
Abstract
Biotechnological applications of microbial pectate lyases (Pels) in plant fiber processing are promising, eco-friendly substitutes for conventional chemical degumming processes. However, to potentiate the enzymes’ use for industrial applications, resolving the molecular structure to elucidate catalytic mechanisms becomes necessary. In this manuscript, we report the high resolution (1.45 Å) crystal structure of pectate lyase (pelN) from Paenibacillus sp. 0602 in apo form. Through sequence alignment and structural superposition with other members of the polysaccharide lyase (PL) family 1 (PL1), we determined that pelN shares the characteristic right-handed β-helix and is structurally similar to other members of the PL1 family, while exhibiting key differences in terms of catalytic and substrate binding residues. Then, based on information from structure alignments with other PLs, we engineered a novel pelN. Our rational design yielded a pelN mutant with a temperature for enzymatic activity optimally shifted from 67.5 to 60 °C. Most importantly, this pelN mutant displayed both higher specific activity and ramie fiber degumming ability when compared with the wild-type enzyme. Altogether, our rational design method shows great potential for industrial applications. Moreover, we expect the reported high-resolution crystal structure to provide a solid foundation for future rational, structure-based engineering of genetically enhanced pelNs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pectin is one of the most widely available polysaccharides, contained in the cell walls of all higher plants, also known as vascular plants (Gummadi and Kumar 2006; Klug-Santner et al. 2006). Accordingly, enzymes that degrade pectin or pectate are present in most plants (Ouattara et al. 2010), as well as in a wide variety of microbes associated with plants (Klug-Santner et al. 2006). One such class of enzymes consists of pectate lyases (EC 4.2.2.2). Pectate lyases belong to the larger class of polysaccharide lyases (PLs) and occupy a key position in the decomposition and recycling of plant material. Importantly, pectate lyases cleave α-1,4-glycosidic bonds between C4 and C5 of pectin or pectic acid, also known as polygalacturonic acid (PGA), to generate an unsaturated double bond oligogalacturonides via the β-transelimination mechanism (Abbott et al. 2010; Jayani et al. 2005).
According to the annotations of the Carbohydrate-Active Enzyme (CAZy) database (http://www.cazy.org), PLs are presently grouped into 21 families (Garron and Cygler 2010). Three dimensional structures have been determined for representatives of most families, including PL1 (Lietzke et al. 1996; Pickersgill et al. 1994; Zheng et al. 2012), PL2 (Abbott and Boraston 2007; McLean et al. 2015), PL3 (Akita et al. 2001; Alahuhta et al. 2011), PL6 (Huang et al. 1999), PL9 (Jenkins et al. 2004), and PL10 (Charnock et al. 2002). PLs exhibit a large variety of fold types, with the right-handed β-helix being the most common type. PL1, PL3, PL6, and PL9 families can be classified as this fold type. Now, while PL1, PL3, and PL9 are bona fide pectate lyase families, PL6 also includes alginate and dermatan sulfate lyase (Huang et al. 1999). The β-jelly roll fold is also well represented among PL families, five of which (PL7, PL13, PL14, PL18, and PL20) share this fold (Yamasaki et al. 2004). A further six families share an (α/α) n toroid fold, and these are further subdivided into proteins with single-domain (α/α) n toroid fold and those containing additional domains other than toroid domains (Abbott and Boraston 2007; Charnock et al. 2002; Yoon et al. 2001). The remaining PL families have a β-sandwich, β-propeller, or triple-stranded β-helix fold. The abundance of different PL folds indicates that PLs have evolved from different ancestral scaffolds and are an example of convergent evolution (Garron and Cygler 2010).
Recently, Li et al. cloned and over-expressed the alkaliphilic PL (pelN) from Paenibacillus sp. 0602 (Li et al. 2014). The pelN gene encodes a polypeptide consisting of 445 amino acid residues with a molecular weight of ~50 kDa, and a specific activity towards methylated pectin of >2060 U/mg, which is the highest among all PLs characterized to date (Li et al. 2014). Of particular relevance, its tolerance to high alkaline conditions makes it a promising candidate for a large number of industrial applications. However, wild-type pelN cannot fully exhaust its potential enzymatic activity at 50 °C, the typical temperature of the water bath in the ramie (Boehmeria nivea) degumming process, due to its optimal temperature for enzymatic activity of 67.5 °C being significantly higher. Thus, modifying the enzyme in a way that shifts its optimal temperature closer to 50 °C would immediately increase the efficiency of the ramie degumming process.
In this work, we contribute to this goal in the following ways: (i) we solved the 3D structure of pelN in apo form. Our results provide much needed information for elucidating and understanding the catalytic mechanism and substrate binding properties of pelN. (ii) Based on this 3D structure, we rationally designed a pelN mutant with an optimal temperature for enzymatic activity of 60 °C, which is considerably closer to the desired value. We anticipate our findings will provide a strong foundation for rational design of enzymes, here aiming to improve PelN’s functional characteristics for industrial applications.
Materials and methods
Protein expression and purification
Recombinant pelN was expressed as described previously (Li et al. 2014), with the exception that in this study, we included a hexahistidine tag at the C-terminus of the protein to facilitate purification under native conditions. The enzyme was purified as described previously (Zhou et al. 2015). The loading buffer was 20 mM phosphate buffer pH 7.0. The elution buffer was 20 mM phosphate buffer pH 7.0 and an additional 400 mM imidazole purified proteins were dialyzed against another loading buffer (20 mM phosphate buffer pH 6.8) and then loaded onto a UNOsphere™ S column (Bio-Rad Laboratories, Inc. Hercules, CA) equilibrated with loading buffer. Proteins were eluted with a linear gradient of NaCl (0–2 M) in elution buffer (20 mM phosphate buffer pH 7.0, with 2 M NaCl). Active fractions were pooled and dialyzed in crystallization buffer (25 mM Tris-HCl, 150 mM NaCl, pH 8.0). The final sample was concentrated to 30 mg/mL.
Crystallization and data collection
Concentrated pelN was subjected to crystallization screening using the sitting drop method as described by Hampton Research (Laguna Niguel, CA, USA). Crystals were grown by mixing 1 μL protein and 1 μL reservoir solution containing 10% PEG3000 and 100 mM Na2HPO4 (the pH was adjusted to 4.0 or 4.2 with citric acid) within 3 days. Subsequently, several crystals suitable for X-ray diffraction experiments were transferred to liquid nitrogen. Synchrotron datasets were then collected at 100 K at the Shanghai Synchrotron Radiation Facility beamlines. Data was processed using the HKL3000 package (Minor et al. 2006). The structure of pelN was then solved using the method of molecular replacement contained in the CCP4 program suite by using pectate lyase (Tmpel) from Thermotoga maritima (PDB: 3ZSC) as a search model (Potterton et al. 2003). We then performed a refinement using Phenix (Adams et al. 2002) and Coot (Emsley and Cowtan 2004). Finally, all structural figures were generated with PyMOL (www.pymol.org).
pelN mutants
Plasmids of pelN mutants were constructed by site-directed mutagenesis using a fast mutagenesis kit. The mutants were expressed and cell lysates were used for thermostability and optimal temperature screening. The selected mutant proteins with 6-His at c-terminus were expressed and purified as previously reported (Zhou et al. 2015). The enzymatic activity and characteristic of pelN mutants were also measured as previously described (Li et al. 2014).
Enzymatic degumming test of ramie fibers
A ramie degumming test was performed using the following protocol: 1 g ramie was soaked in 50 mL glycin-NaOH buffer (pH 9.8). Thirty units per millimeter of purified pelN and pelN mutant were added into the solution and incubated at 50 °C for 12 h (enzyme was omitted in control experiments). Then the ramie was washed and dried at 115 °C. The weight loss of ramie fibers was measured to evaluate degumming efficiency.
The structure of pelN has been submitted to PDB. The PDB ID is 5GT5.
Results
Structure of pelN
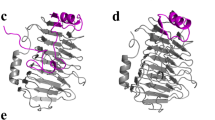
We determined the crystal structure of full-length pelN with a C-terminal His-tag to 1.45 Å resolution by using the method of molecular replacement. Figure 1a shows an overview of the 3D structure that contains two separate and structurally asymmetric pelN molecules in the active form with R work and R free of 0.1484 and 0.1742, respectively (Table 1). Almost all amino acids of pelN were observed clearly in electron density maps. In the crystal structure, the predominant right-handed β-parallel structure formed from three parallel β-sheets (PB1, PB2, and PB3) is readily apparent, and the PBs are connected by three turns. Turn 1 (T1) is between PB1 and PB2, Turn 2 (T2) is between PB2 and PB3, and Turn 3 (T3) is between PB1 and PB3, respectively. T1 is short, while T2 and T3 are longer loops extending from the β-sheets. The tertiary structure of pelN is similar to other PLs from the PL1 family (Lietzke et al. 1996; Pickersgill et al. 1994; Scavetta et al. 1999; Yoder and Jurnak 1995a; Yoder and Jurnak 1995b; Zheng et al. 2012). PB1 has ten ladders (Fig. 1b, c), PB2 has nine, and PB3 has seven ladders.

Overall structure of pelN. a Cartoon representation of pelN. The three β-sheets are denoted PB1, PB2, and PB3. The N-terminus and C-terminus have been marked. b, c Structure comparison of pelN (green) superimposed with Tmpel from Thermotoga maritime (blue; PDB ID: 3ZSC), revealing 10 parallel right-handed β-helices
Enzymatic interactions related residues
To understand the catalytic mechanism of pelN, it is useful to resolve the structure of the complex (pelN bound to substrate). It has been reported that all PLs share a similar enzymatic mechanism (Garron and Cygler 2010; Seyedarabi et al. 2010), but that the amino acid residues in the active regions play different catalytic roles. Thus, we constructed a structural model of pelN bound to its substrate by superimposing the structures of pelN and Tmpel (PDB: 3ZSC) containing pectate trisaccharide substrate, as shown in Fig. 2a. A long groove between PB3 and T2 contains the bound substrate: in pelN several amino acid residues interact with the substrate, including Asp174, Lys245, Leu248, Arg276, Arg279, Arg281, and Arg349, corresponding to Asp139, Lys163, Leu166, Arg197, Arg200, Arg202, and Arg264 in Tmpel. Others residues such as Asp145, Asp147, Lys244, Ala251, Asp275, Asn324, Thr322, and Glu352 in pelN, which also interact with substrate, share no obvious counterparts in Tmpel. Conversely, residues Ala111, Lys112, Asp162, Gln196, Met266, and Ile228 that bind substrate in Tmpel have no homologs in pelN (Fig. 2b).

Substrate recognition and interaction sites. a Model of pelN with bound substrate obtained from superimposition of the structures of pelN (green) and Tmpel. b Interaction residues shown as lines, residues from Tmpel are shown in blue. c Model of pelN with bound hexasaccharide obtained from superimposition of the structures of pelN (green) and Bspel. d Interaction residues shown as lines, residues from Bspel are shown in yellow. e Catalytic activity of wild-type and mutant pelN enzymes
We then performed superimposition of the structures of pelN and Bspel (PDB: 2O17), pectate lyase bound to hexasaccharide, to further investigate substrate binding (Fig. 2c). Additional potential substrate binding residues were identified in the interaction pocket (Fig. 2d), which may explain differences in substrate selectivity between the PLs.
Among the residues involved in the substrate-binding pocket, D152, D174, and D178 form the Ca2+ ion binding sites. The presence of a Ca2+ ion has been shown to be crucial for PL activity in all previous studies (Henrissat et al. 1995; Scavetta et al. 1999; Seyedarabi et al. 2010; Zheng et al. 2012). From the multiple sequence alignment of pelN and other PLs, we predicted three Ca2+ binding sites in pelN, namely Asp152, Asp174, and Asp178, consistent with what was previously reported (Fig. 3). In general, the primary Ca2+ ion binding to the enzyme is independent of substrate (Herron et al. 2003), and the other Ca2+ ions play a different role in facilitating bridging between the enzyme and substrate in the complex. In the model of bound pelN, the primary Ca2+ ion interacts with Asp174 through the carboxylate oxygens. Accordingly, the mutant D174A showed a complete loss of enzymatic activity (Fig. 2e).

Sequence alignment of pelN0602 and other PL family 1 Pels, namely Bsp165PelA (Bacillus sp. N16–5, pectate lyase), BsPel (B. substilus subsp. subtilis str. 168, pectate lyase), EcPelA (Erwinia chrysanthemi, PelA), AcPel (Acidovorax citrulli, pectate lyase), and TmPelC (Thermotoga maritime, PelA). The sequence alignment was performed with ClustalX (Thompson et al. 1997). A red background highlights conserved residues; conservatively substituted residues are boxed. The secondary structural elements (helices-a, strands-b, turns-T, and helices-η) of pelN0602 are indicated above the aligned sequences. The figure was produced using ESPript (Gouet et al. 2003)
The residue concerned with optimal temperature
Comparison of pelN with the two other PL1 pels revealed little differences in the residues for catalytic interaction (Fig. 2). Structure superimposition of pelN and Bspel showed that several residues were involved in the enzyme-substrate interaction, while such involvement was not evident from the structure superimposition of pelN and Tmpel. Tmpel is expressed in thermal bacterium Thermotoga maritima and stable at high temperatures above 90 °C. On the other hand, Bspel is obtained from the mesobacterium Bacillus subtilis and its optimal temperature is 42 °C. We applied the PoPMuSiC algorithm (http://dezyme.com/) to predict how the change of these residues varies the folding free energy (ΔΔG) of pelN. The results showed that K93I mutants decreased the ΔΔG by 1.03 kcal/ml. Therefore, we identified Lys93 as a putative crucial residue to optimize temperature ranges of pelN.
Enzymatic characterization of the Lys93 mutants
We mutated lys93 to other nine amino acids including Ala, Gly, Glu, Phe, Ile, Leu, Gln, Arg, and Val, and analyzed the mutants’ optimal temperatures. Temperature activity profiles of all lys93 mutants were very similar to that of wild-type pelN (around 67.5 °C), except for mutant K93I. Its optimal temperature was measured at 60 °C (Fig. 4a) while its thermostability did not change from that of wild type pelN (Fig. 4b). Moreover, the specific activity of mutant K93I was significantly higher (nearly 1.75-fold) than that of wild-type pelN (Table 2). Also, the K m value of mutant K93I to substrate of PGA was considerably lower than that of wild-type pelN, indicating a high binding ability to PGA substrate of the K93I mutant.

Temperature characteristic of pelN and K93I mutant. a Optimal temperature of the wild-type pelN and K93I mutant. b Kinetic of thermal inactivity of wild-type pelN and K93I mutant at 60 °C
Ramie degumming
The ramie degumming efficiency was evaluated using purified pelN and the K93I mutant. The enzymatic degumming process was performed at 50 °C for 13 h. The degumming efficiency of wild-type enzyme and mutant enzyme was 16.6 and 24.78%, respectively (Fig. 5a). The ramie fibers were soaked in alkaline buffer and we observed a 9.4% weight loss in a control experiment. The released galacturonic acid was also detected during the degumming process (Fig. 5b). These results suggest that our rational design including the K93I mutant shows a superior degumming ability than wild-type pelN at 50 °C.

Ramie degumming experiment. a Percentage of weight loss after ramie degumming treatment by wild-type pelN and K93I mutant. b Production of galacturonic acid from ramie by wild-type pelN and K93I mutant
Discussion
The enzyme pectate lyase degrades pectin. Pectin is a key component in plant cell walls, and plays an important role in the recycling of plants. In this study, we report the crystal structure of pectate lyase pelN from Paenibacillus sp. 0602, which is a member of polysaccharide lyase family 1. Previously, pelN had been successfully cloned (Li et al. 2014), making it an interesting candidate for industrial applications.
The overall protein fold and the active-site region of pelN are similar to those of other family 1 pectate lyases. The structure of pelN adopts a right-handed β-parallel helix formed from three parallel β-sheets, with a basic active-site cleft across the molecular surface between PB2 and T2 structural elements (Fig. 1). Unlike other family 1 pectate lyases, the structure of pelN contains several additional α-helices surrounding the main structure (Fig. 1c). The sequence alignment of pelN with other family 1 pels displays that these α-helices originate from a long sequence insertion before the catalytic residue K244 and several other short sequence insertions in pelN (Fig. 3) (Li et al. 2014). The structure of pelN shows that several conserved arginine residues are involved in interactions with the substrate. In addition, residues Asp152, Asp174, and Asp178 appear to bind Ca2+ and may play an important role in facilitating substrate binding to the active-site cleft of pelN (Fig. 2).
Multiple sequence alignment of PLs shows high homology of pelN to other PL1 family members. For instance, pelN shares 26.98% sequence identity with Tmpel, 26.81% with Bspel, 24.1% with Acpel (PDB: 4HWV), and 24.84% with Ecpel (PDB: 1AIR). Superimposition of pelN’s structure with the structures of Tmpel, Bspel, Acpel, and Ecpel showed that the pel structures were well conserved (Fig. 6a). Moreover, the structure of pelN had a root-mean-square deviation (RMSD) of 1.183 Å with Tmpel, 1.298 Å with Bspel, 1.276 Å with Acpel and 1.573 Å with Ecpel for 174 Cα atoms. Also, pelN has different residues in the substrate binding pockets of Leu248/Ile250 and Arg279/Leu282 from Bspel (Fig. 6d), as well as Arg279/Leu221 from Ecpel (Fig. 6c). Highly conserved residues of Lys245, Arg276, and Arg281 that directly interacted with substrate are shown in the structure alignment (Fig. 6). Microorganisms have evolved a variety of enzymes that degrade pectin, with different carbohydrate specificities and 3D structures (Garron and Cygler 2010). Although structure alignment revealed that the Ca2+ binding residues are identical in most pels, they were totally different in Ecpel (Lietzke et al. 1996).

Structure comparison of pels. a Overlap of cartoon representions of pels: pelN (green), Acpel (pink), Bspel (blue), Ecpel (cyan), Tmpel (red ). b–e Overlap of substrate binding residues between pelN and Acpel, pelN and Ecpel, pelN and Bspel, and pelN and Tmpel
A right-handed parallel β-helix topology, as observed in pelN, can also be found in members of three other polysaccharide lyase families that share a relatively low sequence identity with pelN (<25%). Namely, the PL3 family enzyme pelI from Erwinia chrysanthemi (Creze et al. 2008), family PL6 chondroitinase B from Flavobacterium heparinum (Huang et al. 1999), and family PL9 pel9A from E. chrysanthemi (Jenkins et al. 2004). These enzymes cleave α-1,4 bonds of polygalacturonate via an anti β-elimination reaction. Specifically, the basic residues abstract the proton from C5 to form an enolate intermediate, and then eliminate the glycosidic bond between C-4 and C-5 to generate an unsaturated product. However, structural comparison of these enzymes revealed critical differences. Similar to most PLs in the PL1 and PL9 families, pelN has a long β-helix with 10 turns (Fig. 2). In contrast, PLs in the PL3 family have a shorter β-helix with only eight turns (Creze et al. 2008). In addition, the characteristic ladders of inward-facing aragine residues within regular stacked turns, as observed in the PL1 (Yoder et al. 1993) and PL9 (Jenkins et al. 2004) families, are absent in the PL3 family (Akita et al. 2001; Creze et al. 2008). Importantly, these arginine residues are required for catalysis in PL1 family enzymes. Moreover, superimposition of the structure of pelN with that of Tmpel (PDB: 3ZSC) suggests that R276, R279, and R281 in pelN are involved in catalysis (Fig. 2). However, lysine is known to be the catalytic base in PL3 (Akita et al. 2001; Creze et al. 2008), PL6 (Huang et al. 1999), and PL9 family enzymes (Jenkins et al. 2004). Thus, differences in the catalytic base (arginine or lysine) result in different pK a values in different PL families, which may contribute to differences in the functional specificity between different PL families.
Aside, an alkaline environment and a moderate temperature are key conditions for high degumming efficiency. Therefore, an enzyme with high thermostability and catalytic activity under such conditions is highly desirable for industrial bioscouring applications. In this study, we engineered a pelN mutant based on structure superimpositions, yielding a mutant with an optimal temperature that significantly improved the catalytic activity for ramie degumming as compared with the wild-type form. One the one hand Thermotoga maritima is thermophilic and, accordingly, Tmpel displays high thermostability with optimal temperature of 90 °C. On the other hand, Bacillus subtilis is mesophilic and Bspel displays highest enzymatic activity at 42 °C. Structure superimposition of pelN with Tmpel and Bspel showed that lys93 has different enzymatic interactions with the different substrates. Therefore, we performed site-directed mutagenesis of lys93 and detected the optimal temperature of all mutants. Our results show that the K93I mutant has an optimal temperature of 60 °C, reduced from the wild-type pelN’s temperature of 67.5 °C. Consequently, the K93I mutant displays a higher enzymatic activity than the wild-type pelN at 50 °C, which is a typical temperature used in bioscouring processes. Mutant K93I’s specific activity is significantly higher (nearly 1.75-fold) and its K m value is lower than the corresponding values for wild-type pelN, indicating a high substrate-binding ability of the K93I mutant (Table 2). These improvements may well be explained by the intermolecular interaction of lys93. PelN interacts with substrate via the N atom of lys93 and the O atom of the substrate. In contrast, in Bspel, for instance, there is no such interaction between the corresponding residue Met122 and the substrate (Fig. 7). Therefore, it can be concluded that the interaction between lys93 and the substrate stabilizes the complex and leads to changes in the characteristics of the mutant.

Interaction between residue Lys93 and the substrate. Residue Lys93 is marked in green. The substrate is marked in pink. The corresponding residue Met122 in Bspel is shown in cyan. Their interaction is indicated by a dashed line
The ramie degumming efficiencies of pelN and lys93 mutants were subsequently evaluated. The weight loss of ramie fibers was 24.78% when using the K93I mutant, after incubation at 50 °C for 12 h with 30 U/mL of the enzyme. This is roughly 50% higher than that achieved with wild-type pelN (Fig. 5). This degumming efficiency was comparable to that of the highest reported alkaline and thermostable pectate lyases (Basu et al. 2009; Bruhlmann et al. 2000; Liang et al. 2015; Zou et al. 2013). Importantly, K93I displayed higher specific activity than others. Lastly, the degumming process allows for further improvements by using K93I mutant combined with other kinds of enzymes, such as xylanase or mannanase. Altogether, our results suggest that the K93I mutant has high potential for industrial In summary, we determined the high-resolution crystal structure of pectate lyase pelN from Paenibacillus sp. 0602. Sequence alignment and structural superposition revealed a β-sheet topology with some key differences from other PLs in terms of catalytic and substrate binding residues. Based on this high-resolution structure, we constructed mutants of lys93. Results from our rational design show that the optimal temperature of the K93I mutant is 7.5 °C lower than that of the wild type. Specifically, the optimal temperature reduced from 67.5 to 60 °C. This is much closer to 50 °C, the typical temperature of the water bath in the ramie (Boehmeria nivea) degumming process. Our ramie degumming test also confirmed that K93I mutants display a higher efficiency in bioscouring processes. Altogether, our results provide structural information that significantly increases the understanding of pelN catalytic mechanisms and, consequently, an avenue to guide further rational engineering of pelN for industrial applications.
References
Abbott DW, Boraston AB (2007) A family 2 pectate lyase displays a rare fold and transition metal-assisted beta-elimination. J Biol Chem 282(48):35328–35336. doi:10.1074/jbc.M705511200
Abbott DW, Gilbert HJ, Boraston AB (2010) The active site of Oligogalacturonate lyase provides unique insights into cytoplasmic oligogalacturonate beta-elimination. J Biol Chem 285(50):39029–39038. doi:10.1074/jbc.M110.153981
Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D 58:1948–1954. doi:10.1107/S0907444902016657
Akita M, Suzuki A, Kobayashi T, Ito S, Yamane T (2001) The first structure of pectate lyase belonging to polysaccharide lyase family 3. Acta Crystallogr D 57:1786–1792. doi:10.1107/S0907444901014482
Alahuhta M, Chandrayan P, Kataeva I, Adams MW, Himmel ME, Lunin VV (2011) A 1.5 Å resolution X-ray structure of the catalytic module of Caldicellulosiruptor bescii family 3 pectate lyase. Acta Crystallogr Sect F Struct Biol Cryst Commun 67(Pt 12):1498–1500. doi:10.1107/S1744309111038449
Basu S, Saha MN, Chattopadhyay D, Chakrabarti K (2009) Large-scale degumming of ramie fibre using a newly isolated Bacillus pumilus DKS1 with high pectate lyase activity. J Ind Microbiol Biotechnol 36(2):239–245. doi:10.1007/s10295-008-0490-y
Bruhlmann F, Leupin M, Erismann KH, Fiechter A (2000) Enzymatic degumming of ramie bast fibers. J Biotechnol 76(1):43–50. doi:10.1016/S0168-1656(99)00175-3
Charnock SJ, Brown IE, Turkenburg JP, Black GW, Davies GJ (2002) Convergent evolution sheds light on the anti-beta-elimination mechanism common to family 1 and 10 polysaccharide lyases. P Natl Acad Sci USA 99(19):12067–12072. doi:10.1073/pnas.182431199
Creze C, Castang S, Derivery E, Haser R, Hugouvieux-Cotte-Pattat N, Shevchik VE, Gouet P (2008) The crystal structure of pectate lyase PelI from soft rot pathogen Erwinia chrysanthemi in complex with its substrate. J Biol Chem 283(26):18260–18268. doi:10.1074/jbc.M709931200
Emsley P, Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr D 60:2126–2132. doi:10.1107/S0907444904019158
Garron ML, Cygler M (2010) Structural and mechanistic classification of uronic acid-containing polysaccharide lyases. Glycobiology 20(12):1547–1573. doi:10.1093/glycob/cwq122
Gouet P, Robert X, Courcelle E (2003) ESPript/ENDscript: extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic Acids Res 31(13):3320–3323
Gummadi SN, Kumar DS (2006) Optimization of chemical and physical parameters affecting the activity of pectin lyase and pectate lyase from Debaryomyces nepalensis: a statistical approach. Biochem Eng J 30(2):130–137. doi:10.1016/j.bej.2006.02.014
Henrissat B, Heffron SE, Yoder MD, Lietzke SE, Jurnak F (1995) Functional implications of structure-based sequence alignment of proteins in the extracellular pectate lyase superfamily. Plant Physiol 107(3):963–976. doi:10.1104/Pp.107.3.963
Herron SR, Scavetta RD, Garrett M, Legner M, Jurnak F (2003) Characterization and implications of Ca2+ binding to pectate lyase C. J Biol Chem 278(14):12271–12277. doi:10.1074/jbc.M209306200
Huang WJ, Matte A, Li YG, Kim YS, Linhardt RJ, Su HS, Cygler M (1999) Crystal structure of chondroitinase B from Flavobacterium heparinum and its complex with a disaccharide product at 1.7 Å resolution. J Mol Biol 294(5):1257–1269. doi:10.1006/jmbi.1999.3292
Jayani RS, Saxena S, Gupta R (2005) Microbial pectinolytic enzymes: a review. Process Biochem 40(9):2931–2944. doi:10.1016/j.procbio.2005.03.026
Jenkins J, Shevchik VE, Hugouvieux-Cotte-Pattat N, Pickersgill RW (2004) The crystal structure of pectate lyase Pel9A from Erwinia chrysanthemi. J Biol Chem 279(10):9139–9145. doi:10.1074/jbc.M311390200
Klug-Santner BG, Schnitzhofer W, Vrsanska M, Weber J, Agrawal PB, Nierstrasz VA, Guebitz GM (2006) Purification and characterization of a new bioscouring pectate lyase from Bacillus pumilus BK2. J Biotechnol 121(3):390–401. doi:10.1016/j.jbiotec.2005.07.019
Li X, Wang H, Zhou C, Ma Y, Li J, Song J (2014) Cloning, expression and characterization of a pectate lyase from Paenibacillus sp. 0602 in recombinant Escherichia coli. BMC Biotechnol 14:18. doi:10.1186/1472-6750-14-18
Liang C, Gui X, Zhou C, Xue Y, Ma Y, Tang SY (2015) Improving the thermoactivity and thermostability of pectate lyase from Bacillus pumilus for ramie degumming. Appl Microbiol Biotechnol 99(6):2673–2682. doi:10.1007/s00253-014-6091-y
Lietzke SE, Scavetta RD, Yoder MD, Jurnak F (1996) The refined three-dimensional structure of pectate lyase E from Erwinia chrysanthemi at 2.2 Å resolution. Plant Physiol 111:73–92
McLean R, Hobbs JK, Suits MD, Tuomivaara ST, Jones DR, Boraston AB, Abbott DW (2015) Functional analyses of resurrected and contemporary enzymes illuminate an evolutionary path for the emergence of exolysis in polysaccharide lyase family 2. J Biol Chem 290(35):21231–21243. doi:10.1074/jbc.M115.664847
Minor W, Cymborowski M, Otwinowski Z, Chruszcz M (2006) HKL-3000: the integration of data reduction and structure solution—from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr 62(Pt 8):859–866. doi:10.1107/S0907444906019949
Ouattara HG, Reverchon S, Niamke SL, Nasser W (2010) Biochemical properties of pectate lyases produced by three different Bacillus strains isolated from fermenting cocoa beans and characterization of their cloned genes. Appl Environ Microb 76(15):5214–5220. doi:10.1128/Aem.00705-10
Pickersgill R, Jenkins J, Harris G, Nasser W, Robert-Baudouy J (1994) The structure of Bacillus subtilis pectate lyase in complex with calcium. Nat Struct Biol 1(10):717–723
Potterton E, Briggs P, Turkenburg M, Dodson E (2003) A graphical user interface to the CCP4 program suite. Acta Crystallogr D 59:1131–1137. doi:10.1107/S0907444903008126
Scavetta RD, Herron SR, Hotchkiss AT, Kita N, Keen NT, Benen JA, Kester HC, Visser J, Jurnak F (1999) Structure of a plant cell wall fragment complexed to pectate lyase C. Plant Cell 11(6):1081–1092
Seyedarabi A, To TT, Ali S, Hussain S, Fries M, Madsen R, Clausen MH, Teixteira S, Brocklehurst K, Pickersgill RW (2010) Structural insights into substrate specificity and the anti beta-elimination mechanism of pectate lyase. Biochemistry-Us 49(3):539–546. doi:10.1021/bi901503g
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25(24):4876–4882
Yamasaki M, Moriwaki S, Miyake O, Hashimoto W, Murata K, Mikami B (2004) Structure and function of a hypothetical Pseudomonas aeruginosa protein PA1167 classified into family PL-7: a novel alginate lyase with a beta-sandwich fold. J Biol Chem 279(30):31863–31872. doi:10.1074/jbc.M402466200
Yoder MD, Jurnak F (1995a) Protein motifs. 3. The parallel beta helix and other coiled folds. FASEB journal : Official Publication of the Federation of American Societies for Experimental Biology 9(5):335–342
Yoder MD, Jurnak F (1995b) The refined three-dimensional structure of pectate lyase C from Erwinia chrysanthemi at 2.2 angstrom resolution (implications for an enzymatic mechanism). Plant Physiol 107(2):349–364
Yoder MD, Lietzke SE, Jurnak F (1993) Unusual structural features in the parallel beta-helix in pectate lyases. Structure 1(4):241–251. doi:10.1016/0969-2126(93)90013-7
Yoon HJ, Hashimoto W, Miyake O, Murata K, Mikami B (2001) Crystal structure of alginate lyase A1-III complexed with trisaccharide product at 2.0 Å resolution. J Mol Biol 307(1):9–16. doi:10.1006/jmbi.2000.4509
Zheng YY, Huang CH, Liu WT, Ko TP, Xue YF, Zhou C, Guo RT, Ma YH (2012) Crystal structure and substrate-binding mode of a novel pectate lyase from alkaliphilic Bacillus sp N16-5. Biochem Bioph Res Co 420(2):269–274. doi:10.1016/j.bbrc.2012.02.148
Zhou ZP, Zhao SZ, Wang SQ, Li XM, Su L, Ma YH, Li J, Song JN (2015) Extracellular overexpression of Chitosanase from Bacillus sp TS in Escherichia coli. Appl Biochem Biotech 175(7):3271–3286. doi:10.1007/s12010-015-1494-5
Zou MY, Li XZ, Shi WJ, Guo FF, Zhao J, Qu YB (2013) Improved production of alkaline polygalacturonate lyase by homologous overexpression pelA in Bacillus subtilis. Process Biochem 48(8):1143–1150. doi:10.1016/j.procbio.2013.05.023
Acknowledgements
The authors thank Dr. Yuzhong Zhang for his kind help during data collection and Jie Zhang for assistance with structure determination. This work was supported, in part, by grants from the Hundred Talents Program of the Chinese Academy of Sciences (CAS), the Knowledge Innovation Program of CAS (KSCX2-EW-G-8), and the Tianjin Municipal Science & Technology Commission (10ZCKFSY05600). T.M. L and A.L. would like to thank the Isaac Newton Institute for Mathematical Sciences for support through EPSRC Grant No. EP/K032208/1. J.S. is a recipient of the Hundred Talents Program of CAS. J.L. is an NHMRC Senior Research Fellow.
This article does not contain any studies with human participants or animals.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interest.
Rights and permissions
About this article

Cite this article
Zhou, Z., Liu, Y., Chang, Z. et al. Structure-based engineering of a pectate lyase with improved specific activity for ramie degumming. Appl Microbiol Biotechnol 101, 2919–2929 (2017). https://doi.org/10.1007/s00253-016-7994-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-016-7994-6