaPro-IQ, in which over 120,000 peptides corresponding to 30,000 proteins were identified in a single experiment. Briefly, a three-step database search strategy was performed to identify proteins of the gut microbiota from human and mouse. The first search was performed against the whole gene catalog database to generate a “pseudo-metaproteome” database. Subsequently, the pseudo-metaproteome database was employed for a second search, which was typically a target-decoy database search with strict filtering (e.g., with a false discovery rate < 0.01). Finally, the resulting proteins were mapped to a non-redundant database to generate confidence data. To gather more information about the composition and function of the gut microbiota, the proteins identified were subjected to taxonomic classification and Clusters of Orthologous Groups (COG) categorization, Gene Ontology (GO), and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses.
Applications of metaproteomics in gut microbiota research
Currently, metaproteomics has limited application in gut microbiota research, compared with RNA-seq and metagenomics. With the metaproteomics emerging into the spotlight of research, several metaproteomic projects have been initiated to characterize proteins in the gut microbiota. To illustrate the applicability of metaproteomics in gut microbiota research (Fig. 3), a number of interesting examples are described below. The following section was based on the novelty of the metaproteomic studies of gut microbiota reported to date.

Applications of metaproteomics in the study of the gut microbiota
Characterization of the microbial composition
Increasing evidence suggests that the numerous microorganisms in human gut have important significance for health and disease (Baothman et al. 2016; Kasparovska et al. 2016; Ley et al. 2006; Malaguarnera et al. 2014). Microbial classification has been studied by different methodologies, such as culture, microscopy, and especially, metagenomics. Culture and microscopy techniques can be costly, time-consuming, and biased; therefore, metagenomics and next-generation sequencing were applied broadly to characterize the composition of the gut microbiota. Currently, metaproteomics might provide new insights into the diversity of the gut microbiota, and researchers have worked in the field of characterization of the composition of microbiota using metaproteomics.
Levi Mortera et al. (2016) employed metaproteomics to characterize gut microbiota in mouse models. They compared two different metaproteomic methods, namely MALDI-TOF MS-based culturomic procedures and traditional bottom-up liquid chromatography with subsequent MS/MS shotgun metaproteomic procedures, using a newborn model to dissect the effect of nutrients (e.g., IgA) in maternal milk on the gut microbiota. They found that IgA-deficient milk could cause an increase in the population of opportunistic bacterial pathogens, such as Pasteurella pneumotropica and Staphylococcus xylosus. Notably, the two metaproteomic techniques were used to characterize the gut microbiota from various angles and depth, and both were found to be reliable techniques to describe the gut microbiota.
Identification of therapeutic target in the gut microbiota
MS-based traditional proteomics is an effective tool that is employed in both in vitro and in vivo models to identify therapeutic targets and discover biomarkers and to reveal the molecular mechanisms associated with many diseases, including cancer, cardiovascular disease, and cognitive disease (Wang et al. 2016; Zhang et al. 2016a, b). Recent reports have highlighted the important role of the gut microbiota, which might contribute to the occurrence and development of disease (Tai et al. 2015; Tlaskalová-Hogenová et al. 2011). A study focusing on changes in the metaproteome to understand their influence on disease, to identify new biomarkers in the gut microbiota, and to find new therapeutic targets in the gut microbiota would have the consequent benefits from health and economic aspects. Hence, metaproteomics analysis has been applied gradually to identify the gut microbiota metaproteome in healthy and diseased states, with the aim of identifying specific proteins as targets for treatment.
Liver cirrhosis is the final condition of liver fibrosis, in which the liver fails to work normally because of long-term liver injury. Cirrhosis is characterized by distortion of the liver parenchyma associated with fibrous septae and nodule formation, as well as alterations in blood flow (Bugianesi 2005; Pinter et al. 2016). In recent years, the gut microbiota has been noted to share a close relationship with liver cirrhosis. Wei et al. (2016) used 1D gel electrophoresis and in-gel protein digestion coupled with a high-throughput LC-MS/MS measurement to detect metaproteomic changes in the intestinal microbiota of liver cirrhosis patients. In that study, about 4400 proteins from bacteria were identified. The abundances of 14 proteins, such as chaperone protein DnaK, glutamate dehydrogenase, and elongation factor G, were increased; and seven proteins, such as ketol-acid reductoisomerase, phosphoglycerate kinase, and probable thiol peroxidase, were expressed uniquely in the patients with liver cirrhosis compared with their healthy counterparts. Functionally, these proteins were mainly related to carbohydrate and amino acid transport and metabolism, suggesting that the gut microbiota from patients with liver cirrhosis have higher metabolic activity. In addition, it was found that patients with liver cirrhosis had different biosynthesis of branched chain amino acids, pantothenate, and CoA compared with controls. Overall, this study revealed more comprehensive and specific proteins of gut microbiota in patients with cirrhosis, and therefore provided potential biomarkers and therapeutic targets for the progress and treatment of cirrhosis.
Erickson et al. (2012) used multi-omics (metagenomics/metaproteomics) to reveal the host-microbiota map of Crohn’s disease. They identified the metaproteome of the gut microbiota from different parts, including the ileum and colon. The result showed that proteins of gut microbiota from different parts of the intestine were different. Comparing with the healthy subject, proteins of gut microbiota from the ileum of the subject with Crohn’s disease showed significant differences in COG categories and metabolic levels. Proteins of gut microbiota with functions in carbohydrate transport and metabolism, energy production and conversion, and amino acid transport and metabolism were significantly less abundant in patients. Proteins of gut microbiota related to replication, recombination, and repair were significantly more abundant from the ileum of the subject with Crohn’s disease. Carbohydrate active enzymes, such as glycoside hydrolases and polysaccharide lyase, showed lower abundance compared with the healthy subject. Additionally, the abundance of proteins of gut microbiota involved in butyrate production was lower than in the control. These informations showed a great diversity of microbial function in healthy versus Crohn’s disease subjects, which might lead eventually to the development of new therapeutic targets.
Prediction of drug-induced adverse effect
An adverse drug effect may be referred to as toxic reactions resulting from an intervention of a medicinal product that could cause severe condition and death in patients, which challenge many institutions, such as pharmaceutical companies, regulatory agencies, and healthcare professionals (Lazarou et al. 1998). Approximately 17% patients have an adverse drug reaction (Bohm and Cascorbi 2016). Hence, testing and avoiding adverse drug reactions are mandatory and essential from ethical aspects. Generally, drug-induced toxicities of liver, heart, and renal are tested using in vitro models and experimental animals. Despite the improved detection rate of adverse drug effects at the organ level, some potentially dangerous effects on the gut microbiota are still unavailable. As such, it is vital to predict drug-induced adverse effects according to the dynamic changes of the gut microbiota. The metaproteome of gut microbiota will contribute to understanding the occurrence and development of drug-induced adverse effects, realizing the pharmacological mechanism, and preventing the emergence of drug-induced adverse effects. However, there are few studies on the application of metaproteomics in the study of drug-induced adverse effects.
Perez-Cobas et al. (2013) applied ultra high-performance liquid chromatography coupled with an Orbitrap instrument to investigate the effect of β-lactam therapy by detecting the change in the gut microbiota metaproteome at multiple time points (3, 6, 11, 14, and 40 days). In that study, a total of 3011 proteins were identified. It was discovered that β-lactam treatment could reduce the number of highly abundant proteins of gut microbiota compared with the control sample over time, but increased the number of low-abundance proteins of gut microbiota. Some proteins related to the immune response decreased during β-lactam treatment, which render the bacteria more susceptible to the drug. Some proteins related to glycolysis, pyruvate decarboxylation, the tricarboxylic acid cycle, glutamate metabolism, iron uptake, GTP hydrolysis, and translation termination were increased at the initial stages of β-lactam treatment. These results suggested that β-lactam treatment might affect the metabolic status of the gut microbiota negatively.
Interpretation of host’s gut microbiota adaptation to environmental exposure
The “hygiene hypothesis” is a popular concept that is accepted commonly among scientists and the public. It mainly states that the fewer infections in early childhood, the greater the chance of developing allergic diseases in the future (Strachan 1989). By extension, it has gradually translated to an objective fact that the less microbiota in our body, the higher the chance of developing an allergic disease in the future (von Mutius 2010). For example, there was a lack of understanding as to why beggars seldom get sick and adapt easily to survive in hostile conditions. An experiment relating to the hygiene hypothesis was carried out by observation of the protective effect of growing up on a farm against asthma and allergies (von Mutius 2010). The experiment showed that children growing up on a farm have less chance of getting asthma and allergies, because they have more contact with the microbiota. A large number of studies have used metagenomic approaches to reveal the protection process in individuals (Wong et al. 2016); however, the phenomenon of adaption to long-term microbiota exposure in different environments has been revealed rarely using metaproteomics approaches. Despite this, metaproteomics has broadened our horizons greatly in the study of the symbiotic relationship between humans and their gut microbiota.
Tang et al. (2014) used high-throughput metagenomics integrated with a mass spectrometry-based metaproteomics approach to identify proteins in chicken fecal samples. A total of 3673 proteins were identified. Among them, 155 proteins were from Clostridium spp., 380 were from Lactobacillus spp., and 66 were from Streptococcus spp. The most frequently identified proteins in study were chaperone proteins, including GroEL and DnaK proteins. Cold shock proteins, cytochromes, thioredoxins, and peroxidases, which might be related to the adaptation process, were also identified. However, these stress-associated proteins were identified much less frequently in humans and pigs. The alteration of the central body temperature of the chickens was different to that of humans and pigs. The normal temperature for chicken varies between 39.8 and 43.6 °C at different times of the day; however, the normal temperature for humans is 37 °C and for pigs is 38.8 °C. In addition, birds excrete uric acid, which is another challenging factor for the adaption of gut microbiota. Thus, the main stress factors might be higher than other animals. This result suggested that these proteins might be required for bacteria to adapt to different environments.
Limitations and challenges of metaproteomics analysis
Clarifying the composition and functions of the microbial communities in their corresponding environments can help us understand the mechanism of diseases (Kasparovska et al. 2016; Mangiola et al. 2016; Odamaki et al. 2016). Large metaproteomic sequencing projects that analyze proteins from samples can provide comprehensive data on the diversity of gut microorganisms and their potential roles in specific environments (Tang et al. 2014). However, several challenges remain for metaproteomic analysis. (1) It is difficult to purify and identify microbial proteins from fecal samples that containing the complex metaproteome of gut microbes (Bojanova and Bordenstein 2016). Some potential host proteins (e.g., Secretory IgA) in the sample could influence the identification of the microbial metaproteome (Tang et al. 2014). (2) High diversity in gut microbial composition and variations between individuals might lead to inconsistent outcomes (Bai et al. 2016; Vernocchi et al. 2016), which present many difficulties to the study of metaproteome. Besides, the microbial or protein composition could also be influenced by other factors, such as food (Kasparovska et al. 2016), disease (Maeda et al. 2016), mood (Mangiola et al. 2016), age (Odamaki et al. 2016), and gender (Strati et al. 2016). (3) Current technologies prohibit the detection of low abundance proteins. Although high-performance MS has increased the dynamic range and coverage of proteins significantly, certain low abundance proteins still cannot be detected by the current mass spectrometers (Muth et al. 2013). Thus, a more sensitive, efficient, and sophisticated analytical technology should be developed to study the metaproteome of gut microbiota. (4) It is ambiguous in mapping peptides to distinct proteins, the so-called protein inference, which is especially noticeable in metaproteomics (Kolmeder and de Vos 2014; Nesvizhskii and Aebersold 2005). Generally, MS/MS spectra of peptides, rather than proteins, are matched to search against a database. However, most of the matched peptides are not unique peptides, which are necessary for protein identification. For example, when a peptide is provided, more than one protein could be identified. This is a problem for the specific taxonomical affiliation of detectable peptides and the generation of protein groups. (5) Lack of reference database is another major impediment to the application of metaproteomics in gut microbiota research. Several comprehensive projects, such as the Human Microbiome Project initiated by the NIH, and MetaHIT, which are based on large-scale sequencing, have been developed over the last couple of years and aimed to characterize the composition and function of gut microorganisms. These will help to generate new results from high-performance and universal analyses of complex high-throughput metaproteomic datasets.
Conclusions and prospects
Metaproteomics shows great potential as a universal analytical method that would broaden our knowledge of organisms’ biology within an ecosystem. Ultimately, it should be able to reveal all the proteins expressed by environmental microbial communities at a specific time. Furthermore, it may provide a real-time representation of the microbial activity, function, and signal transduction in the gut microbial community. Moreover, metaproteomics might help to identify the effect of dynamic changes of the gut microbiota. In addition, it could reveal the regulatory mechanisms of the gut microbiota. Above all, metaproteomics plays an essential role in studying the complete biological process of the gut microbiota.
Although the metaproteomics approach has identified complex microbial communities, several challenges restrict its development. Some gut microbial proteome, especially low-abundance gut microbial proteome, have not been identified. It is critical that metaproteomic approach should be significantly improved in throughput in the coming years, and some experiences based on human proteomics strategy of identifying low abundance protein should be shared. Indeed, during the last decade, researchers have applied the metaproteomics strategy to identify the composition of proteins in gut microbial communities and comparatively analyze the differential proteins groups in healthy and sick states. However, how gut microbiota communicate with human and what signaling pathways and mechanisms exist are still the open questions at the moment in the field of intestinal microbiomics, thus, analyzing gut microbiota from a system perspective by applying metaproteomics technology might be the most important research direction to human health care. In addition, using metaproteomics strategy to characterize protein post-translational modifications (PTM) of gut microbiota might be another important direction. Notably, the effectiveness of metaproteomics will depend on the development process of sequencing technologies and computational approaches, and therefore, keeping pace with the progress of metagenomics will contribute to the development of metaproteomics. Meanwhile, metaproteomics could be combined with metagenomics, metatranscriptomics, metabolomics, and other “omics” methods. The integrative omics approaches may generate comprehensive information from genes to RNA to proteins and metabolites. Overall, metaproteomics is a powerful tool with an uncharted potential to reveal disease mechanisms related to the gut microbiota.
References
Aires J, Butel MJ (2011) Proteomics, human gut microbiota and probiotics. Expert Rev Proteomics 8(2):279–288. doi:10.1586/epr.11.5
Apajalahti JH, Sarkilahti LK, Maki BR, Heikkinen JP, Nurminen PH, Holben WE (1998) Effective recovery of bacterial DNA and percent-guanine-plus-cytosine-based analysis of community structure in the gastrointestinal tract of broiler chickens. Appl Environ Microbiol 64(10):4084–4088
Bai Z, Zhang H, Li N, Bai Z, Zhang L, Xue Z, Jiang H, Song Y, Zhou D (2016) Impact of environmental microbes on the composition of the gut microbiota of adult BALB/c mice. PLoS One 11(8):e0160568. doi:10.1371/journal.pone.0160568
Baothman OA, Zamzami MA, Taher I, Abubaker J, Abu-Farha M (2016) The role of gut microbiota in the development of obesity and diabetes. Lipids Health Dis 15:108. doi:10.1186/s12944-016-0278-4
Barker CJ, Gillett A, Polkinghorne A, Timms P (2013) Investigation of the koala (Phascolarctos cinereus) hindgut microbiome via 16S pyrosequencing. Vet Microbiol 167(3–4):554–564. doi:10.1016/j.vetmic.2013.08.025
Betancourt LH, De Bock PJ, Staes A, Timmerman E, Perez-Riverol Y, Sanchez A, Besada V, Gonzalez LJ, Vandekerckhove J, Gevaert K (2013) SCX charge state selective separation of tryptic peptides combined with 2D-RP-HPLC allows for detailed proteome mapping. J Proteome 91:164–171. doi:10.1016/j.jprot.2013.06.033
Bohm R, Cascorbi I (2016) Pharmacogenetics and predictive testing of drug hypersensitivity reactions. Front Pharmacol 7:396. doi:10.3389/fphar.2016.00396
Bojanova DP, Bordenstein SR (2016) Fecal transplants: what is being transferred? PLoS Biol 14(7):e1002503. doi:10.1371/journal.pbio.1002503
Brooks B, Mueller RS, Young JC, Morowitz MJ, Hettich RL, Banfield JF (2015) Strain-resolved microbial community proteomics reveals simultaneous aerobic and anaerobic function during gastrointestinal tract colonization of a preterm infant. Front Microbiol 6:654. doi:10.3389/fmicb.2015.00654
Brunelle JL, Green R (2014) One-dimensional SDS-polyacrylamide gel electrophoresis (1D SDS-PAGE). Methods Enzymol 541:151–159. doi:10.1016/B978-0-12-420119-4.00012-4
Bugianesi E (2005) Review article: steatosis, the metabolic syndrome and cancer. Aliment Pharmacol Ther Suppl 2:40–43. doi:10.1111/j.1365-2036.2005.02594.x
Caprioli RM, Farmer TB, Gile J (1997) Molecular imaging of biological samples: localization of peptides and proteins using MALDI-TOF MS. Anal Chem 69(23):4751–4760
Carrasco-Navarro U, Vera-Estrella R, Barkla BJ, Zuniga-Leon E, Reyes-Vivas H, Fernandez FJ, Fierro F (2016) Proteomic analysis of the signaling pathway mediated by the heterotrimeric Galpha protein Pga1 of Penicillium chrysogenum. Microb Cell Factories 15(1):173. doi:10.1186/s12934-016-0564-x
Chen R, Xiao M, Gao H, Chen Y, Li Y, Liu Y, Zhang N (2016) Identification of a novel mitochondrial interacting protein of C1QBP using subcellular fractionation coupled with CoIP-MS. Anal Bioanal Chem 408(6):1557–1564. doi:10.1007/s00216-015-9228-7
Cheruthazhekatt S, Harding GW, Pasch H (2013) Comprehensive high temperature two-dimensional liquid chromatography combined with high temperature gradient chromatography-infrared spectroscopy for the analysis of impact polypropylene copolymers. J Chromatogr A 1286:69–82. doi:10.1016/j.chroma.2013.02.052
Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R (2009) Bacterial community variation in human body habitats across space and time. Science 326(5960):1694–1697. doi:10.1126/science.1177486
David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, Biddinger SB, Dutton RJ, Turnbaugh PJ (2014) Diet rapidly and reproducibly alters the human gut microbiome. Nature 505(7484):559–563. doi:10.1038/nature12820
Del Chierico F, Petrucca A, Mortera SL, Vernocchi P, Rosado MM, Pieroni L, Carsetti R, Urbani A, Putignani L (2014) A metaproteomic pipeline to identify newborn mouse gut phylotypes. J Proteome 97:17–26. doi:10.1016/j.jprot.2013.10.025
Dhabaria A, Cifani P, Reed C, Steen H, Kentsis A (2015) A high-efficiency cellular extraction system for biological proteomics. J Proteome Res 14(8):3403–3408. doi:10.1021/acs.jproteome.5b00547
Donohoe DR, Garge N, Zhang X, Sun W, O’Connell TM, Bunger MK, Bultman SJ (2011) The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab 13(5):517–526. doi:10.1016/j.cmet.2011.02.018
Douglas DJ, Frank AJ, Mao D (2005) Linear ion traps in mass spectrometry. Mass Spectrom Rev 24(1):1–29. doi:10.1002/mas.20004
Dumpala PR, Lawrence ML, Karsi A (2009) Proteome analysis of Edwardsiella ictaluri. Proteomics 9(5):1353–1363. doi:10.1002/pmic.200800652
Erickson AR, Cantarel BL, Lamendella R, Darzi Y, Mongodin EF, Pan C, Shah M, Halfvarson J, Tysk C, Henrissat B, Raes J, Verberkmoes NC, Fraser CM, Hettich RL, Jansson JK (2012) Integrated metagenomics/metaproteomics reveals human host-microbiota signatures of Crohn’s disease. PLoS One 7(11):e49138. doi:10.1371/journal.pone.0049138
Ficarro SB, Zhang Y, Carrasco-Alfonso MJ, Garg B, Adelmant G, Webber JT, Luckey CJ, Marto JA (2011) Online nanoflow multidimensional fractionation for high efficiency phosphopeptide analysis. Mol Cell Proteomics 10(11):O111.011064. doi:10.1074/mcp.O111.011064
Gao G, Zhao X, Li Q, He C, Zhao W, Liu S, Ding J, Ye W, Wang J, Chen Y, Wang H, Li J, Luo Y, Su J, Huang Y, Liu Z, Dai R, Shi Y, Meng H, Wang Q (2016) Genome and metagenome analyses reveal adaptive evolution of the host and interaction with the gut microbiota in the goose. Sci Rep 6:32961. doi:10.1038/srep32961
Graf D, Di Cagno R, Fak F, Flint HJ, Nyman M, Saarela M, Watzl B (2015) Contribution of diet to the composition of the human gut microbiota. Microb Ecol Health Dis 26:26164. doi:10.3402/mehd.v26.26164
Ho CS, Lam CW, Chan MH, Cheung RC, Law LK, Lit LC, Ng KF, Suen MW, Tai HL (2003) Electrospray ionisation mass spectrometry: principles and clinical applications. Clin Biochem Rev 24(1):3–12
Human Microbiome Jumpstart Reference Strains C, Nelson KE, Weinstock GM, Highlander SK, Worley KC, Creasy HH, Wortman JR, Rusch DB, Mitreva M, Sodergren E, Chinwalla AT, Feldgarden M, Gevers D, Haas BJ, Madupu R, Ward DV, Birren BW, Gibbs RA, Methe B, Petrosino JF, Strausberg RL, Sutton GG, White OR, Wilson RK, Durkin S, Giglio MG, Gujja S, Howarth C, Kodira CD, Kyrpides N, Mehta T, Muzny DM, Pearson M, Pepin K, Pati A, Qin X, Yandava C, Zeng Q, Zhang L, Berlin AM, Chen L, Hepburn TA, Johnson J, McCorrison J, Miller J, Minx P, Nusbaum C, Russ C, Sykes SM, Tomlinson CM, Young S, Warren WC, Badger J, Crabtree J, Markowitz VM, Orvis J, Cree A, Ferriera S, Fulton LL, Fulton RS, Gillis M, Hemphill LD, Joshi V, Kovar C, Torralba M, Wetterstrand KA, Abouellleil A, Wollam AM, Buhay CJ, Ding Y, Dugan S, FitzGerald MG, Holder M, Hostetler J, Clifton SW, Allen-Vercoe E, Earl AM, Farmer CN, Liolios K, Surette MG, Xu Q, Pohl C, Wilczek-Boney K, Zhu D (2010) A catalog of reference genomes from the human microbiome. Science 328(5981):994–999. doi:10.1126/science.1183605
Human Microbiome Project C (2012) Structure, function and diversity of the healthy human microbiome. Nature 486(7402):207–214. doi:10.1038/nature11234
Iraporda C, Errea A, Romanin DE, Cayet D, Pereyra E, Pignataro O, Sirard JC, Garrote GL, Abraham AG, Rumbo M (2015) Lactate and short chain fatty acids produced by microbial fermentation downregulate proinflammatory responses in intestinal epithelial cells and myeloid cells. Immunobiology 220(10):1161–1169. doi:10.1016/j.imbio.2015.06.004
Jansen R, Lachatre G, Marquet P (2005) LC-MS/MS systematic toxicological analysis: comparison of MS/MS spectra obtained with different instruments and settings. Clin Biochem 38(4):362–372. doi:10.1016/j.clinbiochem.2004.11.003
Kane AV, Dinh DM, Ward HD (2015) Childhood malnutrition and the intestinal microbiome. Pediatr Res 77(1–2):256–262. doi:10.1038/pr.2014.179
Karlsson F, Tremaroli V, Nielsen J, Backhed F (2013) Assessing the human gut microbiota in metabolic diseases. Diabetes 62(10):3341–3349. doi:10.2337/db13-0844
Kasparovska J, Pecinkova M, Dadakova K, Krizova L, Hadrova S, Lexa M, Lochman J, Kasparovsky T (2016) Effects of isoflavone-enriched feed on the rumen microbiota in dairy cows. PLoS One 11(4):e0154642. doi:10.1371/journal.pone.0154642
Kawashima Y, Takahashi N, Satoh M, Saito T, Kado S, Nomura F, Matsumoto H, Kodera Y (2013) Enhanced recovery of lyophilized peptides in shotgun proteomics by using an LC-ESI-MS compatible surfactant. Proteomics 13(5):751–755. doi:10.1002/pmic.201200462
Klaassens ES, de Vos WM, Vaughan EE (2007) Metaproteomics approach to study the functionality of the microbiota in the human infant gastrointestinal tract. Appl Environ Microbiol 73(4):1388–1392. doi:10.1128/AEM.01921-06
Kolmeder CA, de Vos WM (2014) Metaproteomics of our microbiome—developing insight in function and activity in man and model systems. J Proteome 97:3–16. doi:10.1016/j.jprot.2013.05.018
Kolmeder CA, de Been M, Nikkilä J, Ritamo I, Mättö J, Valmu L, Salojärvi J, Palva A, Salonen A, de Vos WM (2012) Comparative metaproteomics and diversity analysis of human intestinal microbiota testifies for its temporal stability and expression of core functions. PLoS One 7(1):e29913. doi:10.1371/journal.pone.0029913
Lazarou J, Pomeranz BH, Corey PN (1998) Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA 279(15):1200–1205
Leary DH, Hervey WJ, Li RW, Deschamps JR, Kusterbeck AW, Vora GJ (2012) Method development for metaproteomic analyses of marine biofilms. Anal Chem 84(9):4006–4013. doi:10.1021/ac203315n
Levi Mortera S, Del Chierico F, Vernocchi P, Rosado MM, Cavola A, Chierici M, Pieroni L, Urbani A, Carsetti R, Lante I, Dallapiccola B, Putignani L (2016) Monitoring perinatal gut microbiota in mouse models by mass spectrometry approaches: parental genetic background and breastfeeding effects. Front Microbiol 7:1523. doi:10.3389/fmicb.2016.01523
Ley RE, Peterson DA, Gordon JI (2006) Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 124(4):837–848. doi:10.1016/j.cell.2006.02.017
Lipecka J, Chhuon C, Bourderioux M, Bessard MA, van Endert P, Edelman A, Guerrera IC (2016) Sensitivity of mass spectrometry analysis depends on the shape of the filtration unit used for filter aided sample preparation (FASP). Proteomics 16(13):1852–1857. doi:10.1002/pmic.201600103
Lopez JL, Marina A, Alvarez G, Vazquez J (2002) Application of proteomics for fast identification of species-specific peptides from marine species. Proteomics 2(12):1658–1665. doi:10.1002/1615-9861(200212)2:12<1658::AID-PROT1658>3.0.CO;2-4
Maeda Y, Kumanogoh A, Takeda K (2016) Altered composition of gut microbiota in rheumatoid arthritis patients. Nihon Rinsho Meneki Gakkai Kaishi 39(1):59–63. doi:10.2177/jsci.39.59
Magrone T, Jirillo E (2013) The interaction between gut microbiota and age-related changes in immune function and inflammation. Immun Ageing 10(1):31. doi:10.1186/1742-4933-10-31
Malaguarnera G, Giordano M, Nunnari G, Bertino G, Malaguarnera M (2014) Gut microbiota in alcoholic liver disease: pathogenetic role and therapeutic perspectives. World J Gastroenterol 20(44):16639–16648. doi:10.3748/wjg.v20.i44.16639
Mangiola F, Ianiro G, Franceschi F, Fagiuoli S, Gasbarrini G, Gasbarrini A (2016) Gut microbiota in autism and mood disorders. World J Gastroenterol 22(1):361–368. doi:10.3748/wjg.v22.i1.361
Martin C, Zhang Y, Tonelli C, Petroni K (2013) Plants, diet, and health. Annu Rev Plant Biol 64:19–46. doi:10.1146/annurev-arplant-050312-120142
Mawuenyega KG, Kaji H, Yamuchi Y, Shinkawa T, Saito H, Taoka M, Takahashi N, Isobe T (2003) Large-scale identification of Caenorhabditis elegans proteins by multidimensional liquid chromatography-tandem mass spectrometry. J Proteome Res 2(1):23–35
Mayne J, Ning Z, Zhang X, Starr AE, Chen R, Deeke S, Chiang CK, Xu B, Wen M, Cheng K, Seebun D, Star A, Moore JI, Figeys D (2016) Bottom-up proteomics (2013-2015): keeping up in the era of systems biology. Anal Chem 88(1):95–121. doi:10.1021/acs.analchem.5b04230
Muth T, Benndorf D, Reichl U, Rapp E, Martens L (2013) Searching for a needle in a stack of needles: challenges in metaproteomics data analysis. Mol BioSyst 9(4):578–585. doi:10.1039/c2mb25415h
Nesvizhskii AI, Aebersold R (2005) Interpretation of shotgun proteomic data: the protein inference problem. Mol Cell Proteomics 4(10):1419–1440. doi:10.1074/mcp.R500012-MCP200
Noordin R, Othman N (2013) Proteomics technology—a powerful tool for the biomedical scientists. Malays J Med Sci 20(2):1–2
Odamaki T, Kato K, Sugahara H, Hashikura N, Takahashi S, Xiao JZ, Abe F, Osawa R (2016) Age-related changes in gut microbiota composition from newborn to centenarian: a cross-sectional study. BMC Microbiol 16:90. doi:10.1186/s12866-016-0708-5
O’Farrell PH (1975) High resolution two-dimensional electrophoresis of proteins. J Biol Chem 250(10):4007–4021
O’Hara AM, Shanahan F (2006) The gut flora as a forgotten organ. EMBO Rep 7(7):688–693. doi:10.1038/sj.embor.7400731
Peach M, Marsh N, Miskiewicz EI, MacPhee DJ (2015) Solubilization of proteins: the importance of lysis buffer choice. Methods Mol Biol 1312:49–60. doi:10.1007/978-1-4939-2694-7_8
Perez-Cobas AE, Gosalbes MJ, Friedrichs A, Knecht H, Artacho A, Eismann K, Otto W, Rojo D, Bargiela R, von Bergen M, Neulinger SC, Däumer C, Heinsen FA, Latorre A, Barbas C, Seifert J, dos Santos VM, Ott SJ, Ferrer M, Moya A (2013) Gut microbiota disturbance during antibiotic therapy: a multi-omic approach. Gut 62(11):1591–1601. doi:10.1136/gutjnl-2012-303184
Pinter M, Trauner M, Peck-Radosavljevic M, Sieghart W (2016) Cancer and liver cirrhosis: implications on prognosis and management. ESMO Open 1(2):e000042. doi:10.1136/esmoopen-2016-000042
Prauchner CA, Kozloski GV, Farenzena R (2013) Evaluation of sonication treatment and buffer composition on rumen bacteria protein extraction and carboxymethylcellulase activity. J Sci Food Agric 93(7):1733–1736. doi:10.1002/jsfa.5959
Quan Q, Szeto SS, Law HC, Zhang Z, Wang Y, Chu IK (2015) Fully automated multidimensional reversed-phase liquid chromatography with tandem anion/cation exchange columns for simultaneous global endogenous tyrosine nitration detection, integral membrane protein characterization, and quantitative proteomics mapping in cerebral infarcts. Anal Chem 87(19):10015–10024. doi:10.1021/acs.analchem.5b02619
Redon E, Loubiere P, Cocaign-Bousquet M (2005) Role of mRNA stability during genome-wide adaptation of Lactococcus lactis to carbon starvation. J Biol Chem 280(43):36380–36385. doi:10.1074/jbc.M506006200
Rose C, Parker A, Jefferson B, Cartmell E (2015) The characterization of feces and urine: a review of the literature to inform advanced treatment technology. Crit Rev Environ Sci Technol 45(17):1827–1879. doi:10.1080/10643389.2014.1000761
Satija A, Bhupathiraju SN, Rimm EB, Spiegelman D, Chiuve SE, Borgi L, Willett WC, Manson JE, Sun Q, Hu FB (2016) Plant-based dietary patterns and incidence of type 2 diabetes in US men and women: results from three prospective cohort studies. PLoS Med 13(6):e1002039. doi:10.1371/journal.pmed.1002039
Schneider T, Keiblinger KM, Schmid E, Sterflinger-Gleixner K, Ellersdorfer G, Roschitzki B, Richter A, Eberl L, Zechmeister-Boltenstern S, Riedel K (2012) Who is who in litter decomposition? Metaproteomics reveals major microbial players and their biogeochemical functions. ISME J 6(9):1749–1762. doi:10.1038/ismej.2012.11
Slebos RJ, Brock JW, Winters NF, Stuart SR, Martinez MA, Li M, Chambers MC, Zimmerman LJ, Ham AJ, Tabb DL, Liebler DC (2008) Evaluation of strong cation exchange versus isoelectric focusing of peptides for multidimensional liquid chromatography-tandem mass spectrometry. J Proteome Res 7(12):5286–5294. doi:10.1021/pr8004666
Soltwisch J, Souady J, Berkenkamp S, Dreisewerd K (2009) Effect of gas pressure and gas type on the fragmentation of peptide and oligosaccharide ions generated in an elevated pressure UV/IR-MALDI ion source coupled to an orthogonal time-of-flight mass spectrometer. Anal Chem 81(8):2921–2934. doi:10.1021/ac802301s
Sonnenburg JL, Backhed F (2016) Diet-microbiota interactions as moderators of human metabolism. Nature 535(7610):56–64. doi:10.1038/nature18846
Stephen AM, Cummings JH (1980) The microbial contribution to human faecal mass. J Med Microbiol 13(1):45–56. doi:10.1099/00222615-13-1-45
Strachan DP (1989) Hay fever, hygiene, and household size. BMJ 299(6710):1259–1260
Strati F, Di Paola M, Stefanini I, Albanese D, Rizzetto L, Lionetti P, Calabrò A, Jousson O, Donati C, Cavalieri D, De Filippo C (2016) Age and gender affect the composition of fungal population of the human gastrointestinal tract. Front Microbiol 7:1227. doi:10.3389/fmicb.2016.01227
Tai N, Wong FS, Wen L (2015) The role of gut microbiota in the development of type 1, type 2 diabetes mellitus and obesity. Rev Endocr Metab Disord 16(1):55–65. doi:10.1007/s11154-015-9309-0
Tanca A, Palomba A, Pisanu S, Addis MF, Uzzau S (2015) Enrichment or depletion? The impact of stool pretreatment on metaproteomic characterization of the human gut microbiota. Proteomics 15(20):3474–3485. doi:10.1002/pmic.201400573
Tang Y, Underwood A, Gielbert A, Woodward MJ, Petrovska L (2014) Metaproteomics analysis reveals the adaptation process for the chicken gut microbiota. Appl Environ Microbiol 80(2):478–485. doi:10.1128/AEM.02472-13
Tlaskalová-Hogenová H, Stěpánková R, Kozáková H, Hudcovic T, Vannucci L, Tučková L, Rossmann P, Hrnčíř T, Kverka M, Zákostelská Z, Klimešová K, Přibylová J, Bártová J, Sanchez D, Fundová P, Borovská D, Srůtková D, Zídek Z, Schwarzer M, Drastich P, Funda DP (2011) The role of gut microbiota (commensal bacteria) and the mucosal barrier in the pathogenesis of inflammatory and autoimmune diseases and cancer: contribution of germ-free and gnotobiotic animal models of human diseases. Cell Mol Immunol 8(2):110–120. doi:10.1038/cmi.2010.67
Trufelli H, Palma P, Famiglini G, Cappiello A (2011) An overview of matrix effects in liquid chromatography-mass spectrometry. Mass Spectrom Rev 30(3):491–509. doi:10.1002/mas.20298
Verberkmoes NC, Russell AL, Shah M, Godzik A, Rosenquist M, Halfvarson J, Lefsrud MG, Apajalahti J, Tysk C, Hettich RL, Jansson JK (2009) Shotgun metaproteomics of the human distal gut microbiota. ISME J 3(2):179–189. doi:10.1038/ismej.2008.108
Vernocchi P, Del Chierico F, Putignani L (2016) Gut microbiota profiling: metabolomics based approach to unravel compounds affecting human health. Front Microbiol 7:1144. doi:10.3389/fmicb.2016.01144
von Mutius E (2010) 99th Dahlem conference on infection, inflammation and chronic inflammatory disorders: farm lifestyles and the hygiene hypothesis. Clin Exp Immunol 160(1):130–135. doi:10.1111/j.1365-2249.2010.04138.x
Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10(1):57–63. doi:10.1038/nrg2484
Wang HB, Zhang ZX, Li H, He HB, Fang CX, Zhang AJ, Li QS, Chen RS, Guo XK, Lin HF, Wu LK, Lin S, Chen T, Lin RY, Peng XX, Lin WX (2011) Characterization of metaproteomics in crop rhizospheric soil. J Proteome Res 10(3):932–940. doi:10.1021/pr100981r
Wang J, Cunningham R, Zetterberg H, Asthana S, Carlsson C, Okonkwo O, Li L (2016) Label-free quantitative comparison of cerebrospinal fluid glycoproteins and endogenous peptides in subjects with Alzheimer’s disease, mild cognitive impairment and healthy individuals. Proteomics Clin Appl 10(12):1225–1241. doi:10.1002/prca.201600009
Wei X, Jiang S, Chen Y, Zhao X, Li H, Lin W, Li B, Wang X, Yuan J, Sun Y (2016) Cirrhosis related functionality characteristic of the fecal microbiota as revealed by a metaproteomic approach. BMC Gastroenterol 16(1):121. doi:10.1186/s12876-016-0534-0
Westergaard D, Li J, Jensen K, Kouskoumvekaki I, Panagiotou G (2014) Exploring mechanisms of diet-colon cancer associations through candidate molecular interaction networks. BMC Genomics 15:380. doi:10.1186/1471-2164-15-380
Whitehouse CM, Dreyer RN, Yamashita M, Fenn JB (1985) Electrospray interface for liquid chromatographs and mass spectrometers. Anal Chem 57(3):675–679
Wilmes P, Bond PL (2004) The application of two-dimensional polyacrylamide gel electrophoresis and downstream analyses to a mixed community of prokaryotic microorganisms. Environ Microbiol 6(9):911–920. doi:10.1111/j.1462-2920.2004.00687.x
Wisniewski JR, Zougman A, Nagaraj N, Mann M (2009) Universal sample preparation method for proteome analysis. Nat Methods 6(5):359–362. doi:10.1038/nmeth.1322
Wong K, Shaw TI, Oladeinde A, Glenn TC, Oakley B, Molina M (2016) Rapid microbiome changes in freshly deposited cow feces under field conditions. Front Microbiol 7:500. doi:10.3389/fmicb.2016.00500
Xie H, Guo R, Zhong H, Feng Q, Lan Z, Qin B, Ward KJ, Jackson MA, Xia Y, Chen X, Chen B, Xia H, Xu C, Li F, Xu X, Al-Aama JY, Yang H, Wang J, Kristiansen K, Wang J, Steves CJ, Bell JT, Li J, Spector TD, Jia H (2016) Shotgun metagenomics of 250 adult twins reveals genetic and environmental impacts on the gut microbiome. Cell Syst 3(6):572–584. doi:10.1016/j.cels.2016.10.004
Xiong W, Giannone RJ, Morowitz MJ, Banfield JF, Hettich RL (2015) Development of an enhanced metaproteomic approach for deepening the microbiome characterization of the human infant gut. J Proteome Res 14(1):133–141. doi:10.1021/pr500936p
Xu X, Shi L, Wang M (2016) Comparative quantitative proteomics unveils putative mechanisms involved into mercury toxicity and tolerance in Tigriopus japonicus under multigenerational exposure scenario. Environ Pollut 218:1287–1297. doi:10.1016/j.envpol.2016.08.087
Zhang M, Yang XJ (2016) Effects of a high fat diet on intestinal microbiota and gastrointestinal diseases. World J Gastroenterol 22(40):8905–8909. doi:10.3748/wjg.v22.i40.8905
Zhang W, Zhong T, Chen Y (2016a) LC-MS/MS-based targeted proteomics quantitatively detects the interaction between p53 and MDM2 in breast cancer. J Proteome 152:172–180. doi:10.1016/j.jprot.2016.11.002
Zhang X, Hou HT, Wang J, Liu XC, Yang Q, He GW (2016b) Plasma proteomic study in pulmonary arterial hypertension associated with congenital heart diseases. Sci Rep 6:36541. doi:10.1038/srep36541
Zhang X, Ning Z, Mayne J, Moore JI, Li J, Butcher J, Deeke SA, Chen R, Chiang CK, Wen M, Mack D, Stintzi A, Figeys D (2016c) MetaPro-IQ: a universal metaproteomic approach to studying human and mouse gut microbiota. Microbiome 4(1):31. doi:10.1186/s40168-016-0176-z
Zhao X, Jiang Z, Yang F, Wang Y, Gao X, Wang Y, Chai X, Pan G, Zhu Y (2016) Sensitive and simplified detection of antibiotic influence on the dynamic and versatile changes of fecal short-chain fatty acids. PLoS One 11(12):e0167032. doi:10.1371/journal.pone.0167032
Zhou Y, Meng Z, Edman-Woolcott M, Hamm-Alvarez SF, Zandi E (2015) Multidimensional separation using HILIC and SCX pre-fractionation for RP LC-MS/MS platform with automated exclusion list-based MS data acquisition with increased protein quantification. J Proteomics Bioinform 8(11):260–265. doi:10.4172/jpb.1000378
Zhu W, Gregory JC, Org E, Buffa JA, Gupta N, Wang Z, Li L, Fu X, Wu Y, Mehrabian M, Sartor RB, McIntyre TM, Silverstein RL, Tang WH, DiDonato JA, Brown JM, Lusis AJ, Hazen SL (2016) Gut microbial metabolite TMAO enhances platelet hyperreactivity and thrombosis risk. Cell 165(1):111–124. doi:10.1016/j.cell.2016.02.011
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was supported by the National Science and Technology Major Projects for “Major New Drugs Innovation and Development” (grant numbers 2015ZX09J15102-004-004) and the Tianjin Support Plans for the Top of the Notch Youth Talents.
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
About this article

Cite this article
Xiao, M., Yang, J., Feng, Y. et al. Metaproteomic strategies and applications for gut microbial research. Appl Microbiol Biotechnol 101, 3077–3088 (2017). https://doi.org/10.1007/s00253-017-8215-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-017-8215-7