
Abstract
Intense rainfall is one of the most serious and common natural events, causing the excessive inflow of rainwater into wastewater treatment plants. However, little is known about the impacts of rainwater dilution on the structure and function of the sludge microorganisms. Here, high-throughput sequencing of 16S ribosomal RNA (rRNA) genes was implemented to describe the microbial community dynamics during the simulated intense rainfall situation (event i) in which approximately 45 % of the sludge biomass was artificially overflowed by massive water supply in a pilot-scale membrane bioreactor. Thereafter, we investigated the functional and structural responses of the perturbed microbial communities to subsequent conditional changes, i.e., an increase in organic loading rate from 225 to 450 mg chemical oxygen demand (COD) l−1 day−1 (event ii) and an addition of a microbiota activator (event iii). Due to the event i, the COD removal declined to 78.2 %. This deterioration coincided with the decreased microbial diversity and the proliferation of the oligotrophic Aquabacterium sp. During the succeeding events ii and iii, the sludge biomass increased and the COD removal became higher (86.5–97.4 %). With the apparent recovery of the reactor performance, microbial communities became diversified and the compositions dynamically changed. Notably, various bacterial micropredators were highly enriched under the successive conditions, most likely being involved in the flexible reorganization of microbial communities. These results indicate that the activated sludge harbored functionally redundant microorganisms that were able to thrive and proliferate along with the conditional changes, thereby contributing to the functional maintenance of the membrane bioreactor.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Membrane bioreactors (MBRs) have been widely used as an advanced wastewater treatment technology for gaining better quality of treated water compared with that obtained from the conventional activated sludge method. An important advantage of MBRs is its ability to maintain high concentrations of biomass (i.e., the sludge microorganisms), which permits the stable and efficient degradation of high-strength organic wastewaters (Le-Clech 2010). During the practical operation for municipal and industrial wastewater treatment, MBRs are often subjected to various conditional changes such as hydraulic shock, variable wastewater composition, and organic load fluctuation within short time periods, potentially affecting reactor performances and microbial communities in the activated sludge (Yang et al. 2006; Sozen et al. 2014).
Intense rainfall is one of the most serious and common natural events, inducing the excessive inflow of rainwater into wastewater treatment plants (Kessler 2011; Veltmann et al. 2011). Due to this situation, the sludge concentration is diluted and feed to microorganisms (F/M) ratio suddenly decreases. There have been reports investigating the effect of rain events on wastewater treatment efficiency, but most of which have focused mainly on the relationship between reactor operation and architectural countermeasure (Chen and Beck 2001; Wilen et al. 2006). Little is known about the impacts of rainwater dilution on the structure and function of the sludge microbial communities in wastewater treatment plants. More specifically, it is yet to be clarified whether the rainwater–diluted sludge microbial communities can maintain their degradation ability and how the once functionally disrupted microbial communities will respond to the other environmental stimuli such as the changes in organic loading and feed composition, which can occur in actual wastewater treatment plants. These evaluations are of particular interest in closed MBR systems wherein activated sludge washout is prevented, resulting in the low physical replacement of the sludge microorganisms.
Recently, high-throughput sequencing of 16S ribosomal RNA (rRNA) gene amplicons has been employed to study the microbial community structure and function in various natural ecosystems (Aoyagi et al. 2015a; Navarro et al. 2015). We have so far revealed that the combined use of Illumina sequencing and process analyses was effective for the sensitive and precise assessment of the microbial community dynamics under successive conditions in a pilot-scale MBR (Sato et al. 2015a; Sato et al. 2015b). Here, the MBR was subjected to the artificial overflow of its activated sludge by massive water supply in order to simulate the rainwater dilution of the sludge biomass during intense rainfall. Our main objective was to investigate the microbial community dynamics during the water-dilution perturbation (event i) in a pilot-scale MBR by high-throughput Illumina sequencing of 16S rRNA genes. Furthermore, in order to gain a better understanding of the functional stability and structural flexibility of the perturbed microbial communities, we also aimed to clarify their transition behaviors in response to the subsequent conditional changes, i.e., an increase in organic loading rate (OLR) from 225 to 450 mg chemical oxygen demand (COD) l−1 day−1 (event ii) followed by the addition of a microbiota activator (MA) (event iii).
Materials and methods
Setup and operation of a pilot-scale MBR
Schematic configuration of the pilot-scale MBR used in this study is shown in Fig S1. The reactor has three compartments with operating volumes of 92.0, 80.5, and 57.5 L. A 0.24-m2 flat polyacrylonitrile membrane module (M-fine; Awa Paper Mfg, Co., Tokushima, Japan) with 0.07-μm pore size was fixed in the rightmost compartment. The membrane module was operated with a cycle of permeate extraction for 9 min and a pause for 1 min. Air was provided through an air diffuser set in each compartment at a flow rate of 12.5–30.0 l min−1 for mixing the activated sludge and for controlling dissolved oxygen (DO). The submerged membrane surface was also aerated continuously to reduce membrane fouling. The employed activated sludge was obtained from a municipal wastewater treatment plant (Kinu aqua-station, Ibaraki, Japan). Throughout the experimental period, the bioreactor was constantly fed with a synthetic wastewater stored in a 20-l feed tank at 4 °C. The flow rates of both the input wastewater and the output membrane-filtered treated water were 115 l day−1, resulting to a hydraulic retention time (HRT) of 2 days. The flow of the return sludge from the rightmost to leftmost compartments was also set at 115 l day−1. No sludge was withdrawn from the reactor, except for the washout from the simulated intensive rainfall as well as from periodic sampling. The concentration of the original synthetic wastewater was set at 450 mg COD l−1 and this contained (g l−1) CH3COONa, 2.65; NH4Cl, 0.376; KH2PO4, 0.109; peptone, 0.706; as well as trace elements (mg l−1): FeCl3·6H2O, 0.782; CaCl2, 1.56; MgSO4, 1.56; KCl, 1.56; NaCl, 1.56.
Operational conditions of the MBR
The artificial changes in operational conditions are as described below. Before sampling at day 1, the MBR was pre-operated for 12 days and it was confirmed that the reactor performance had reached steady state. From days 1 to 5, the MBR was operated continuously using 450 mg COD l−1 (1130 TOC mg l−1) of synthetic wastewater. After sampling at day 5, a heavy rainfall event was simulated by the excessive feeding of running water, resulting in the overflow of approximately 45 % of activated sludge (event i). From days 5 to 22, the MBR was operated at the original conditions to monitor the response of microbial communities to the simulated water-dilution disturbance. Just after the sampling point at day 22, the concentration of organic matter in the synthetic wastewater was adjusted to 900 mg COD l−1 (event ii) by simply doubling the amount of each component. This condition was maintained until the end of the experiment (day 39). As an additional event, 5 kg of MA placed in a perforated cylindrical metal container (BRcnt-5E; CNT Corporation, Kagawa, Japan) was supplemented at day 31 to the second compartment of the MBR to further stimulate the sludge microbial communities (event iii). The MA is composed of both porous pumice stones for microbial habitants and mixed organic-mineral pellets for biostimulation. The pellet contains 27.50–31.68 % organic substances, 0.93–1.42 % total nitrogen, 50.83–52.00 % SiO2, 2.80–4.73 % Fe, 0.19–0.71 % CaO, 0.09–0.16 % K2O, 0.15–0.24 % MgO, <0.14 % P2O5, and others (10.16–16.17 %). In this regard, events ii and iii were carried out based on our previous report that confirmed their ability to improve MBR performance, albeit in the absence of the water-dilution perturbation (Sato et al. 2015a; Sato et al. 2015b). The mixed liquor suspended solids (MLSS), temperature, DO, and pH in all three compartments and the transmembrane pressure (TMP) of the membrane module were monitored throughout the experimental period. Fifteen-milliliter samples of the activated sludge in the first and second compartments of the MBR as well as the effluent (i.e., treated water) were taken at daily intervals for chemical and microbial analyses.
Analytical procedures for chemical analyses
The samples were centrifuged (15,300×g, 15 min, 4 °C) and the resulting supernatant was further filtered using a cellulose acetate membrane (Φ, 0.20 μm, C020A025A; ADVANTEC, Tokyo, Japan). The supernatant and treated water were analyzed according to the previous reports (Navarro et al. 2015; Sato et al. 2015b). Briefly, COD was measured with a COD analyzer (DR2800 and DRB200; Hach, Loveland, CO, USA) using appropriate kits (TNT820 or TNT821, Hach). The concentrations of NH4 +, NO2 −, and NO3 − were determined using capillary electrophoresis (CE; Agilent, Santa Clara, CA, USA). All data were represented as the mean values from at least two different sampling points of the reactor.
DNA extraction and PCR amplification
Fifty-six activated sludge samples periodically obtained from the two different sampling points (i.e., first and second compartments) of the MBR were washed once with a 50 mM sodium phosphate buffer (pH 7.0) and stored at −20 °C as a pellet until use. DNA was extracted from a 50 mg of activated-sludge pellet according to a direct lysis protocol that includes chemical lysis, bead-beating, phenol–chloroform extraction, and ethanol precipitation (Noll et al. 2005). RNA was digested with RNase (Type II-A; Sigma-Aldrich, St. Louis, MO, USA). Purified DNA was quantified using NanoDrop Lite (Thermo Fisher Scientific, Waltham, MA, USA), and was used as template for PCR amplification with a high-fidelity DNA polymerase (Q5; NEB, Ipswich, MA, USA). The V4 region of 16S rRNA genes was amplified using the universal primers 515F and 806R (Caporaso et al. 2012). Both primers were modified to contain an Illumina adapter region, and the reverse primer contained a 12-bp barcode for multiplex sequencing (Bartram et al. 2011). The thermal condition of PCR was the same as that in a previous report (Hori et al. 2014), except for the 30–35 cycle numbers employed.
High-throughput Illumina sequencing of 16S rRNA gene amplicons
High-throughput sequencing was performed as described previously (Aoyagi et al. 2015b). Briefly, the PCR products were purified using an AMPure XP kit (Beckman Coulter, Brea, CA, USA). The resulting DNA solution was subjected to agarose-gel electrophoresis and the target DNA band was excised. Recovery of DNA in the gel slice was performed with a Wizard SV Gel and PCR Clean-Up System (Promega, Madison, WI, USA). The DNA concentration was determined spectrophotometrically using a Quant-iT PicoGreen dsDNA reagent and kit (Life Technologies, Carlsbad, CA, USA). An appropriate amount of the 16S rRNA gene segments and an internal control (PhiX Control V3; Illumina, San Diego, CA, USA) were subjected to paired-end sequencing with a 300-cycle MiSeq reagent kit (Illumina) and a MiSeq sequencer (Illumina). Removal of PhiX, low-quality (Phred value score [Q], <30), and chimeric sequences and assembly of the paired-end sequences were performed according to a previous report (Itoh et al. 2014). Contaminated PhiX sequences in the Illumina sequence libraries were detected using a homology search against the Greengenes database (DeSantis et al. 2006) using the Burrows–Wheeler Aligner, version 4.0.5 (Li and Durbin 2009). The PhiX sequences were then removed from the library by self-written scripts. The paired-end sequences were joined using a fastq-join tool in the ea-utils software package (https://code.google.com/p/ea-utils/downloads/list), version 1.1.2-301 (Aronesty 2013). The joined sequences with Q scores of ≥30 were collected using the software package QIIME, version 1.7.0 (Caporaso et al. 2010) and aligned using the program mothur, version 1.31.2 (Schloss et al. 2009); thereafter, the chimeric sequences were detected and excluded from the library. The sequences in each library were characterized phylogenetically using the QIIME software package (Caporaso et al. 2010). The α-diversity indices (i.e., Chao1, Shannon, and Simpson reciprocal) and the weighted UniFrac distances for principal coordinate analysis (PCoA) were calculated using the QIIME software package. For the calculations, a total of 56 libraries with more than 15,406 sequences were used. Two libraries were ruled out from the diversity analyses due to the low sequences obtained. The closest cultured relatives of the identified operational taxonomic units (OTUs) were further determined based on the BLAST search (http://blast.ncbi.nlm.nih.gov/Blast.cgi) by comparing the sequenced 16S rRNA genes with those in the nucleotide sequence database. The raw sequence data in this study were deposited in the MG-RAST database (http://metagenomics.anl.gov/) as a “Microbial community dynamics in a perturbed MBR 2015” project under the following IDs: 4675246.3, 4675249.3, 4675251.3, 4675253.3, 4675256.3, 4675258.3–4675280.3.
Results
Changes in reactor performance throughout the operation of a pilot-scale MBR
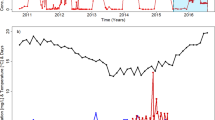
The MLSS decreased from 3770 mg l−1 at day 4 to 2067 mg l−1 at day 8 (Fig. 1a), indicating approximately 45 % reduction of the total biomass within the MBR. Thereafter, the reactor was stably operated without any artificial stimuli and its MLSS increased to 4270 mg l−1 at day 19. Nonetheless, the COD removal ratio gradually decreased due to the event i, ranging from 90.6 % at day 4 to 78.2 % at day 19 (Fig. 1c). The response capability of the once functionally disrupted microbial communities to short-term conditional changes was then evaluated. The concentration of synthetic wastewater was doubled from 450 to 900 mg COD l−1 after sampling at day 22 (event ii). Furthermore, an MA was supplemented into the MBR after the sampling at day 31 (event iii). Both events resulted to substantial increases in MLSS, reaching 7067 mg l−1 at day 26 (after the event ii) and 13,300 mg l−1 at day 39 (after the event iii). These imply that the high organic loading and the MA addition facilitated the microbial growth in the activated sludge. Although the addition of this MA was demonstrated to be effective to reduce the accumulated ammonium concentration in the reactor (Sato et al. 2015a), such phenomenon was not observed in the present operation (Fig. S2). The DO dropped to 0.11 mg l−1 at day 24 (i.e., just after the event ii) and afterwards remained at <0.87 mg l−1 (Fig. 1b), suggesting that the sludge microorganisms consumed most of the supplied oxygen to degrade the high-strength organic wastewater so that the reactor conditions became microaerobic and/or presumably partially anaerobic. During the operation after events ii and iii, the COD removal ratios were maintained at high levels (86.5–97.4 %) even under the high OLR conditions. The values of TMP prominently increased from 9 kPa at day 36 to 30 kPa at day 39, during which the effluent flow rate rapidly decreased (Fig. 1d), indicating the initiation of membrane fouling in this period.

Changes in physicochemical parameters of the membrane bioreactor during the operation. a MLSS in the sludge. The arrows indicate the timing of the three events. b DO in the sludge. c COD in the effluent. d Closed circle, TMP of the membrane module; open circle, effluent flow rate
Changes in the microbial diversity indices throughout the operation
The sludge microbial communities in the MBR were examined by means of high-throughput Illumina sequencing of 16S rRNA genes. The total number of the sequences obtained from a total of 56 different sludge samples was around 1.65 million, corresponding to an average of 29,388 sequences per library (minimum, 3739; maximum, 60,137; and standard deviation, 11,869). Because the structural patterns of the microbial communities from two sampling points were similar (Fig. S3a, b), the data from the second compartment was used as a representative for presentation. Alpha diversity indices (i.e., Chao1, Shannon, and Simpson reciprocal) based on the Illumina sequence data are shown in Fig. 2. The Chao1 index denotes the richness of microbial species to predict the total number of phylogenetically distinct species, while the indices of Shannon and Simpson reciprocal give indications of the evenness as well as the richness of microbial communities, emphasizing the effect from rare species and predominant species, respectively (Hill 1973). These diversity indices showed different trends in their variation with time. Just after the artificial water dilution (event i), the Chao1 values were kept relatively stable, albeit on a declining trend after day 15 (Fig. 2a), whereas the microbial evenness indices drastically decreased (Fig. 2b, c). Compared to those at day 5 (before the event i; representative of stable operation), the values of Chao1, Shannon, and Simpson reciprocal at day 22 decreased by 50.7, 46.2, and 84.7 %, respectively. These indicate that the simulated intense rainfall situation reduced the microbial diversity (i.e., both the richness and evenness). Despite their gradual increase after events ii and iii, the values of Chao1, Shannon, and Simpson reciprocal were recovered not fully but only to some degree, accounting for 94.2, 80.5, and 58.3 % at the end of the operation as compared to their respective values at day 5.

Alpha diversity indices calculated based on the obtained sequence data. a Chao1, b Shannon, and c Simpson reciprocal. Each index was calculated based on an equal amount of sequences (15,406) sub-sampled ten times from original libraries (mean ± SD). For each index, higher values represent more diverse microbial communities. The symbols denote the operational days: closed circles, days 1–5; open circles, days 8–22; closed triangles, days 23–31; open triangles, days 32–39. The arrows indicate the timing of the three events
Comparison of whole microbial community structure throughout the operation
Community similarities were evaluated through the operation by principal coordinate analysis (PCoA) of the Illumina sequence data based on weighted UniFrac distances (Fig. 3). The PCoA scatter plot indicated the marked shifts of microbial communities associated with the artificial conditional changes, i.e., events i to iii. In detail, the operational periods with stable reactor performance (i.e., days 1–5) were clustered closely at the upper left on the plot. After the artificial water dilution of the activated sludge (event i), the microbial community structure drastically shifted to the middle right on the plot. After implementing the high organic loading (event ii), we found the reorganization of microbial communities at days 30–31 that were more similar to those observed before the event i (days 1–5) than those observed before event ii (days 8–22) in terms of the value of PC1. The operational periods after adding an MA (event iii), i.e., days 32–38, were clustered in a group with days 30–31 on the plot, suggesting that the MA supplementation had less impact on the changes in the microbial community structure. The final time point, i.e., day 39, at which the TMP drastically increased, was located rather far from days 30–38.

PCoA scatter plot of 16S rRNA sequences based on the weighted UniFrac obtained by Illumina sequencing of 26 samples from activated sludge. The symbols denote the operational days: closed circles, days 1–5; open circles, days 8–22; closed triangles, days 23–31; open triangles, days 32–39. The numbers beside the plots indicate their respective sampling time
Compositional changes of microbial communities throughout the operation
The class- and genus-level phylogenetic analyses of Illumina sequence data using the QIIME and mothur software were conducted to clarify the changes in composition of the sludge microbial communities throughout the operation (Fig. 4). Alpha-, β- and γ-proteobacteria, Bacteroidetes, and Sphingobacteriia dominated the initial activated sludge, accounting for 11.6, 44.2, 8.4, 4.0, and 23.0 %, respectively, of the total sludge biomass (Fig. 4a). This composition is similar to those found in the other activated sludge treatment plants (Hu et al. 2012; Wang et al. 2012). Firstly, the artificial water dilution (event i) at day 5 affected differently these predominant microbial groups. Beta-proteobacteria increased to a relative abundance of 66.1 % at day 8; the most dominant genus of this class was Aquabacterium (39.1 %) (Fig. 4b). Afterwards, Aquabacterium spp. continued to increase and accounted for 62.8 % of the total at day 22. Though Azospira spp. also dominated the class β-proteobacteria, its relative abundance decreased remarkably from 10.9 % at day 8 to 0.07 % at day 22. With respect to the dominant microbial groups other than β-proteobacteria, the relative abundances of Sphingobacteriia and Bacteroidetes decreased to 0.1 and 3.6 %, respectively, at day 22. Meanwhile, the abundance of γ-proteobacteria and α-proteobacteria remained rather stable from day 5 to 22. Aside from these groups, the class Flavobacteriia increased to a relative abundance of 7.2 % at day 22. Secondly, after employing the high OLR conditions (event ii), γ-proteobacteria consisting mainly of the genus Luteimonas increased to a relative abundance of 21.5 % at day 31. Although Sphingobacteriia declined during the water-dilution perturbation (after the event i; days 8–22), it recovered and dominated again the microbial communities, reaching 15.7 % of the total at day 31. Flexibacteraceae was the major family of the increased Sphingobacteriia population. The third conditional change (event iii), i.e., supplementation of an MA, triggered the increases in the classes Clostridia (mainly composed by Fusibacter spp. and Clostridiaceae bacteria) and Bacteroidia (mainly the Porphyromonadaceae family). Within the class β-proteobacteria, the genus Aquabacterium representing the largest proportion during events i and ii (days 8–31) was notably reduced after the event iii, and eventually its initial predominance was comparable with that of the other beta-proteobacterial genus Acidovorax.

Structural changes of the microbial community during the membrane bioreactor operation. Microbial community structure in activated sludge sample was analyzed by high-throughput sequencing of 16S rRNA genes. Relative distributions of the sequences at class (a) and genus (b) level are shown. The classes and genera which showed more than 3 % in relative abundance are displayed individually. The higher taxonomic category is listed for class or genus that QIIME analysis was unable to identify. The arrows indicate the timing of the three events. The colored version of this figure is available online
Transition of the OTUs throughout the operation
The OTU-level phylogenetic analysis of Illumina sequence data demonstrated microbial species responding to the three different conditional changes, i.e., artificial water dilution (event i), high OLR application (event ii), and addition of an MA (event iii). The top three most dominant OTUs were extracted from the Illumina sequence library at days 5, 22, 31, and 39, and these were considered as the representative OTUs before event i and after events i, ii, and iii, respectively. As a result, a total of eight dominant OTUs were identified and a couple of these (OTUs II and III) were present at different events (Fig. 5). The top 3 most abundant microbial species at day 5 were OTUs I (relative abundance 18.3 %, closest relative Azospira restricta), II (8.8 %, Leadbetterella byssophila), and III (8.1 %, Aquabacterium sp. Aqua2); those at day 22 were OTUs III (61.0 %), IV (6.8 %, Solitalea canadensis), and V (6.0 %, Paracoccus marcusii); those at day 31 were OTUs III (31.4 %), VI (18.2 %, Luteimonas marina), and II (14.5 %); those at day 39 were OTUs VII (20.7 %, Acidovorax ebreus), III (19.3 %), and VIII (8.42 %, Rhodobacter sp. 16-62). Based on the time-course change in relative abundances of these dominant OTUs, each of the genera Azospira, Aquabacterium, Luteimonas, and Acidovorax, the family Flexibacteraceae, and the class Flavobacteriia that we monitored with a special focus in the preceding section are mostly represented by these individual OTUs. Moreover, the sum of the relative abundances of these 8 OTUs ranged from 67.8 to 84.6 % after events i to iii, while this was only 39.2 % before the event i. This agreed with the microbial diversity data (Fig. 2b, c) that showed a drastic decrease in evenness just after the event i and which even at the end of the operation did not fully recovered. On the other hand, Table S1 summarizes the top five highly increased OTUs after each event. After the event i (days 8–22), the OTU IV (Solitalea-related Flavobacteriia bacterium) exhibited a large, i.e., 141-fold, increase in its relative abundance at day 22 (6.8 %) as compared to that at day 5, while the most major OTU III (Aquabacterium sp.) increased 8-fold during this period. The high OLR conditions (after the event ii; days 23–31) stimulated the growth of the OTUs II (Leadbetterella-related Flexibacteraceae bacterium) and VI (L. marina); they showed 27-fold and 8-fold increases (relative abundance 14.5 and 18.2 % at day 31), respectively. In addition to these dominant OTUs, the minor OTU XII (Bdellovibrio-related delta-proteobacterium) increased approximately 100-fold after the event ii, accounting for 2.2 % of the total microorganisms at day 31. The MA supplementation (event iii; days 32–39) had an impact on the dynamics of the minor OTUs. In particular, the obligatory anaerobic bacterial OTUs XV (Tissierella sp.), XVI (Petrimonas sp.), and XVIII and XIX (Fusibacter spp.) affiliated within the classes Clostridia and Bacteroidia exhibited remarkable changes, i.e., 127–440-fold increases, during this period when microaerobic and/or partially anaerobic conditions were established (Fig. 1b).

The OTUs responding to the three different conditional changes. The top three most dominant OTUs were extracted from the sequence library at days 5 (before event i), 22 (after event i), 31 (after event ii), and 39 (after event iii). Each bar denotes relative abundance of the OTU corresponding to the table on the right. The most closely related species and the similarity score, as predicted by BLAST analysis, are shown in the table. Class, order, and family of each OTU were predicted by QIIME. The colored version of this figure is available online
Discussion
In this study, we investigated the high-resolution dynamics of the sludge microbial communities along with the three conditional changes, i.e., water dilution (event i), high organic loading (event ii), and supplementation of an MA (event iii), in a pilot-scale MBR using a combination of chemical analyses and high-throughput Illumina sequencing of 16S rRNA genes. During the event i, a deteriorated reactor performance was artificially induced by the overflow of its activated sludge through intensive water supply (i.e., the simulated intense rainfall situation). Hereby, we were able to unveil the structural and functional responses of the once functionally disrupted sludge microbial communities to the subsequent environmental stimuli, i.e., events ii and iii. Recently, the non-perturbed community responses to these two events have been reported (Sato et al. 2015a; Sato et al. 2015b). Using these experimental data as controls, distinct phenomena and response characteristics that are specific to the perturbed communities are discussed.
The effects of the water-dilution perturbation employed (event i) can be divided into two types. One is the washout (i.e., physical removal) of the sludge biomass and the other is the subsequent decrease in F/M ratios. The former most likely caused the dramatic decrease in the microbial evenness, i.e., Shannon and Simpson reciprocal indices (Fig. 2b, c), and the considerable changes in the composition of the sludge microbial communities (Fig. 4a, b) in the immediate aftermath of the event i. This was also reflected by the long distance between data points at days 5 and 8 on the PCoA plot (Fig. 3). On the other hand, the effect of the latter was continuous but rather gradual. Specifically, the decline of the COD removal ratio (Fig. 1d), the slow decrease in microbial richness and evenness, i.e., Chao1 and Shannon indices (Fig. 2a, b), and the disappearance and reappearance of certain bacteria (e.g., OTU I [Azospira sp.] and OTU IV [Solitalea-related Flavobacteriia bacterium]) (Fig. 4b) stemmed conceivably from the decrease in F/M ratio. These changes in physicochemical and microbiological parameters were not found in the non-perturbed MBRs (Sato et al. 2015b). Therefore, the deterioration processes observed were truly associated with the water-dilution perturbation (event i). Furthermore, employing the high OLR conditions (event ii) facilitated the recovery of the COD removal, and the stable reactor performance continued. The supplementation of the MA (event iii), which was effective to reduce ammonium concentration for the non-perturbed MBR (Sato et al. 2015a), seemed not to work in the present study, probably due to the preceding water-dilution effect on microbial communities. During these phases, all the three microbial diversity indices gradually increased and ultimately recovered to some degree (Fig. 2). Remarkably, the PCoA plots in Fig. 3 clearly showed that the points representing the deteriorative periods (after the event i; days 8–22) were closely clustered together, while after the event ii, the whole microbial community structure shifted towards the initial situation before the event i, which is characterized as the stable performance. Although distinctly clustered, the stable operational phases (days 1–5 and 30–38) were located with the similar PC1 values. The data points for days 24–26 were adjacent to those for the deteriorative phase (i.e., days 8–22) in the plot (Fig. 3), which indicates that there was a lag time to reestablish the microbial communities in response to the event ii. It has been reported that operational periods of at least threefold of the HRT are required for the turnover of the supplied wastewater (Ueno et al. 2006, Hori et al. 2015). Taken together, the dynamics of the microbial diversity indices and fine-scale microbial community structure based on the weighted UniFrac distances were apparently correlated with the reactor performance, especially the COD removal ratio.
The structural responses of the sludge microbial communities varied from one conditional change to another. The water-dilution perturbation (event i) produced a sharp increase in the relative abundance of OTU II (Aquabacterium sp.) (Fig. 4b). Aquabacterium spp. have been isolated from a drinking water system, and their cells were found to contain polyalkanoate and polyphosphate as nutrient storage polymers (Kalmbach et al. 1999). Such metabolic strategies may be effective for these species to survive in oligotrophic environments such as the situation observed during the event i. Our previous study also confirmed the dominance of related microorganisms under low OLR conditions in the MBR (Sato et al. 2015b). On the contrary, OTU I (A. restricta) decreased dramatically after the event i (days 8–22) and it was eventually reduced to a quite low population level (<0.25 %) (Fig. 4b). In the non-perturbed MBR (Sato et al. 2015b), its predominance was observed under not only low- but also high-OLR conditions, indicating that the water dilution initiated the elimination of this dominant microbial constituent from the MBR. Azospira restricta was originally isolated from groundwater, and it has been also known as an oligotroph capable of producing nutrient storage polymers including poly-beta-hydroxybutyrate (Bae et al. 2007). It remains to be clarified which environmental factors select and shape the oligotrophic bacterial populations in the perturbed MBR. Nevertheless, such functional redundancy and niche differentiation are essential for the maintenance of the microbial community function, i.e., the stabilization of reactor performances, as reported in the other wastewater treatment plants (Allison and Martiny 2008; Vuono et al. 2015). On the other hand, OTU IV (Solitalea-related Flavobacteriia bacterium) showed a drastic increase after the event i, so that became the second most abundant OTU at day 22 (relative abundance 6.8 %; Fig. 5 and Table S1). Its cultured relatives, Solitalea spp., have been reported to be involved in the hydrolysis of gelatin and starch (Weon et al. 2009). Furthermore, they were identified as the bacterial micropredators metabolically active in soil environments (Lueders et al. 2006). In this regard, the other highly increased OTU X (Myxococcales bacterium; 69-folds; relative abundance 1.0 % at day 22) is closely related to the facultative micropredator Sorangium cellulosum (Pasternak et al. 2013). OTU VI (L. marina), possessing a high increasing ratio after the event i (81-folds; relative abundance 2.4 % at day 22) and the second most dominant after the event ii (18.2 % at day 31), is also phylogenetically close to one of the well-characterized micropredators, Lysobacter spp. (Lueders et al. 2006; Vasilyeva et al. 2008; Seccareccia et al. 2015). During days 23–31 (after the event ii), the most highly increased OTU XII (Bdellovibrio bacteriovorus; 98-folds; 2.2 % at day 31) has been reported to be an obligate predatory bacterium that preys on gram-negative bacteria as a substrate for growth (Rendulic et al. 2004). Other than these OTUs, the major OTU II (Leadbetterella-related Flexibacteraceae bacterium; 14.5 % at day 31) and minor OTU XI (Lewinella-related Saprospiraceae bacterium; 0.4 % at day 31) are also possible micropredator candidates owing to their previously reported genomic insights (Pasternak et al. 2013; Abt et al. 2011). As a consequence, although viruses and protozoa have been regarded as crucial players in controlling the sludge biomass (Barr et al. 2010; Ni et al. 2010), the results of this study indicate that the bacterial micropredators may play a prominent role in the flexible reorganization of the sludge microbial communities, thus contributing to the functional stability in the closed MBR system.
Finally, it is noteworthy to point out the membrane fouling that occurred at the end of the operation (Fig. 1e). Although the excessive cumulation of the sludge biomass (MLSS >12,000 mg l−1) was considered as its main cause based on previous reports (Trussell et al. 2007), the dynamic shift of the microbial communities from day 38 to 39 might also contribute (Fig. 3). The top 10 highly increased OTUs during days 38–39 are listed in Table S2. The OTU XX, which exhibited the highest increasing ratio (55-fold increase; relative abundance 0.33 % at day 39), was phylogenetically related to the parasitic Acholeplasmatales bacterium. Acholeplasmatales bacteria are known to parasitize a variety of organisms such as plants, mammals, crustaceans, and insects. Among them, Acholeplasma laidlawii that was originally isolated from wastewater is a cultured species exclusively holding both a parasitic and free-living existence (Lazarev et al. 2011). Only fairly recently, these bacteria were reported capable of actively incorporating dead bacterial cells and their components including cell wall and membrane, and intracellular materials (e.g., amino acids and nucleic acids) (Hanajima et al. 2015). This suggests that the release and accumulation of these dead cell components may be one of the causes for the membrane fouling in the MBR. The other highly increased OTUs XXI–XXIX were all affiliated with anaerobic bacteria (Table S2).
Due to the prevention of the sludge washout that allows the low replacement of the sludge microorganisms, the MBR environment is ideal for studying the structure and function of microbial communities in closed systems (Miura et al. 2007; Le-Clech 2010). In order to gain deeper insight into the functional stability and structural flexibility of the sludge microorganisms, we employed high-throughput Illumina sequencing of 16S rRNA genes to describe the fine-scale dynamics of microbial communities in response to the three conditional changes, i.e., the water dilution resulting to the decrease in F/M ratio, the increase in OLR, and the MA supplementation. The results in this study showed that the diversity and overall structure of microbial communities were tightly associated with the reactor performance, i.e., the COD removal ratio. In this context, co-existence and niche differentiation of physiologically similar microorganisms and the dynamic proliferation of bacterial micropredators took pivotal roles in the flexible reorganization of the microbial communities. Environmental transcriptomics (Morita et al. 2014) and ultra-high-sensitivity stable isotope probing (Aoyagi et al. 2015a) targeting on these key microbial groups should further demonstrate their ecophysiology and metabolic network within the sludge microbial communities.
References
Abt B, Teshima H, Lucas S, Lapidus A, Del Rio TG, Nolan M, Tice H, Cheng JF, Pitluck S, Liolios K, Pagani I, Ivanova N, Mavromatis K, Pati A, Tapia R, Han C, Goodwin L, Chen A, Palaniappan K, Land M, Hauser L, Chang YJ, Jeffries CD, Rohde M, Goker M, Tindall BJ, Detter JC, Woyke T, Bristow J, Eisen JA, Markowitz V, Hugenholtz P, Klenk HP, Kyrpides NC (2011) Complete genome sequence of Leadbetterella byssophila type strain (4M15(T)). Stand Genomic Sci 4(1):2–12
Allison SD, Martiny JBH (2008) Resistance, resilience, and redundancy in microbial communities. P Natl Acad Sci USA 105:11512–11519
Aoyagi T, Hanada S, Itoh H, Sato Y, Ogata A, Friedrich MW, Kikuchi Y, Hori T (2015a) Ultra-high-sensitivity stable-isotope probing of rRNA by high-throughput sequencing of isopycnic centrifugation gradients. Env Microbiol Rep 7(2):282–287
Aoyagi T, Kimura M, Yamada N, Navarro RR, Itoh H, Ogata A, Sakoda A, Katayama Y, Takasaki M, Hori T (2015b) Dynamic transition of chemolithotrophic sulfur-oxidizing bacteria in response to amendment with nitrate in deposited marine sediments. Front Microbiol 6:426
Aronesty E (2013) Comparison of sequencing utility programs. Open Bioinforma J 7:1–8
Bae HS, Rash BA, Rainey FA, Nobre MF, Tiago I, da Costa MS, Moe WM (2007) Description of Azospira restricta sp nov., a nitrogen-fixing bacterium isolated from groundwater. Int J Syst Evol Micr 57:1521–1526
Barr JJ, Slater FR, Fukushima T, Bond PL (2010) Evidence for bacteriophage activity causing community and performance changes in a phosphorus-removal activated sludge. FEMS Microbiol Ecol 74(3):631–642
Bartram AK, Lynch MD, Stearns JC, Moreno-Hagelsieb G, Neufeld JD (2011) Generation of multimillion-sequence 16S rRNA gene libraries from complex microbial communities by assembling paired-end Illumina reads. Appl Environ Microbiol 77(11):3846–3852
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7(5):335–336
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, Gormley N, Gilbert JA, Smith G, Knight R (2012) Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6(8):1621–1624
Chen J, Beck MB (2001) Operational control of storm sewage at an activated sludge process. Water Sci Technol 43(7):131–138
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL (2006) Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72(7):5069–5072
Hanajima D, Aoyagi T, Hori T (2015) Survival of free-living Acholeplasma in aerated pig manure slurry revealed by 13C-labeled bacterial biomass probing. Front Microbiol 6:1206
Hill MO (1973) Diversity and evenness: a unifying notation and its consequences. Ecology 54(2):427–432
Hori T, Kimura M, Aoyagi T, Navarro RR, Ogata A, Sakoda A, Katayama Y, Takasaki M (2014) Biodegradation potential of organically enriched sediments under sulfate- and iron-reducing conditions as revealed by the 16S rRNA deep sequencing. J Water Environ Technol 12(4):357–366
Hori T, Haruta S, Sasaki D, Hanajima D, Ueno Y, Ogata A, Ishii M, Igarashi Y (2015) Reorganization of the bacterial and archaeal populations associated with organic loading conditions in a thermophilic anaerobic digester. J Biosci Bioeng 119(3):337–344
Hu M, Wang X, Wen X, Xia Y (2012) Microbial community structures in different wastewater treatment plants as revealed by 454-pyrosequencing analysis. Bioresour Technol 117:72–79
Itoh H, Navarro R, Takeshita K, Tago K, Hayatsu M, Hori T, Kikuchi Y (2014) Bacterial population succession and adaptation affected by insecticide application and soil spraying history. Front Microbiol 5:457
Kalmbach S, Manz W, Wecke J, Szewzyk U (1999) Aquabacterium gen. nov., with description of Aquabacterium citratiphilum sp. nov., Aquabacterium parvum sp. nov. and Aquabacterium commune sp. nov., three in situ dominant bacterial species from the Berlin drinking water system. Int J Syst Bacteriol 49:769–777
Kessler R (2011) Stormwater strategies: cities prepare aging infrastructure for climate change. Environ Health Persp 119(12):A514–A519
Lazarev VN, Levitskii SA, Basovskii YI, Chukin MM, Akopian TA, Vereshchagin VV, Kostrjukova ES, Kovaleva GY, Kazanov MD, Malko DB, Vitreschak AG, Sernova NV, Gelfand MS, Demina IA, Serebryakova MV, Galyamina MA, Vtyurin NN, Rogov SI, Alexeev DG, Ladygina VG, Govorun VM (2011) Complete genome and proteome of Acholeplasma laidlawii. J Bacteriol 193(18):4943–4953
Le-Clech P (2010) Membrane bioreactors and their uses in wastewater treatments. Appl Microbiol Biotechnol 88(6):1253–1260
Li H, Durbin R (2009) Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25(14):1754–1760
Lueders T, Kindler R, Miltner A, Friedrich MW, Kaestner M (2006) Identification of bacterial micropredators distinctively active in a soil microbial food web. Appl Environ Microbiol 72(8):5342–5348
Miura Y, Hiraiwa MN, Ito T, Itonaga T, Watanabe Y, Okabe S (2007) Bacterial community structures in MBRs treating municipal wastewater: relationship between community stability and reactor performance. Water Res 41(3):627–637
Morita T, Koike H, Hagiwara H, Ito E, Machida M, Sato S, Habe H, Kitamoto D (2014) Genome and transcriptome analysis of the basidiomycetous yeast Pseudozyma antarctica producing extracellular glycolipids, mannosylerythritol lipids. PLoS One 9(2):e86490
Navarro RR, Aoyagi T, Kimura M, Koh H, Sato Y, Kikuchi Y, Ogata A, Hori T (2015) High-resolution dynamics of microbial communities during dissimilatory selenate reduction in anoxic soil. Environ Sci Technol 49(13):7684–7691
Ni BJ, Rittmann BE, Yu HQ (2010) Modeling predation processes in activated sludge. Biotechnol Bioeng 105(6):1021–1030
Noll M, Matthies D, Frenzel P, Derakshani M, Liesack W (2005) Succession of bacterial community structure and diversity in a paddy soil oxygen gradient. Environ Microbiol 7(3):382–395
Pasternak Z, Pietrokovski S, Rotem O, Gophna U, Lurie-Weinberger MN, Jurkevitch E (2013) By their genes ye shall know them: genomic signatures of predatory bacteria. ISME J 7(4):756–769
Rendulic S, Jagtap P, Rosinus A, Eppinger M, Baar C, Lanz C, Keller H, Lambert C, Evans KJ, Goesmann A, Meyer F, Sockett RE, Schuster SC (2004) A predator unmasked: life cycle of Bdellovibrio bacteriovorus from a genomic perspective. Science 303(5658):689–692
Sato Y, Hori T, Navarro RR, Habe H, Ogata A (2015a) Effect of a microbiota activator on accumulated ammonium and microbial community structure in a pilot-scale membrane bioreactor. J Gen Appl Microbiol 61(4):132–138
Sato Y, Hori T, Navarro RR, Habe H, Yanagishita H, Ogata A (2015b) Fine-scale monitoring of shifts in microbial community composition after high organic loading in a pilot-scale membrane bioreactor. J Biosci Bioeng. doi:10.1016/j.jbiosc.2015.10.003
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75(23):7537–7541
Seccareccia I, Kost C, Nett M (2015) A quantitative analysis of Lysobacter predation. Appl Environ Microbiol 81(20):7098–7105
Sozen S, Cokgor EU, Basaran ST, Aysel M, Akarsubasi A, Ergal I, Kurt H, Pala-Ozkok I, Orhon D (2014) Effect of high loading on substrate utilization kinetics and microbial community structure in super fast submerged membrane bioreactor. Bioresour Technol 159:118–127
Trussell RS, Merlo RP, Hermanowicz SW, Jenkins D (2007) Influence of mixed liquor properties and aeration intensity on membrane fouling in a submerged membrane bioreactor at high mixed liquor suspended solids concentrations. Water Res 41(5):947–958
Ueno Y, Sasaki D, Fukui H, Haruta S, Ishii M, Igarashi Y (2006) Changes in bacterial community during fermentative hydrogen and acid production from organic waste by thermophilic anaerobic microflora. J Appl Microbiol 101(2):331–343
Vasilyeva NV, Tsfasman IM, Suzina NE, Stepnaya OA, Kulaev IS (2008) Secretion of bacteriolytic endopeptidase L5 of Lysobacter sp XL1 into the medium by means of outer membrane vesicles. FEBS J 275(15):3827–3835
Veltmann K, Palmowski LM, Pinnekamp J (2011) Modular operation of membrane bioreactors for higher hydraulic capacity utilisation. Water Sci Technol 63(6):1241–1246
Vuono DC, Benecke J, Henkel J, Navidi WC, Cath TY, Munakata-Marr J, Spear JR, Drewes JE (2015) Disturbance and temporal partitioning of the activated sludge metacommunity. ISME J 9(2):425–435
Wang X, Hu M, Xia Y, Wen X, Ding K (2012) Pyrosequencing analysis of bacterial diversity in 14 wastewater treatment systems in China. Appl Environ Microbiol 78(19):7042–7047
Weon HY, Kim BY, Lee CM, Hong SB, Jeon YA, Koo BS, Kwon SW (2009) Solitalea koreensis gen. nov., sp nov and the reclassification of [Flexibacter] canadensis as Solitalea canadensis comb. nov. Int J Syst Evol Micr 59:1969–1975
Wilen BM, Lumley D, Mattsson A, Mino T (2006) Rain events and their effect on effluent quality studied at a full scale activated sludge treatment plant. Water Sci Technol 54(10):201–208
Yang WB, Cicek N, Ilg J (2006) State-of-the-art of membrane bioreactors: worldwide research and commercial applications in North America. J Memb Sci 270(1–2):201–211
Acknowledgments
We would like to acknowledge the Kinu-aqua station for kindly providing the activated sludge. We thank Mr. Takayuki Iijima and Ms. Yumiko Kayashima (National Institute of Advanced Industrial Science and Technology (AIST)) for operating the reactor and analyzing the water samples.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Yuya Sato and Tomoyuki Hori contributed equally to this work.
Electronic supplementary material
ESM 1
(PDF 265 kb)
Rights and permissions
About this article

Cite this article
Sato, Y., Hori, T., Navarro, R.R. et al. Functional maintenance and structural flexibility of microbial communities perturbed by simulated intense rainfall in a pilot-scale membrane bioreactor. Appl Microbiol Biotechnol 100, 6447–6456 (2016). https://doi.org/10.1007/s00253-016-7466-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-016-7466-z