
Abstract
Comprehensive studies of the biodiversity of the microbial epilithic community on monuments may provide critical insights for clarifying factors involved in the colonization processes. We carried out a high-throughput investigation of the communities colonizing the medieval church of San Leonardo di Siponto (Italy) by Illumina-based deep sequencing. The metagenomic analysis of sequences revealed the presence of Archaea, Bacteria, and Eukarya. Bacteria were Actinobacteria, Proteobacteria, Bacteroidetes, Cyanobacteria, Chloroflexi, Firmicutes and Candidatus Saccharibacteria. The predominant phylum was Actinobacteria, with the orders Actynomycetales and Rubrobacteriales, represented by the genera Pseudokineococcus, Sporichthya, Blastococcus, Arthrobacter, Geodermatophilus, Friedmanniella, Modestobacter, and Rubrobacter, respectively. Cyanobacteria sequences showing strong similarity with an uncultured bacterium sequence were identified. The presence of the green algae Oocystaceae and Trebuxiaceae was revealed. The microbial diversity was explored at qualitative and quantitative levels, evaluating the richness (the number of operational taxonomic units (OTUs)) and the abundance of reads associated with each OTU. The rarefaction curves approached saturation, suggesting that the majority of OTUs were recovered. The results highlighted a structured community, showing low diversity, made up of extremophile organisms adapted to desiccation and UV radiation. Notably, the microbiome appeared to be composed not only of microorganisms possibly involved in biodeterioration but also of carbonatogenic bacteria, such as those belonging to the genus Arthrobacter, which could be useful in bioconservation. Our investigation demonstrated that molecular tools, and in particular the easy-to-run next-generation sequencing, are powerful to perform a microbiological diagnosis in order to plan restoration and protection strategies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Stone monuments, due to their prevalently outdoor location, are exposed to the effects of a complex series of physical, chemical, and biological factors. The inorganic mineral surface of the building stones provides the habitat for the growth and development of microorganisms. The textural and mineralogical characteristics of the stones, together with the surrounding environmental conditions, determine the bioreceptivity, i.e., the ability of a material to be colonized by living organisms (Guillitte 1995). Bacteria, fungi, algae, and lichens are the microorganisms classically found on stone surfaces, forming a heterogeneous biofilm (Ragon et al. 2012). In particular, bacteria that develop on outdoor monuments are mostly phototrophs and chemolithoautotrophs, which are characterized by relatively simple nutritional (inorganic minerals, atmospheric ammonia, etc.) and ecological needs (presence of light, CO2, and water). The microorganisms developing on the surface of monuments, with time, are frequently involved in the biodeterioration of the stone by secreting enzymes and activating other metabolic activities (Warscheid and Braams 2000; Hueck 2001; Scheerer et al. 2009; Dakal and Cameotra 2012). However, microorganisms, although often inducing aesthetical changes, could also have a role in the bioconservation of stones, for example, in their consolidation through the process of biomineralization (Mann 2001; Santhosh et al. 2001; Jroundi et al. 2012).
Thus, an in-depth characterization of the microbial diversity flourishing on the stone surfaces of a monument is a prerequisite to plan useful strategies for a correct restoration and conservation (Sterflinger and Piñar 2013). To this aim, the knowledge of the microbial communities composition may also orient the development of new more selective engineered materials with anti-biofouling activity, an issue recently addressed by some of the authors of this paper (Ditaranto et al. 2011; Ditaranto et al. 2015; van der Werf et al. 2015).
Until about a decade ago, classical microbiology methods were the only approaches that were used to obtain information on microorganisms inhabiting cultural assets. However, those culture-dependent methods may underestimate the microbial diversity thus becoming inadequate for studying the whole microbial community (Ward et al. 1990). Therefore, DNA-based typing methods have been developed to study the microbial diversity from various ecological niches. These methods were revealed to be complementary to conventional microbiological techniques (Scheerer et al. 2009).
Advances in high-throughput analysis, mainly based on sequencing of the bacterial 16S rRNA gene (Chakravorty et al. 2007; Tringe and Hugenholtz 2008), have provided new tools for the identification of microorganisms present in complex systems. At present, next-generation sequencing (NGS) technologies have amazingly expanded the possibility of characterizing large populations of microbial communities co-existing in many different ecosystems (de Castro et al. 2013). In spite of these evidences, so far, NGS has been scarcely used to analyze the microbial colonization of ancient stone monuments. To the best of our knowledge, just one paper refers to NGS of wood and brick specimens of the historic buildings of Auschwitz (Gutarowska et al. 2015).
In this study, we applied Illumina-based deep sequencing on 16S rDNA amplicons to reach the following goals: (a) to characterize the microorganisms colonizing the stones of the medieval church of San Leonardo di Siponto (Manfredonia, FG, Italy) and (b) to evaluate differences in the distribution pattern of the microbiota which are possibly associated with different niches of the historical building.
Materials and methods
Site description
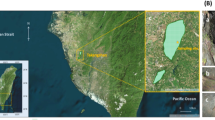
The Abbey of San Leonardo di Siponto is a big monumental settlement located near Manfredonia (latitude 41° 36′ 0″ N; longitude 15° 54′ 0″ E; 0 m at sea level), Apulia, south east of Italy. This area shows a Mediterranean climate with an annual temperature ranging from 5 to 29 °C, often reaching maximum peaks over 35 °C in summer, a mean yearly rainfall of 48.8 mm, mostly during the winter season, and a 3- to 5-dry month period in summer. The complex comprehends the church of San Leonardo, a monastery, and a hospital. It was founded presumably by Benedectine monks in the twelfth century. Augustinian monks and then the Teutonic Knights continued its construction and remodeling during the thirteenth century and beyond. The monastic complex and the interior of the church were repaired in several occasions. In 1995, the exterior northern portal of the church was restored, while the other outdoor parts were not recently subjected to restoration treatments and show evident signs of microbial colonization (Fig.1a).

Sampling areas. a General view of the Abbey of San Leonardo di Siponto, with permission from Laboratorio Centro Aerofotografico, Università degli Studi di Bari “Aldo Moro,” 1989. b East front of the church, with the apse. c–f Details of sampling areas. c The pear of the small arches in the east apse (samples SanLeo1, SanLeo2, SanLeo3). d The archivolt of the window in the east apse (samples SanLeo5 and SanLeo6). e The pear in the west front (samples SanLeo4 and SanLeo7). f The lion in the north portal (sample SanLeo8)
Sampling
Authorizations for sampling were obtained from the Italian “Soprintendenza per i Beni Artistici, Storici ed Etnoantropologici della Puglia.” Sampling was carried out in 2013, from areas where the operation was minimally invasive. Complex samples (ten specimens for each sampling area were pooled to give a composite sample that was representative of an area of 15 × 15 cm2) were aseptically collected by carefully scraping off from the surface small quantities of the stone material. Eight sites of sampling (SanLeo1–8) were selected on the basis of cardinal directions, macroscopic evidence of microbial colonization, apparently different petrographic composition, and surface morphology of the stones (smooth or carved).
All samples but one were taken from the exterior walls, namely, SanLeo1, SanLeo2, SanLeo3, SanLeo5, and SanLeo6, were from the east side of the monument, in particular from the major apse. SanLeo1, SanLeo2, SanLeo3 were from one of the pears of the small arches (Fig. 1, b and c), whereas SanLeo5 and SanLeo6 were from the archivolt of the window (Fig. 1 , b and d); SanLeo4 and SanLeo7 were taken from one of the pears of the west portal (Fig.1e). Finally, SanLeo8 was sampled from one of the lion statues in the north portal (Fig. 1f). Samples were stored at −80 °C until use.
Petrographic study
Small stone fragments (ca. 10 × 5 × 2 mm3) were sampled from all sampling areas (SanLeo 1–8) (see above). Thin sections were prepared for optical microscopic analysis. A detailed analysis of minerals and micro-texture was performed by observing the sections using a Zeiss Axioscop 40 Pol in transmitted light with polars and crossed polars. This microscopic analysis was also efficiently used for an esteem of the total porosity and pore size classification (Andriani 2003), as a complementary or alternative method of mercury intrusion porosimetry (MIP), which could not be performed requiring large stone samples.
DNA extraction
Samples were powdered using sterile tubes and pestles. DNA was directly extracted from 0.1 g of each of the homogenized samples using the Fast Prep System (BIO 101) and the commercial kit Fast DNA Spin Kit for Soil (MP Biomedicals, CA, United States), according to the manufacturer’s instructions. The final product was 100 μl of application ready DNA. The quality and concentration of the DNA extracts were determined in 0.7 % agarose-1× TAE gels stained with Gel Red ™ 10,000× (Biotium, Inc.) and by spectrophotometric measurements at 260, 280, and 230 nm using the NanoDrop® ND-1000 Spectrophotometer (ThermoFisher Scientific Inc., MI, Italy). DNA samples were stored at −20 °C until further analyses.
Amplicon library preparation and Illumina-based sequencing
Preparation of amplicon libraries of the V5-V6 hypervariable regions of 16S rRNA gene was performed as described previously (Manzari et al. 2015). In brief, for each metagenomic DNA sample, two amplification steps were performed to obtain a final amplicon having, to each end, an Illumina adapter (P7 or P5) and an Illumina Nextera index sequence, required by the dual index sequencing approach. The use of the robust Illumina sequencing technology allowed us to avoid replicating PCRs (Smith and Peay 2014). Purified amplicons were pooled in equimolar ratio and subjected to a 2 × 250 bp paired-end sequencing on the Ilumina MiSeq platform. Raw sequences were deposited in the NCBI Sequence Read Archive (SRA) database (accession number: SRP073496).
Pre-processing
Illumina paired-end reads (PE reads) were processed using the mothur v.1.33.0 software (Schloss et al. 2009) following the standard operating procedure (http://www.mothur.org/wiki/MiSeq_SOP). Contigs between read pairs were assembled and differences in base calls in the overlapping region were solved using the ΔQ parameter (Kozich et al. 2013); primer sequences were removed, no ambiguous bases were allowed, and maximum homopolymer size was 8 bp. The remaining sequences were de-replicated and screened for chimeras using UCHIME in de novo mode (Edgar et al. 2011). Since 16S rRNA gene sequences from chloroplast were also amplified, we built a customized reference database consisting of 14,956 bacterial and archaeal sequences from Silva database, plus 1022 plastidial 16S rRNA gene sequences of photosynthetic eukaryotes retrieved from PhytoREF database (Decelle et al. 2015). Reads were then classified using this reference database and a naïve Bayesian classifier with a pseudo-bootstrap threshold of 80 %, (method = wang) in mothur. This initial classification served to screen our dataset and to split prokaryotic and eukaryotic communities. Prokaryotic and eukaryotic datasets were then aligned in mothur, using kmer searching for finding the template sequence and Needleman-Wunsch algorithm to make a pairwise alignment between candidates and bacterial and eukaryotic reference alignments provided inside mothur (http://www.mothur.org/wiki/Alignment_database). PE reads were then clustered in operational taxonomic units (OTUs) at 98 % of similarity using the average neighbor method, and one representative read per OTU (the most abundant) was selected. Taxonomy was assigned to bacterial representative reads with the Ribosomal Database Project classifier (http://rdp.cme.msu.edu/classifier/) and using a pseudo-bootstrap threshold of 80 %, whereas the eukaryotic sequences were classified using the blast server in the PhytoREF website (http://phytoref.fr).
Diversity analyses
All analyses were performed separately for the bacterial community alone and for the total community, Eukarya plus Bacteria, and were run in the R environment using R-package vegan (Oksanen et al. 2013) (http://CRAN.R-project.org/package=vegan).
Rarefaction curves of the observed OTUs, alpha diversity indices (Shannon, Berger-Parker, Simpson-evenness), and the richness estimator (Chao1) were calculated. Community dissimilarity data matrices from normalized data were computed (vegdist function in vegan) using the Bray–Curtis index (abundance data) and were then used in ordination and cluster analyses. Non-metric multi-dimensional scaling was performed to map samples on simplified two-dimensional spaces using the metaMDS function, which performs multiple NMDS runs and retains the best solution. Dendrograms presenting hierarchically organized samples were built using hclust and UPGMA methods. Normalization was performed by randomly sub-sampling OTUs to the number of reads present in the sample with the lowest amount of reads. Moreover, the communities were evaluated by the 2D ordination NMDS (envfit function from the vegan R package), for correlation with location, petrographic composition, porosity, and substrate morphology of the stone substrate samples. The fit statistic goodness was reported as r 2, i.e., the percentage of variance, and as a permutation test assessing significance.
Results
In order to define the microbiota flourishing outdoor the medieval church of San Leonardo di Siponto (Manfredonia, Apulia, Italy), samples were collected from eight different areas of the exterior carbonate stone surfaces (Table 1).
The petrographic composition and an evaluation of the porosity (total porosity and pore size classification) of the stone substrates were performed by optical microscopy. These appeared to be mainly composed of locally sourced fossiliferous carbonate stones. Although adjacent, some stones showed different petrographic composition; this was the case of the samples SanLeo5 and SanLeo6, localized side by side in the archivolt of the apse window. This is a common feature in medieval buildings, which were often built using recycled materials. Estimated porosity ranged from <1 % to almost 20 %. In all sampling areas, macroscopic alterations consisting in the diffused presence of differently colored crusts and patinas were observed.
DNA was extracted, PCR amplified with rDNA universal primers, and subjected to Illumina-based deep sequencing. One of the collected samples, SanLeo4, was proved to be un-amplifiable and was thus excluded from the subsequent analyses. A general description of the dataset is shown in Table 2, both as single and pooled results. A total of 1,399,490 raw PE reads were generated, resulting after denoising in 873,322 high-quality PE reads, belonging to the three domains of life, Archaea, Bacteria and Eukarya. Archaea showed to be a minor component (0.01 %), whereas Bacteria were the major identified component (57.1 %), together with a substantial number of eukaryotic sequences (42.9 %). Due to the similarity between bacterial and chloroplast 16S rDNA regions, the amplicon-based approach detected also the eukaryotic microbial photosynthetic community. Thus, in this study, data were analyzed both as bacterial community alone and as total community, Bacteria plus Eukarya. Clustering at 98 % of similarity produced 3785 OTUs (0.1 % Archaea, 86.7 % Bacteria, and 13.2 % Eukarya).
As reported in Table 2, the relative proportion of archaeal, bacterial, and eukaryotic PE reads varied among the different samples: archaeal sequences were present only in SanLeo6 and bacterial sequences were the major component in SanLeo5, SanLeo6, and SanLeo7, whereas eukaryotic sequences were prevalent in SanLeo1, SanLeo2, SanLeo3, and SanLeo8.
Taxonomy overview
The taxonomic pattern was explored in terms of number of PE reads (abundance) and number of OTUs (richness). PE reads belonging to seven different bacterial phyla, namely Actinobacteria, Proteobacteria, Bacteroidetes, Cyanobacteria, Chloroflexi, Firmicutes, and Candidatus Saccharibacteria were identified across the analyzed sites. Regarding the relative abundances, the predominant phylum was Actinobacteria (45 %), mainly represented by the orders Actynomycetales (29 %) and Rubrobacteriales (14 %), followed by the phylum of Bacterioidetes (10 %) with the order Cytophagales (10 %), and the phylum of Proteobacteria (28 %), essentially represented by the order of Oceanospirillales (9 %).
Within Actynomycetales, 40 % of the sequences were members of the genus Pseudokineococcus, followed by the genera Sporichthya (10 %), Blastococcus (9 %), Arthrobacter (9 %), Geodermatophilus (7 %), Friedmanniella (6 %), and Modestobacter (2 %). The order Rubrobacteriales was represented exclusively by the genus Rubrobacter, whereas the genera affiliated to the order Cytophagales were Adhaeribacter (38 %), Hymenobacter (35 %), and Roseivirga (12 %). Within Oceanospirillales, 96 % of the sequences were assigned to the genus Halomonas (Online Resource Table S1).
Figure 2 and Fig. 3 show the relative proportions of PE reads and OTUs in the seven analyzed sites, as referred to the bacterial community and to the total community, respectively.

Relative abundances of the bacterial orders, in terms of sequences and OTUs, in each sample. Taxonomic groups accounting for <1 % of all sequences are summarized in the artificial group “Others”

Relative abundances of bacterial and eukaryotic orders, in terms of sequences and OTUs, in each sample. Eukaryotic orders are boxed. Taxonomic groups accounting for <1 % of all sequences are summarized in the artificial group “Others”
Concerning the kingdom of Archaea, sequences belonging to the class of Euryarchaeota, order Halobacteriales, were found only in sample SanLeo6 (0.1 %) (Fig. 2).
Among the Bacteria, the order Actynomycetales was dominant in most of the samples: (SanLeo1, 63 %; SanLeo3, 30 %; SanLeo5, 36 %; SanLeo6, 29 %; SanLeo7, 30 %), with the greatly represented genus Pseudokineococcus proportionately distributed along the sites. PE reads belonging to the others more represented genera of Actynomycetales were mainly found in sample SanLeo6, which accounted for almost 98 % of all PE reads belonging to Sporichthya and for 81 % and 62 % of those referred to Blastococcus and Arthrobacter, respectively. Besides, SanLeo6 accounted for 63 % of Geodermatophilaceae found, represented by three genera: Blastococcus (81 % of the PE reads belonging to this genus were found in SanLeo6), Geodermatophilus (about half of the sequences belonging to this genus were recovered from this sample), and Modestobacter, which, on the contrary, appeared to be distributed along the sites. In SanLeo2, the orders Oceanospirillales (55 %) and Alteromonadales (27 %) were prevailing, while in SanLeo8, Cytophagales (35 %) and Rhodospirillares (30 %) were predominant. The phototrophic phylum of Cyanobacteria represented only 6 % of the total bacterial sequences. This group was peculiar of sample SanLeo6, where it represented 17 % of the total bacterial community. Cyanobacteria reads showed a strong level of similarity, about 99 %, with an uncultured bacterium sequence (DQ990940.1) (Kuhlman et al. 2006) (Fig. 2 and Online Resource Table S1).
As for the kingdom of Eukarya, PE reads belonging to the Archaeplastida Chlorophyta group, with the families Oocystaceae and Trebuxiaceae, were identified across the analyzed sites. In terms of relative abundance, 30 % of the eukaryotic sequences were assigned to Oocystaceae and 7 % to Trebouxiaceae.
As shown in Fig.3, SanLeo3 was the sample which was most dominated by Eukarya, being 88 % of the sequences assigned to Oocystaceae, followed by SanLeo1 (65 %), SanLeo2 (58 %), and SanLeo5 (35 %). In SanLeo8 and SanLeo7 the percentages of Oocystaceae were low (15 % and 2 %, respectively), but the group of Trebouxiaceae increased by 40 % of the total PE reads in SanLeo8 and by 11 % in SanLeo7. In SanLeo6, the eukaryotic component was almost absent, representing less than 1 % of the sequences.
Furthermore, about half of Eukarya OTUs (corresponding to about 5000 sequences) were assigned to Archaeplastida Streptophyta, with the two families of plants Geraniales and Graminae (data not shown). These sequences are not actually part of the microbial communities and were thus excluded from downstream analyses.
Diversity analyses
As shown in Fig.4a, rarefaction curves approached saturation, suggesting that the majority of the OTUs were recovered in this study; however, since no stationarity was reached, more reads would be required to capture all the diversity. The curves gave identical results for the bacteria alone (left) and the total community (right), with the highest diversity in SanLeo7, followed by SanLeo6, and the lowest diversity in SanLeo2, followed by SanLeo3.

Alpha-diversity analyses. a Rarefaction curves of OTUs clustered at 98 % sequence similarity in the bacterial community alone (left) and total (Bacteria and Eukarya) community (right) across samples. b Observed and estimated OTU richness of bacterial (left) and total community (right) for each sample. The bars show the number of observed (left colored part) and estimated OTUs (right transparent part). Estimations based on Chao1 richness estimator. c Non parametric indexes of richness and diversity of bacterial alone and total community for each sample
Chao richness estimator was applied in order to predict undetected OTUs. As can be observed in Fig. 4b, for both communities, the results evidenced a scenario which is congruent with the rarefaction curve analyses. Nonparametric indicators (Shannon, Simpsoneven and Bergerparker indexes) were used to extrapolate the total richness from the observed OTUs, both as the prokaryotic only and the total components, in the seven sites (Fig. 4c). The bacterial community showed a higher richness than the total microbial population, all over the analyzed areas. Moreover, the Shannon index of the bacterial community was higher as compared with the same index referred to the total community, with the exception of SanLeo6 and SanLeo7, which are characterized by a reduced prevalence of eukaryotic sequences. The simpsoneven index highlighted the lowest value of diversity concerning the total community in SanLeo3, which showed strongly different patterns regarding the total and the bacterial components. The Berger parker index showed a low diversity of the bacterial community in sample SanLeo2, which is heavily characterized by the presence of the two groups of Oceanospirillales and Alteromonadales.
In conclusion, all the used indexes highlighted that the OTU richness varied among the sites, with higher values in SanLeo6 and SanLeo7 and lower ones in SanLeo2, thus confirming the trends that were observed with rarefaction analyses.
The variation in composition and abundance across the samples, as explored by hierarchical clustering analyses, displayed different patterns both for bacteria alone and for the total community (Fig. 5a). Although for the bacterial community no structure in terms of clusters of samples could be revealed, the SanLeo5–SanLeo7 pair could be evidenced. On the other hand, for the total community, two groups could be distinguished. In the first group consisting of SanLeo1, SanLeo3, and SanLeo2, Oocystaceae were dominant, and in the second one containing SanLeo5 and SanLeo7, the percentage of sequences belonging to Actynomycetales and Rubrobacteriales exceeded 50 %. By contrast, SanLeo8 and SanLeo6 were linked with high (>0.70) dissimilarity levels (values ranging from 0 for identical communities to 1 for different communities), suggesting a lack of biological meaning of these links.

Beta diversity analyses. a Cluster tree for the bacterial community (left) and total (Bacteria and Eukaria) community (right). b NMDS analyses for the bacterial community (left) and total community (right)
To compare the community structures of the different sampling sites, we applied non-metric multi-dimensional scaling (NMDS). Separated ordination plots for the bacterial and the total community (Fig. 5b) generally showed scattered patterns; however, a strong similarity in the distribution of OTUs in SanLeo2 and SanLeo3 appeared when the total community was considered. The envfit test showed no significant correlation of the bacterial community with any of the analyzed parameters (location, petrographic composition, porosity, and substrate morphology of the stone substrate sample); on the other hand, when the total community was considered, the porosity (r 2 = 0.7902, p = 0.048) and the petrographic composition (r 2 = 0.8666, p = 0.015) were shown to be significantly related to the variance of the community structures among the sites.
Discussion
This study was performed to reveal the genetic diversity of the epilithic microbial community living on the outdoor carbonate stones of the medieval church of San Leonardo di Siponto (Manfredonia, FG, Italy). The characterization of the microbiome might be useful in order to define the most appropriate treatment for its conservation.
Molecular characterization of the stone-autochthonous microbial community using DNA-based techniques have already been performed, mainly by random amplification of polymorphic DNA (RAPD) fingerprinting (Suihko et al. 2007), denaturing gradient gel electrophoresis (DGGE) analysis (Piñar et al. 2009; Jroundi et al. 2015), amplified 16S rDNA restriction analysis (ARDRA) (Urzì et al. 2001), or sequencing of libraries (Ragon et al. 2012). The NGS technology has been widely used to define the microbial diversity of communities from various ecological niches mainly related to soil (Nesme and Simonet 2015) and human gut (Modi et al. 2014), but limited data are available concerning artistic buildings (Gutarowska et al. 2015).
The results of this study demonstrated that the Illumina-based deep sequencing used here is able to clearly identify the bacterial and the photosynthetic eukaryotic components of the microbiota. The co-amplification of bacterial and eukaryotic sequences using already validated bacterial primer pairs was not surprising, being already reported in the literature (Manzari et al. 2015; Walker and Pace 2007; Starke et al. 2013).
The analyses of the metagenomic samples revealed low bacterial diversity, since PE reads belonging to only seven phylotypes were identified, confirming that stone surfaces are oligotrophic substrata (Berdoulay and Salvado 2009). Besides, the Mediterranean climate, characterized by 3 to 5 months of high temperature and dry conditions, appears to exert a strong selective pressure which limits diversity and determines the ecology of the microbial community made up of extremophile organisms adapted to desiccation and UV radiation. Indeed, this evidence is in agreement with previous reports (Ragon et al. 2012).
This study shows that Archaea represent a minor component of the epilithic community, endorsing the indication that sunlight-exposed surfaces are not the preferential habitat of Archaea (Ragon et al. 2011). We detected reads belonging to the halophilic order Halobacteriales. Actually, this order has been reported as a component of the microbiota of a sub-surface medieval chapel (Piñar et al. 2009). The presence of these organisms, often found in ancient salt deposits, in such different habitats endorses the idea that they already inhabited the materials the monuments were made of (Piñar et al. 2009).
The most abundant bacterial phylotype inhabiting the outdoor stones of San Leonardo was Actinobacteria. This phylum, frequently found on carbonate stones (Berdoulay and Salvado 2009; Jroundi et al. 2010; Jroundi et al. 2015, Sghaier et al. 2016), comprehends heterotrophic bacteria well adapted to severe conditions: indeed Actinobacteria almost show the characteristics of desiccation-related life strategies useful for the survival in extremely dry environments (Mohammadipanah and Wink 2016; Barnard et al. 2013). Our data showed the presence of Geodermatophilaceae, thus confirming the affinity of these organisms to the stone surfaces in the Mediterranean basin area (Urzì et al. 2001). Members of this family have been found to account for almost the totality of cultivable bacteria in highly deteriorated stone monuments, where they contribute not only to formation of differently colored patinas and spots but also to the deterioration by bio-pitting and powdering (Urzì and Realini 1998). However, it is noteworthy to consider that the phylum of Actinobacteria also comprehends carbonatogenic bacteria, which can be a useful tool for environmentally friendly stone consolidation thanks to their high bio-mineralization capacity (Jroundi et al. 2015). In this study, among the more abundant sequences, those belonging to the genus Arthrobacter, able to precipitate calcium carbonate (Jroundi et al. 2012), were identified.
Moreover, sequences belonging to the moderate halophilic, dessication-, UV- and ionizing radiation-resistant genus Rubrobacter (Ragon et al. 2011) were also abundant in our samples. These organisms are commonly found in biodeteriorated monuments (Ettenauer et al. 2014), where they play a significant role in the aesthetical changes due to the secretion of biogenic pigments (Laiz et al. 2009).
These results endorse the importance of a deeper knowledge of the composition of the microbial community in order to evaluate the possible roles of the different components in the deterioration or even in the conservation of heritage stone (Pinna 2014).
As for the characterization of the epilithic community in terms of ecological interactions, the Illumina-based sequencing used in this study has clearly shown a structured community, where the photoautotrophic cyanobacteria and green algae have the role of pioneering primary producers, providing organic carbon to the heterotrophic members of the community. In all but one of the sites, the microbial photosynthetic eukaryotic component was present. In particular, SanLeo3 was almost exclusively dominated by photosynthetic green algae. It is worth of note that such a kind of dominance seems to be peculiar for this site, since the other samples showed a diversity relying on a certain number of different prokaryotic and eukaryotic OTUs at intermediate frequencies. Among the algae, Chlorophyta are the ones most frequently found on monument stones (Ortega-Calvo et al. 1993). In this study, only sequences belonging to the families Oocystaceae and Trebouxiaceae, with the classes Chlorellales and Trebouxiales, respectively, were observed. Chlorellales are indeed among the most common green algae present in monuments worldwide, without any specific preference for a substratum or a climate (Macedo et al. 2009). On the other hand, the algae Trebouxiaceae are extremophile organisms, frequently found as phycobionts of lichens, well adapted to harsh environments such as building stones characterized by desiccation and exposure to radiations (Ragon et al. 2012; Ragon et al. 2011). The hierarchical analyses clearly showed an Oocystaceae-dominated cluster and a less defined group characterized by the presence of Trebouxiaceae. The different kinds of domination that were found among the samples are in agreement with the notion that the physical characteristics of the substrate are the driving force for the colonization of Trebouxiaceae (Tiano et al. 1995), although a significant role has also been attributed to sun exposition (Hallmann et al. 2013).
Moreover, in this study, the presence of photosynthetic Cyanobacteria, in combination with the absence of green algae, was evidenced in only one of the sampled sites, which shows peculiar petrographic characteristics and texture. Cyanobacteria PE reads produced in this study showed 99 % similarity with an uncultured bacterium sequence (DQ990940.1), closely related to the radiation-resistant Chroococcidiopsis sp. (Kuhlman et al. 2006). Species belonging to the genus Chroococcus are among the most common and ubiquitous cyanobacteria inhabiting building stones in the Mediterranean Basin and beyond (Macedo et al. 2009).
Indeed, the investigation of alpha- and beta-microbial diversity highlighted a small-scale variety of epilithic communities. According to what has been described in a previous study (Portillo et al. 2009), the inhospitable external macro-conditions allow few specific taxa to drive microbial colonization of the spatially different micro-environments. It appears that each spot, although at a short distance from the others, may form an insulated and protected niche able to produce a very dissimilar microcosm (de Wit and Bouvier 2006; Friedmann and Ocampo-Friedmann 1984). Thus, our findings suggested a physiological adaptation of the microbial populations to the environment (light exposure, high temperatures, low water availability, and absence of organic carbon sources) resulting in multiple survival strategies, with a significant influence of the petrographic characteristics and porosity of the stones on the structuration of the communities, as highlighted by the correlation analysis. The low amount of stone material that could be sampled from the historic monument was a limitation for a more comprehensive characterization of the stone.
Our results clearly emphasize the effectiveness of the spot approach here used, which is analyzing the representative complex samples separately, more than getting the whole composition of a pooled composite sample.
In conclusion, our investigation demonstrated that molecular tools, and in particular the easy-to-run next generation sequencing, are important to accomplish an accurate microbiological investigation, which can be explored further to plan better strategies for restoration and protection of historical and artistic monuments.
This pivotal study did not analyze the fungal component; however, additional analyses are in progress in order to characterize also this important portion of the microbial community.
References
Andriani G, Walsh N (2003) Fabric, porosity and water permeability of calcarenites from Apulia (SE Italy) used as building and ornamental stone. Bull Eng Geol Environ 62:77–84
Barnard RL, Osborne CA, Firestone MK (2013) Responses of soil bacterial and fungal communities to extreme desiccation and rewetting. ISME J 7(11):2229–2241. doi:10.1038/ismej.2013.104
Berdoulay M, Salvado JC (2009) Genetic characterization of microbial communities living at the surface of building stones. Lett Appl Microbiol 49(3):311–316. doi:10.1111/j.1472-765X.2009.02660.x
Chakravorty S, Helb D, Burday M, Connell N, Alland D (2007) A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria. J Microbiol Methods 69(2):330–339
Dakal TC, Cameotra SS (2012) Microbially induced deterioration of architectural heritages: routes and mechanisms involved. Environmental Sciences Europe 24:36
de Castro AP, Sartori da Silva MR, Quirino BF, Kruger RH (2013) Combining “omics” strategies to analyze the biotechnological potential of complex microbial environments. Curr Protein Pept Sci 14(6):447–458
de Wit R, Bouvier T (2006) ‘Everything is everywhere, but, the environment selects’; what did baas Becking and Beijerinck really say? Environ Microbiol 8(4):755–758
Decelle J, Romac S, Stern RF, Bendif el M, Zingone A, Audic S, Guiry MD, Guillou L, Tessier D, Le Gall F, Gourvil P, Dos Santos AL, Probert I, Vaulot D, de Vargas C, Christen R (2015) PhytoREF: a reference database of the plastidial 16S rRNA gene of photosynthetic eukaryotes with curated taxonomy. Mol Ecol Resour 15(6):1435–1445. doi:10.1111/1755-0998.12401
Ditaranto N, Loperfido S, van der Werf I, Mangone A, Cioffi N, Sabbatini L (2011) Synthesis and analytical characterisation of copper-based nanocoatings for bioactive stone artworks treatment. Anal Bioanal Chem 399:473–481. doi:10.1007/s00216-010-4301-8
Ditaranto N, van der Werf ID, Picca RA, Sportelli MC, Giannossa LC, Bonerba E, Tantillo G, Sabbatini L (2015) Characterization and behaviour of ZnO-based nanocomposites designed for the control of biodeterioration of patrimonial stoneworks. New J Chem 39:6836–6843. doi:10.1039/C5NJ00527B
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27(16):2194–2200. doi:10.1093/bioinformatics/btr381
Ettenauer JD, Jurado V, Piñar G, Miller AZ, Santner M, Saiz-Jimenez C, Sterflinger K (2014) Halophilic microorganisms are responsible for the rosy discolouration of saline environments in three historical buildings with mural paintings. PLoS One 9(8):e103844. doi:10.1371/journal.pone.0103844
Friedmann EI, Ocampo-Friedmann R (1984) The Antarctic cryptoendolithic ecosystem: relevance to exobiology. Orig Life 14(1–4):771–776
Guillitte O (1995) Bioreceptivity: a new concept for building ecology studies. Sci Total Environ 167:215–220
Gutarowska B, Celikkol-Aydin S, Bonifay V, Otlewska A, Aydin E, Oldham AL, Brauer JI, Duncan KE, Adamiak J, Sunner JA, Beech IB (2015) Metabolomic and high-throughput sequencing analysis-modern approach for the assessment of biodeterioration of materials from historic buildings. Front Microbiol 6:979. doi:10.3389/fmicb.2015.00979
Hallmann C, Stannek L, Fritzlar D, Hause-Reitner D, Friedl T, Hoppert M (2013) Molecular diversity of phototrophic biofilms on building stone. FEMS Microbiol Ecol 84(2):355–372. doi:10.1111/1574-6941.12065
Hueck HJ (2001) The biodeterioration of materials—an appraisal. Int Biodeterior Biodegrad 48:5–11
Jroundi F, Fernández-Vivas A, Rodriguez-Navarro C, Bedmar EJ, González-Muñoz MT (2010) Bioconservation of deteriorated monumental calcarenite stone and identification of bacteria with carbonatogenic activity. Microb Ecol 60(1):39–54. doi:10.1007/s00248-010-9665-y
Jroundi F, Gómez-Suaga P, Jimenez-Lopez C, González-Muñoz MT, Fernandez-Vivas MA (2012) Stone-isolated carbonatogenic bacteria as inoculants in bioconsolidation treatments for historical limestone. Sci Total Environ 425:89–98. doi:10.1016/j.scitotenv.2012.02.059
Jroundi F, Gonzalez-Muñoz MT, Sterflinger K, Piñar G (2015) Molecular tools for monitoring the ecological sustainability of a stone bio-consolidation treatment at the Royal Chapel, Granada. PLoS One 10(7):e0132465. doi:10.1371/journal.pone.0132465
Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD (2013) Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol 79(17):5112–5120. doi:10.1128/AEM.01043-13
Kuhlman KR, Fusco WG, La Duc MT, Allenbach LB, Ball CL, Kuhlman GM, Anderson RC, Erickson IK, Stuecker T, Benardini J, Strap JL, Crawford RL (2006) Diversity of microorganisms within rock varnish in the Whipple Mountains, California. Appl Environ Microbiol 72(2):1708–1715. doi:10.1128/AEM.72.2.1708-1715.2006
Laiz L, Miller AZ, Jurado V, Akatova E, Sanchez-Moral S, Gonzalez JM, Dionísio A, Macedo MF, Saiz-Jimenez C (2009) Isolation of five Rubrobacter strains from biodeteriorated monuments. Naturwissenschaften 96(1):71–79. doi:10.1007/s00114-008-0452-2
Macedo MF, Miller AZ, Dionísio A, Saiz-Jimenez C (2009) Biodiversity of cyanobacteria and green algae on monuments in the Mediterranean Basin: an overview. Microbiology 155(Pt 11):3476–3490. doi:10.1099/mic.0.032508-0
Mann S (2001) Biomineralization: principles and concepts in bioinorganic materials chemistry. Oxford University Press, Oxford, United Kingdom
Manzari C, Fosso B, Marzano M, Annese A, Caprioli R, D’Erchia AM, Gissi C, Intranuovo M, Picardi E, Santamaria M, Scorrano S, Sgaramella G, Stabili L, Piraino S, Pesole G (2015) The influence of invasive jellyfish blooms in a coastal lagoon (Varano, SE Italy) detected by an Illumina-based deep sequencing strategy. Biol Invasions 17:923–940. doi:10.1007/s10530-014-0810-2
Modi SR, Collins JJ, Relman DA (2014) Antibiotics and the gut microbiota. J Clin Invest 124(10):4212–4218. doi:10.1172/JCI72333
Mohammadipanah F, Wink J (2016) Actinobacteria from arid and desert habitats: diversity and biological activity. Front Microbiol 6:1541. doi:10.3389/fmicb.2015.01541
Nesme J, Simonet P (2015) The soil resistome: a critical review on antibiotic resistance origins, ecology and dissemination potential in telluric bacteria. Environ Microbiol 17(4):913–930. doi:10.1111/1462-2920.12631
Oksanen J, Blanchet FG, Kindt R, Legendre P, O’Hara RB, Simpson GL, Solymos P, Stevens MHH, Wagner H (2013) Vegan: community ecology package. R Packag Version 2:7
Ortega-Calvo JJ, Hernandez-Marine M, Saiz-Jimenez C (1993) Cyanobacteria and algae on historic buildings and monuments. In Naya Prokash (ed) Recent advances in biodeterioration and degradation 1: biodeterioration of cultural heritage, pp 173–203
Piñar G, Ripka K, Weber J, Sterflinger K (2009) The micro-biota of a sub-surface monument the medieval chapel of St. Virgil (Vienna, Austria). Int Biodeterior Biodegrad 63:851–859. doi:10.1016/j.ibiod.2009.02.004
Pinna D (2014) Biofilms and lichens on stone monuments: do they damage or protect? Front Microbiol 5:133. doi:10.3389/fmicb.2014.00133
Portillo MC, Alloza R, Gonzales JM (2009) Three different phototrophic microbial communities colonizing a single natural shelter containing prehistoric paintings. Sci Total Environ 407(17):4876–4881. doi:10.1016/j.scitotenv.2009.05.038
Ragon M, Restoux G, Moreira D, Møller AP, López-García P (2011) Sunlight-exposed biofilm microbial communities are naturally resistant to Chernobyl ionizing-radiation levels. PLoS One 6(7):e21764. doi:10.1371/journal.pone.0021764
Ragon M, Fontaine MC, Moreira D, López-García P (2012) Different biogeographic patterns of prokaryotes and microbial eukaryotes in epilithic biofilms. Mol Ecol 21(15):3852–3868. doi:10.1111/j.1365-294X.2012.05659.x
Santhosh K, Ramachandran SK, Ramakrishnan V, Bang SS (2001) Remediation of concrete using microrganisms. Am Conc Inst Mater J 98:3–9
Scheerer S, Ortega-Morales O, Gaylarde C (2009) Microbial deterioration of stone monuments—an updated overview. Adv Appl Microbiol 66:97–139. doi:10.1016/S0065-2164(08)00805-8
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75(23):7537–7541. doi:10.1128/AEM.01541-09
Sghaier H, Hezbri K, Ghodhbane-Gtari F, Pujic P, Sen A, Daffonchio D, Boudabous A, Tisa LS, Klenk HP, Armengaud J, Normand P, Gtari M (2016) Stone-dwelling actinobacteria Blastococcus saxobsidens, Modestobacter marinus and Geodermatophilus obscurus proteogenomes. ISME J 10(1):21–29. doi:10.1038/ismej.2015.108
Smith DP, Peay KG (2014) Sequence depth, not PCR replication, improves ecological inference from next generation DNA sequencing. PLoS One 9(2):e90234. doi:10.1371/journal.pone.0090234
Starke V, Kirshtein J, Fogel ML, Steele A (2013) Microbial community composition and endolith colonization at an Arctic thermal spring are driven by calcite precipitation. Environ Microbiol Rep 5(5):648–659. doi:10.1111/1758-2229.12063
Sterflinger K, Piñar G (2013) Microbial deterioration of cultural heritage and works of art—tilting at windmills? Appl Microbiol Biotechnol 97(22):9637–9646. doi:10.1007/s00253-013-5283
Suihko ML, Alakomi HL, Gorbushina A, Fortune I, Marquardt J, Saarela M (2007) Characterization of aerobic bacterial and fungal microbiota on surfaces of historic Scottish monuments. Syst Appl Microbiol 30(6):494–508
Tiano P, Accolla P, Tomaselli L (1995) Phototrophic biodeteriogens on lithoid surfaces: an ecological study. Microb Ecol 29(3):299–309. doi:10.1007/BF00164892
Tringe SG, Hugenholtz P (2008) A renaissance for the pioneering 16S rRNA gene. Curr Opin Microbiol 11(5):442–446. doi:10.1016/j.mib.2008.09.011
Urzì C, Realini M (1998) Colour changes of Noto’s calcareous sandstone as related to its colonisation by microorganisms. Int Biodeterior Biodegrad 42:45–54. doi:10.1016/S0964-8305(98)00045-6
Urzì C, Brusetti L, Salamone P, Sorlini C, Stackebrandt E, Daffonchio D (2001) Biodiversity of Geodermatophilaceae isolated from altered stones and monuments in the Mediterranean basin. Environ Microbiol 3(7):471–479
van der Werf ID, Ditaranto N, Picca RA, Sportelli MC, Sabbatini L (2015) Development of a novel conservation treatment of stone monuments with bioactive nanocomposites. Heritag Sci 3:29. doi:10.1186/s40494-015-0060-3
Walker JJ, Pace NR (2007) Phylogenetic composition of Rocky Mountain endolithic microbial ecosystems. Appl Environ Microbiol 73(11):3497–3504
Ward DM, Weller R, Bateson MM (1990) 16S rRNA sequences reveal numerous uncultured microorganisms in a natural community. Nature 345(6270):63–65
Warscheid T, Braams J (2000) Biodeterioration of stone: a review. Int Biodeterior Biodegrad 46(4):343–368. doi:10.1016/S0964-8305(00)00109-8
Acknowledgments
The authors would like to thank Prof. P. Acquafredda (Dept. Scienze della Terra e Geoambientali, University of Bari “Aldo Moro”) for the optical microscopy study of the stone samples and Architect Antonello D’Ardes (Manfredonia, Italy) for providing logistic support. This work was performed in the framework of the PRIN 2010–2011 “Sustainability in cultural heritage: from diagnosis to the development of innovative systems for consolidation, cleaning and protection” code 2010329WPF, funded by MIUR (Italy) and used the facilities of the Molecular Biodiversity Laboratory (MoBiLab) of LifeWatch-Italy. I.D. van der Werf is supported by the Future in Research program financed by the Apulian Region (Italy).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 367 kb)
Rights and permissions
About this article

Cite this article
Chimienti, G., Piredda, R., Pepe, G. et al. Profile of microbial communities on carbonate stones of the medieval church of San Leonardo di Siponto (Italy) by Illumina-based deep sequencing. Appl Microbiol Biotechnol 100, 8537–8548 (2016). https://doi.org/10.1007/s00253-016-7656-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-016-7656-8