
Abstract
While glutamate and glutamate-rich compounds are widely used for culturing Streptomyces sp., little is known regarding glutamate catabolism at molecular level. Noting the presence of two distinct putative glutamate dehydrogenases (GDH), we constructed knockout mutants of each gene with Streptomyces coelicolor M145 and examined the functionality related to antibiotic production. Out of the two, the sco2999 knockout (ΔgdhB, NAD+-specific) showed outstanding effects; it decreased the growth sevenfold but initiated the undecylprodigiosin (RED) production in complex Difco nutrient media which otherwise does not support the production from M145. With glucose supplementation, the growth difference by ΔgdhB disappeared but we could obtain significantly increased actinorhodin (ACT) and RED biosynthesis with the mutant by limiting the glucose content (0.5∼1.0 %, w/v). Complementing the gene to the knockout mutant inhibited the production, confirming its gene specificity. Along with the extended impacts on overall nitrogen metabolism based on the intracellular metabolite analysis and enzyme assays, GdhB and glutamate utilization were shown to interfere with N-acetylglucosamine metabolism and the activity of its associated global transcriptional regulator (DasR). Taken together, GdhB—subjected to the nutritional context-dependent regulation—is proposed as a key member of central nitrogen metabolism to control the secondary metabolism initiation in exploiting the organic nitrogen sources.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Streptomycetes are soil-inhabiting filamentous bacteria that produce numerous therapeutic agents for treatment of bacterial infection or cancer. They undergo a developmental cycle, spanning the multi-nuclear stages (from the vegetative to aerial mycelial growth stages) to a uni-nuclear sporulation stage. Underlying the biological features, complex genetic regulatory networks and signaling cascades operate with the interplay of plural transcriptional factors (TFs; Swiatek et al. 2013) and key metabolites. Streptomyces coelicolor A3(2) M145 is the most well-studied model organism of the genus, and its genome has numerous gene clusters for secondary metabolites, which include the characterized blue colored type-II polyketide actinorhodin (ACT), the red colored hybrid polyketide-tripyrrole undecylprodigiosin (RED), the lipopeptide calcium-dependent antibiotic (CDA), and the cryptic yellow pigmented type-I polykdetide (yCPK; Bentley et al. 2002; Gottelt et al. 2010).
As profiles of secondary metabolites produced from an organism depend on nutritional composition of the chosen culture media, primary metabolism must pave ways to initiate secondary metabolism. Still, many annotated components of the genome sequences for the primary metabolism need to be experimentally evaluated of their functions in the metabolism system. Traditionally, components of glucose metabolism and the tricarboxylic acid (TCA) cycle have been studied, such as glyceraldehyde 3-phosphate dehydrogenase citrate synthase, aconitase, polyphosphate kinase, and phosphofructokinase (Borodina et al. 2008; Chouayekh and Virolle 2002; Li and Townsend 2006). Either deletion of the gene(s) or overexpression was opted to direct metabolic flux toward the secondary metabolism. For example, overexpression of acetyl-CoA carboxylase was aimed to increase malonyl-CoA, the major building block of polyketide compounds (Ryu et al. 2006). Similar approach can be applied to the other primary metabolic processes, and we have looked on the potential of manipulating the glutamate utilization.
At the practical background of the study, streptomycetes are routinely cultured with protein (or amino acids)-rich complex media in which glutamate is most abundant. In addition, glutamate is frequently supplemented to minimal media for improving the cell growth. Inside the cells, it is also the most abundant free intracellular metabolite in S. coelicolor (Borodina et al. 2005; Nieselt et al. 2010; Wentzel 2012) and becomes the direct precursor of many other important metabolites. Its metabolic and physiological significance is inseparable from its de-aminated product, α-ketoglutarate, which is involved in diverse oxidation reactions and in oxygen and nitrogen sensing (Ninfa and Jiang 2005; Ratcliffe 2013; Velvizhi et al. 2002). Glutamate can be synthesized in vivo, mainly by reductive glutamate dehydrogenase (GDH) and by glutamine oxoglutarate amino transferase (GOGAT),·glutamine synthetase (GS). Previous studies with Streptomyces have been focused on the NADPH-dependent glutamate biosynthesis, since glutamate-mediated ammonium assimilation is responsible for 85 % of total nitrogen assimilation (Laycock et al. 1985). By contrast, glutamate catabolism has been little understood at the molecular level. Based on genome annotation, we noticed the presence of two different types of GDH for bidirectional conversion between α-ketoglutarate and glutamate (Vancurova et al. 1989). We initiated this study with an aim to distinguish the two distinct types of GDHs via in-frame deletion mutants of each gene with S. coelicolor A3(2) M145. And, this study reports the significant phenotypic and metabolic changes observed with a knockout mutant of sco2999 for the NAD+-dependent GDH (to be called as GdhB, EC.1.4.1.2) from the complex media-based cultures. We confirm GdhB as the active member of the central nitrogen metabolism and show its importance in initiating secondary metabolism.
Materials and methods
Bacterial cell culture S. coelicolor A3(2) M145 and its derivatives are listed in Supplementary Table 1. Following the general methods of Practical Streptomyces Genetics (Kieser T et al. 2000) and our previous study, the bacterial cells and spores were maintained and used. Difco™ nutrient medium (DN, Becton, Dickinson and Company, France) is the chosen complex medium. DN broth is made of 3-g beef extract and 5-g peptone. DN agar contains 15 % agar in addition. For stable cell growth and antibiotic production in broth, 50 g of polyethylene glycol 6000, 25 mM TES buffer (2-[[1,3-dihydroxy-2-(hydroxymethyl)propan-2-l]amino] ethanesulfonic acid), trace element solution (equivalent to R5 media), and 0.5 % (w/v) glucose were added (named as DN_Glc). Modifying the supplemented liquid minimal medium (SMM; Strauch et al. 1991), we reduced the glucose content from 1.0 to 0.1 % (w/v) but added 0.1 % N-acetylglucosamine (GlcNAc, w/v; mSMM_GlcNAc). Without casamino acids and glucose, the basic minimal medium (BMM) was prepared modifying SMM. Solid basic minimal medium (BMM) was made with 10 g agar and 0.5 g K2HPO4, 0.2 g MgSO4·7H2O, and 0.01 g FeSO4·7H2O per a liter of solution. Sterile stock solutions of glucose, GlcNAc, casamino acids, ammonium sulfate, alanine, glutamine, and glutamate were separately prepared and combined to make each medium prior to cell cultivation. Cell growth and the production of total ACT RED were measured as previously described (Kim et al. 2012). Absorbance was scanned in a 96-well plate format (Thermo Scientific, USA).
Gene manipulations Each of gdhB ( sco2999) and gdhA (sco4683) was deleted from the chromosome using REDIRECT system (Gust et al. 2003). Successful exconjugants were screened by antibiotic resistance against apramycin and kanamycin (AprR KanS). Targeted open-reading frame (ORF) and its vicinity were PCR amplified and then sequenced (Cosmogenetech, Korea). For gdhB complementation, pSETneo was used as previously reported and the gdhB complemented strain, SK01, was maintained with neomycin (10 μg/ml). Primers used in this study are listed in Supplementary Table 2.
Preparation of the crude cell-free lysates for enzyme assays S. coelicolor mycelia were collected by centrifugation. Routinely, 50 mM phosphate-buffered saline (PBS, pH 7.2) was used for washing and rupturing the cells with an exception of GS assay that was done with imidazole buffer solution (25 mM, pH 7.2). Cells were broken by sonication in an ice bath (repeated cycles of 2-s pulse and 2-s pause). Clear soluble cell lysates were collected by removing the cell debris with centrifugation (13,000× rpm, for 15 min, at 4 °C) and filtration (0.2 μm pore size). Protein concentration was determined by Bradford assay (Bio-Rad).
Enzyme assays Reductive and oxidative changes of cofactors were assessed by monitoring the absorbance at 340 nm. Specifically, the increase rate of NADH for oxidative GdhB was measured in the presence of 15 mM L-glutamate, NAD+ (2 mM), and essential activator AMP (1 mM) in PBS (pH 7.2) (Brana et al. 1986). For NADH production from other amino acids, the GdhB assay was modified by replacing glutamate with each of specific L-amino acids. For GdhA and GOGAT assays, consumption rates of NADPH (5 mM α-ketoglutarate and 0.25 mM NADPH) and NADH (5 mM α-ketoglutarate·glutamine and 0.25 mM NADH) were calculated (Fisher 1989). The earlier method for the GS assay was followed (Bressler SaA 1984) with an exception of Malachite Green Phosphate Assay Kits (BioAssay Systems, USA) used for measuring phosphate contents. To consider the autonomous changes, each control reaction (without substrate) was prepared and its value was subtracted from that of corresponding experimental reaction. Equal protein amounts of M145 and BG20 lysates were used in each assay (within a range of 20∼200 μg).
Zymogram (in-gel dehydrogenase assay) Crude cell-free lysates were separated in a 5 % non-denaturing acrylamide gel by electrophoresis (at constant 100 V), and the dehydrogenase reaction was visualized by color development after immersing the gel in a reaction buffer (50 mM PBS (pH 7.2), 20 mM amino acid, nitroblue tetrazolium (0.3 mg/ml), phenazine methosulfate (0.05 mg/ml), and 0.5 mM NAD+; Han et al. 2012). Equal protein amounts of M145 and BG20 lysates were compared.
Intracellular quantitation Ten-milliliter S. coelicolor mycelium pellets was collected at indicated time points (DN_Glc broth) for analyzing 20 amino acids and the cofactors NAD(P)H of different reducing status. Metabolic reactions of the harvested cells were quenched with cold methanol and washed with cold 60 % methanol (v/v) by centrifugation (13,000× rpm, 1 min; de Koning and van Dam 1992). Quenching and washing processes were completed within 10 min. Resuspended in 60 % ethanol, cells were ruptured by repeated cycles of freezing and thawing, and particles were removed by centrifugation (13000× rpm for 20 min at 4 °C). To the clear extract, equal volume of chloroform was mixed and the aqueous fraction was collected by centrifugation (7 min, 13,000× rpm, 4 °C). The final aqueous fraction after two more repeats of chloroform extraction was subjected to hydrophilic interaction chromatography-tandem mass spectrometer (HILIC-MS/MS) analysis (Bajad et al. 2006). Hypersil GOLD HILIC (dimension 100 × 2.1 mm; Thermo Scientific, USA) was used as an ion exchange column. The LC-MS/MS system was composed of Accela auto-sampler, Accela 1250 pump, and triple-stage quadrupole (TSQ) Quantum Access MASS. The glutamate peak (a parent ion of 148.1 Da and its daughter ion of 84.2 Da) and glucosamine-6-phosphate peak (GlcN6p, a parent ion of 260.1 Da and its daughter ion of 126.1 Da) were identified from a positive mode run. For the liquid chromatography operation, metabolites were separated through the linear gradient flow of solvent A (95 % H2O, 5 % acetonitrile, and 20 mM ammonium acetate, pH 9.0) and solvent B (acetonitrile 100 %) at 400 μl/min. Cell mass-normalized (wet weights) fold changes were reported for BG20 compared to M145.
Reverse transcription and real-time quantitative PCR Total cellular RNA was obtained using acidic phenol and chloroform (Sigma). Quality of RNA and its concentration were determined by A260 nm/A280 nm ratio (within the range of 1.9–2.1 in DEPC-treated water). NanoDrop spectrophotometer was used (ND-1000, Thermo Scientific, USA). RNA integrity was confirmed with formaldehyde agarose gel electrophoresis. And, complementary DNA (cDNA) was obtained by reverse transcription with Molony Murine Leukemia Virus-RTase (MBiotech, Korea) and random hexameric nucleotides. The cDNA was properly diluted to ensure the linear range of the following PCR reaction. Quantitative real-time PCR was performed using TOPreal™ qPCR 2× PreMIX solution (Enzynomics, Korea). Bio-Rad C100 Thermocycler was used (CFX96 Real-Time System, Bio-Rad). Triplicate reactions for each gene expression were averaged and normalized to the internal control of rrnB expression. The relative induction level in BG20 compared to M145 was calculated (ΔΔCtBG20-M145; Livak and Schmittgen 2001) and reported in log2 ratio.
Results
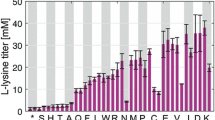
ACT·RED production by glutamate in minimal media and the construction of gdhB knockout With M145 (wild-type strain), glutamate was superior to the other amino acids as a boost for ACT and RED production in a solid state of minimal media. All 20 L- amino acids were used individually as a sole carbon and nitrogen (C/N) source in the minimal medium (BMM; Fig. 1a).

Boosted ACT·RED production by glutamate and the in-frame deletion of gdhB. a ACT·RED production from M145 on the solid minimal media (BMMs). Each amino acid was used as a sole C/N source in each well. Image was acquired on day 3. b PCR confirmation of sco2999 ORF knockout. The 4962 bp was replaced with an apramycin resistance cassette. Multiple isolates were obtained. c Decreased relative specific activity of GdhB by gdhB knockout in DN_Glc broth. d Increased intracellular glutamate by the knockout (TSQ metabolite analysis). Y axis represents the cell mass-normalized fold change of BG20 compared to M145. Inserted box is the standard curve of glutamate for quantification. Three independent experimental measurements of samples at 45 h were averaged
SCO2999 is annotated as the putative oxidative glutamate dehydrogenase in S. coelicolor and is expected for glutamate degradation based on its cofactor specificity (KEGG gene ontology pathway). BG20 was obtained by PCR targeting the gene, sco2999, using M145 as the parental strain (Gust et al. 2003; Fig. 1b). With the crude cell lysate-based analysis, gdhB knockout was found to decrease the NADH production from the use of glutamate. However, the difference of M145 and BG20 in the catabolic GdhB activity could be detected only when the cell growth stopped ca. 36 h in the glucose-supplemented complex liquid media (DN_Gl, 0.5 %; Fig. 1c). At earlier time points of growth, M145 and BG20 were not significantly different each other in the activity. At 45 h after growth, the mutant showed sixfold more of intracellular glutamate than M145 (LC-MS/MS, Fig. 1d).
Glucose-dependent effects on growth and ACT·RED production Growth of BG20 was inferior to M145 especially in the sugar-free DN broth (Fig. 2a). Glucose supplementation—as low as 0.2 % (w/v) —improved the growth of BG20 and made it comparable to M145 in growth (Fig. 2b, c). Still, BG20 yielded much less aerial mycelia than M145 after more than 5 days of delay on the glucose-supplemented DN agar (Fig. 2d).

Phenotypic characteristics of BG20 (ΔgdhB) contrasting with M145. a, b Growth curve of M145 and BG20 in complex DN broth (a −glucose and b +2 % glucose). Reduced growth of BG20 in the absence of glucose was overcome with glucose supplementation. c Comparison of wet-weight gain (48 h, two independent experiments). Of the glucose supplementation, 0.2 % was enough for improving the growth of BG20 in the DN broth. d Outstanding increase of ACT·RED production, compared to M145 (parental strain) and BG40 (ΔgdhA) on the glucose-supplemented DN agar. e Increased production in DN_Glc broth (0.5 % Glc). Three independent cultures were averaged. f The gdhB gene-specific characteristics of production in DN broth
Normally, DN media do not support the ACT·RED production from M145. But, BG20 showed outstanding production both on the solid and in the liquid DN media (Fig. 2d, e), thereby contrasting itself with the gdhA knockout mutant. Considering the 7-fold decreased growth (by wet-weight gain), this equals to 60-fold increase of RED in terms of specific production. In general, ACT production from M145 and BG20 proportionately increased with the glucose content in the DN agar. Still, ACT production from BG20 was much more significant and premature than M145. Liquid cultures mitigated the effect of gdhB knockout on the ACT production, compared to the solid state culture. And, glucose content over 2.0 % (w/v) in the broth showed no difference in the production by the knockout (data not shown). We routinely added 0.5 or 1.0 % glucose to the DN broth to get the clear catabolic GdhB activity as well as the difference in the production by the gene knockout, while keeping the two strains’ cell mass comparable.
The increased production by gdhB knockout was found to be gene specific as we obtained the complemented strain without the production in DN media (Fig. 2f).
Global impacts on nitrogen metabolism From a metabolic perspective, the loss of function to deaminate glutamate by ΔgdhB might be compensated by the use of other amino acids so as to maintain the internal pools of nitrogen and NADH. However, our observation described below did not support this idea. Cells grown in the glucose-supplemented complex medium (DN_Glc, 1 %) showed synchronous decreases of amino acid usage in BG20 compared to the parental strain, M145, at 40 h (transition phase). According to the crude whole-cell lysate assay, BG20 made less NADH from the amino acids (Fig. 3a). NADH from BG20 with branched chain amino acids (BCAAs) was less than M145, which does not parallel with the previous finding of its positive relation to the ACT production (Sprusansky et al. 2005). Decreased NADH production from alanine was confirmed with in-gel activity staining (Fig. 3a, insert). Supporting the changes of the enzyme activities, comparative intracellular metabolite analysis showed that BG20 yielded more free intracellular amino acids than M145, particularly, glutamate (Fig. 3b). With NAD+ being more prevalent than NADH (>average 1000-fold) at the tested time point, the NADH content of BG20 was 13-fold less than that of M145 (Fig. 3c). Decreased ammonium assimilation via glutamate and glutamine is likely, since the substrate-dependent activities of GdhA and GS were approximately 40 % less in BG20 than M145 (p < 0.05 t test, ≥n = 3; Fig. 3d).

Systemic decrease of nitrogen metabolism by gdhB knockout (at 40 h, DN_Glc broth). a Decreased NADH production from the use of each L-amino acid. Crude cell-free lysates of M145 and BG20 were assayed with 15mM individual amino acid. Y axis indicates the difference of relative specific activity, Δ(BG20-M145). Inserted is the zymogram for the alanine dehydrogenase activity. b Relative abundance of intracellular amino acids in BG20 compared to M145. c Fold change of NAD+ and NADH by gdhB knockout. Y axis represents the cell mass-normalized fold change of BG20 compared to M145 (in log2 scale). d Decreased central nitrogen metabolism by gdhB knockout. Relative enzyme activities of GdhA, GOGAT, and GS (percentile of BG20 compared to M145) are shown. More than three independent experiments were averaged
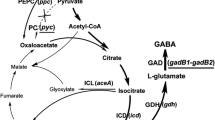
Association to GlcNAc metabolism via DasR Glutamate is closely associated with N-acetylglucosamine (GlcNAc) metabolism for cell wall and for the use of chitin in the environment (see Fig. 6). GlcNAc is a monomeric unit of chitin and bacterial cell wall component. And, its metabolic intermediate, glucosamine-6-phophate (GlcN6p), modulates the DasR activity to control GlcNAc uptake degradation and subsequent ACT·RED production in a nutritional context-dependent manner (Rigali et al. 2008). GlcN6p can affect the growth (Kawada-Matsuo et al. 2012) and antibiotic production (Rigali et al. 2008). To find out possible overlap or interference of GdhB-mediated glutamate metabolism with the GlcNAc-dependent production, we examined the key transcriptional regulator DasR along with its effector, GlcN6p. Although we could detect only a low level of GlcN6p in the cells, it was present more in BG20 than M145 (Fig. 4a). Subsequent de-repression (i.e., induction) of otherwise inhibitory DasR activity was confirmed; DasR regulon was up-regulated in BG20 compared to M145 (Fig. 4b). Quantitative real-time PCR (qRT-PCR) was performed with nagE (for uptaking GlcNAc), nagB (for de-amination of GlcN6p), and dasA (neighboring gene of dasR) along with actII (the transcriptional activator for ACT). The transcriptional level of difference was manifested at later time points of examination. As a non-member of DasR regulon, GlmS did differ in the expression pattern from the others. GlmS is the enzyme working in opposite direction to NagB. Together, they decide the dynamics of cell wall metabolism. Of the two glmS genes, one showed decreased expression and the other showed no significant change by gdhB knockout.

Parallel induction of DasR regulon with the increased ACT·RED production. a Intracellular level of GlcN6p comparing BG20 relative to M145 (40 h). b Induction of DasR-regulon in BG20 (real-time qRT-PCR). Y axis represents the relative induction (RI) level of genes in BG20 compared to M145 (log2 scale). More than three independent experiments were performed
Further direct association of glutamate and GlcNAc metabolism was tested by using GlcNAc-supplemented minimal defined media. Both M145 and BG20 showed GlcNAc-dependent ACT·RED production on the solid state cultures with the minimal media (1 %, BMMs_GlcNAc; Fig. 5b), agreeing with the previous finding (Rigali et al. 2008). However, they differed in response to the addition of glutamate. When we added glutamate (4 mM or more) to the media, it resulted in the inhibited production from M145 (BMMs_GlcNAc + Glu; Fig. 5a, c). Alanine did not affect the production by contrast (Fig. 5a). Interestingly, additional overload of α-ketoglutarate (BMMs_Glu + GlcNAc + α-KG) could restore the production from M145, seemingly canceling the inhibitory effect of glutamate. Pyruvate did not affect the production, supporting the uniqueness of α-ketoglutarate in counteracting the effect of glutamate over the GlcNAc usage (Fig. 5d). Unlike M145, BG20 was immune to the glutamate-mediated inhibition of the GlcNAc-dependent production on the solid state (BMMs_GlcNAc + Glu; Fig. 5b, c). With a liquid state culture, gdhB knockout also enhanced ACT·RED production in a GlcNAc-supplemented minimal medium if the glucose content was lowered to 0.1 % from the usual 1.0 % (mSMM_GlcNAc; Fig.5e).

Counteractive effects of glutamate and α-ketoglutarate over the GlcANc-dependent production. a Glutamate (4 mM, the second well) suppressed the GlcNAc-dependent induction of ACT·RED (BMMs_GlcNAc, 1 %, the first well), while alanine did not (4 mM, the third well). b, top GlcNAc-dependent production on the minimal agar. bottom Glutamate (40 mM) repressed the production from M145 but not from BG20. Each well of the media contains the twofold serially diluted GlcNAc from 2.0 %. c Streaked cultures of M145 and BG20 on BMMs with GlcNAc (1 %) and glutamate (40 mM). d M145 cultured in the presence of excess exogenous α-KG (100–150 mM) to the minimal medium, BMMs_1% GlcNAc + 40 mM Glu. Concentrations (mM) of α-KG are indicated. e Increased ACT·RED production by gdhB knockout in the liquid minimal medium, mSMM_0.1 % GlcNAc + 0.1 % Glc. Two independent experiments were averaged and represented

Proposed GdhB activity. Inability to shunt glutamate to the TCA cycle in complex media leads to ACT·RED production in the rich media (production from the “feast”)
Discussion
The ability to sense nutrient availability of carbon/nitrogen (C/N) is essential to maintain life and appears to have evolved different adjustment mechanisms to harness networks of genes. With functional characterization of gene knockout, we propose that NAD+-dependent GdhB can play a dual role on the growth and show the complex regulatory effects of the nutritional context. Central nitrogen metabolism and the related metabolites of glutamate/glutamine/α-ketoglutarate are widely assumed to play key roles in initiating the production. However, there is little supportive experimental evidence pinpointing the responsible enzyme(s) for the direct metabolic control over the secondary metabolism induction. Unlike the assimilatory pathway of GDH/GOGAT/GS (D’Alia et al. 2010; Fink et al. 2002; Fisher and Wray 1989; Jin and Jiao 1998; Kormanec and Sevcikova 2002; Penyige et al. 2009; Rexer et al. 2006; Sola-Landa et al. 2013), the two putative GDHs likely for the reversible conversion between glutamate and α-ketoglutarate have not been characterized in vivo and remained functionally un defined regarding the glutamate catabolism with streptomycetes. Complexity of glutamate association in any biological system is challenging for describing the physiological impacts of glutamate utilization. Although Escherichia coli systems have far less capacity for secondary metabolism as Streptomyces and do not have the enzyme as GdhB, there are 64 glutamate-associated reactions compared to 37 and 50 reactions associated with aspartate and alanine, respectively (Metacyc.org).
GdhB of Streptomyces is very large with its monomer to be ∼180 kDa. And, it was firstly characterized in vitro with the cloned gene of S. clavuligerus (Minambres et al. 2000). It is highly conserved across Streptomyces and also found in Mycobacterium, Pseudomonas, Caulobacter, and Vibrio. It is an open question if GdhB can contribute to any of their intriguing physiological features with the bacterial genera, e.g., performing secondary metabolism or the ability to cellular differentiation or the interaction with higher eukaryotic organism. And, this study is the first showing its impacts on the secondary metabolism induction with the industrial organism.
Using the crude whole-cell lysates, the success of detecting the catabolic GdhB activity of S. coelicolor depends not only on the composition of the media but also on the sampling time points as previously described. This suggests yet-identified regulatory mechanism(s) being implemented over the activity and/or expression of the enzyme. While we are currently hunting for additional regulatory mechanisms over GdhB and its gene, glucose was the first nutritional factor that showed up the physiological effects of the gdhB knockout on growth and the production. And, the growth defect by the gdhB knockout can be manifested in the absence of glucose. We have observed the reduced gdhB transcription in the presence of glucose in the complex media (not reported data). However, at the metabolic level, glucose and its metabolic derivative(s) must minimize the requirement of GdhB in growth regardless of its expression. As an alternative route for nitrogen acquisition, transaminases are likely to replace GdhB. Different from GdhB, they demand the organic acids to transfer the released amine group from the substrates. GdhB contributes significantly to the growth and is proposed to participate in halting the production meanwhile. To remove such a barrier to the production without affecting the growth, reducing the glucose content in the media was effective. By limiting the glucose content to 0.5∼1.0 %, clear catabolic GdhB activity was demonstrated with M145 and contrasted with BG20. Only after growth stopped, production of BG20 was dramatically enhanced. With another commonly used complex media, R5, we observed such a phenomenon with lowering its glucose content to 0.5 % from the suggested 1.0 % (data not shown) (Kieser T et al. 2000).
We consider several possibilities that explain the GdhB association to the production.
First, gdhB knockout may cause “nitrogen limitation,” when the cells must depend on the organic nitrogen sources. Systemic changes of the intracellular amino acid levels and enzyme activities suggest the decreased ammonium assimilation to glutamate and glutamine (Fig. 3). This agrees with the notion that nitrogen limitation is better than carbon limitation for higher specific ACT production rate with lower maintenance energy (Avignone Rossa et al. 2002). Along with other central nitrogen metabolism, the decreased GS activity should be sound and reasonable, because it demands the use of both ammonium ion and ATP energy. GS is known to be post-transcriptionally modified in Streptomyces (Hesketh et al. 2002).
Second, the increased RED production can be a mechanism to relieve stress from untimely or excess intracellular accumulation of glutamate. In vitro, glutamate is a competitive inhibitor to cysteine/aspartate/lysine/BCAA degradation as well as the cell wall biosynthesis posing certain level of stress to the cells depending on its concentration (product inhibition). The threshold level of intracellular glutamate for such inhibition in vivo is not known; the dramatic increase by the gdhB knockout must have crossed the line. Glutamate is converted to proline by three successive reactions and further to RED. Proline and serine are two amino acid precursors of RED.
And finally, GdhB directly relates with the GlcNAc metabolism for initiating the antibiotic production. As mentioned above, the decreased central nitrogen metabolism may have triggered the GlcNAc degradation pathway. From the crisscross of glutamate and GlcNAc metabolism, the GntR-type master regulator, DasR, is assumed to take part in the control directly or indirectly (Fig. 4b). Recapitulating the previous study (Rigali et al. 2008), M145 does not produce the antibiotics when GlcNAc is added to the rich complex media (representing the “feast” condition) but it does when GlcNAc is added to minimal media (representing the “famine” condition). The feast and famine for GlcNAc-dependent production may be arbitrary terms, as we show here, and the production-supportive medium can be switched to the non-supportive one with the glutamate supplementation (BMMs_1% GlcNAc + 40 mM glutamate; Fig. 5b, c). Emphasizing the close link of glutamate to the GlcNAc-dependent production, alanine did not affect the production. And, this made a sharp contrast with our earlier observation stating that glutamate can promote the production promotion when used as the sole carbon/nitrogen source (Fig. 1a). And, this inhibition shown with GlcNAc therefore indicates its double-edged effects over the production and raises a next question how coexisting organic nitrogen sources are metabolized and sensed in a complex media to determine the production. With α-ketoglutarate counteracting with glutamate-dependent production inhibition from M145 (Fig. 5d), the ratio of glutamate and α-ketoglutarate might be important for the production. Or considering the possibility of in vivo GdhB inhibition by α-ketoglutarate in line with its in vitro inhibition by TCA intermediates (Minambres et al. 2000), the decreased GdhB activity per se may result in inducing the ACT·RED production as found with the gdhB knockout. Additional study should follow to unveil the mechanism.
Taken together, NAD+-dependent GdhB (EC.1.4.1.2) a is shown to have extended physiological impacts over the nitrogen metabolism as a key member of the central nitrogen metabolism and its glutamate shunt to the TCA cycle (Fig. 6; Miflin and Habash 2002) affects the initiation of the secondary metabolism.
References
Avignone Rossa C, White J, Kuiper A, Postma PW, Bibb M, Teixeira de Mattos MJ (2002) Carbon flux distribution in antibiotic-producing chemostat cultures of Streptomyces lividans. Met Eng. 4(2):138–150. doi:10.1006/mben.2001.0217
Bajad SU, Lu W, Kimball EH, Yuan J, Peterson C, Rabinowitz JD (2006) Separation and quantitation of water soluble cellular metabolites by hydrophilic interaction chromatography-tandem mass spectrometry. J Chromato A 1125(1):76–88. doi:10.1016/j.chroma.2006.05.019
Bentley SD, Chater KF, Cerdeno-Tarraga AM, Challis GL, Thomson NR, James KD, Harris DE, Quail MA, Kieser H, Harper D, Bateman A, Brown S, Chandra G, Chen CW, Collins M, Cronin A, Fraser A, Goble A, Hidalgo J, Hornsby T, Howarth S, Huang CH, Kieser T, Larke L, Murphy L, Oliver K, O’Neil S, Rabbinowitsch E, Rajandream MA, Rutherford K, Rutter S, Seeger K, Saunders D, Sharp S, Squares R, Squares S, Taylor K, Warren T, Wietzorrek A, Woodward J, Barrell BG, Parkhill J, Hopwood DA (2002) Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 417(6885):141–147. doi:10.1038/417141a
Borodina I, Krabben P, Nielsen J (2005) Genome-scale analysis of Streptomyces coelicolor A3(2) metabolism. Gen Res. 15(6):820–829. doi:10.1101/gr.3364705
Borodina I, Siebring J, Zhang J, Smith CP, van Keulen G, Dijkhuizen L, Nielsen J (2008) Antibiotic overproduction in Streptomyces coelicolor A3(2) mediated by phosphofructokinase deletion. J Bio Chem 283(37):25186–25199. doi:10.1074/jbc.M803105200
Brana A, Paiva N, Demain A (1986) Pathways and regulation of ammonium assimilation in Streptomyces clavuligerus. J Gen Microbiol 132:1305–1317
Bressler SaA S (1984) Detection of glutamine synthetase activity in marine phytoplankton: optimization of biosynthetic assay. Marin Eco-progress series 14:207–217
Chouayekh H, Virolle MJ (2002) The polyphosphate kinase plays a negative role in the control of antibiotic production in Streptomyces lividans. Mol Microbiol 43(4):919–930
D’Alia D, Nieselt K, Steigele S, Muller J, Verburg I, Takano E (2010) Noncoding RNA of glutamine synthetase I modulates antibiotic production in Streptomyces coelicolor A3(2). J Bacteriol 192(4):1160–1164. doi:10.1128/JB.01374-09
Fink D, Weissschuh N, Reuther J, Wohlleben W, Engels A (2002) Two transcriptional regulators GlnR and GlnRII are involved in regulation of nitrogen metabolism in Streptomyces coelicolor A3(2). Mol Microbiol 46(2):331–347
Fisher SH (1989) Glutamate synthesis in Streptomyces coelicolor. J Bacteriol 171(5):2372–2377
Fisher SH, Wray LV Jr (1989) Regulation of glutamine synthetase in Streptomyces coelicolor. J Bacteriol 171(5):2378–2383
Gottelt M, Kol S, Gomez-Escribano JP, Bibb M, Takano E (2010) Deletion of a regulatory gene within the CPK gene cluster reveals novel antibacterial activity in Streptomyces coelicolor A3(2). Microbiol. 156(Pt 8):2343–2353. doi:10.1099/mic.0.038281-0
Gust B, Challis GL, Fowler K, Kieser T, Chater KF (2003) PCR-targeted Streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin. PNAS 100(4):1541–1546. doi:10.1073/pnas.0337542100
Han S, Auger C, Appanna VP, Lemire J, Castonguay Z, Akbarov E, Appanna VD (2012) A blue native polyacrylamide gel electrophoretic technology to probe the functional proteomics mediating nitrogen homeostasis in Pseudomonas fluorescens. J Microbiol Methods 90(3):206–210. doi:10.1016/j.mimet.2012.05.006
Hesketh A, Fink D, Gust B, Rexer HU, Scheel B, Chater K, Wohlleben W, Engels A (2002) The GlnD and GlnK homologues of Streptomyces coelicolor A3(2) are functionally dissimilar to their nitrogen regulatory system counterparts from enteric bacteria. Mol Microbiol 46(2):319–330
Jin Z, Jiao R (1998) Purification and properties of glutamate synthase from Streptomyces lincolnensis. Sci in China C 41(1):37–46. doi:10.1007/BF02882704
Kawada-Matsuo M, Mazda Y, Oogai Y, Kajiya M, Kawai T, Yamada S, Miyawaki S, Oho T, Komatsuzawa H (2012) GlmS and NagB regulate amino sugar metabolism in opposing directions and affect Streptococcus mutans virulence. PLoS One 7(3):e33382. doi:10.1371/journal.pone.0033382
Kieser T BM, Buttner MJ, Chater KF, and Hopwood DA (2000) Practical Streptomyces genetics. Norwich, UK: The John Innes Foundation
Kim SH, Lee BR, Kim JN, Kim BG (2012) NdgR, a common transcriptional activator for methionine and leucine biosynthesis in Streptomyces coelicolor. J Bacteriol 194(24):6837–6846. doi:10.1128/JB.00695-12
de Koning W, van Dam K (1992) A method for the determination of changes of glycolytic metabolites in yeast on a subsecond time scale using extraction at neutral pH. Anal Biochem 204(1):118–123
Kormanec J, Sevcikova B (2002) Stress-response sigma factor sigma (H) directs expression of the gltB gene encoding glutamate synthase in Streptomyces coelicolor A3(2). Biochim et Biophys Acta. 1577(1):149–154
Laycock M, McInnes AG, Walter JA, Shapiro S, Vining LC (1985) Pathway of ammonium assimilation in Streptomyces venezuelae examined by amino acid analyses and 15 N nuclear magnetic resonance spectroscopy. Can J Microbiol 31(7):629–634
Li R, Townsend CA (2006) Rational strain improvement for enhanced clavulanic acid production by genetic engineering of the glycolytic pathway in Streptomyces clavuligerus. Met Eng. 8(3):240–252. doi:10.1016/j.ymben.2006.01.003
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−delta delta C(T)) method. Methods 25(4):402–408. doi:10.1006/meth.2001.1262
Miflin BJ, Habash DZ (2002) The role of glutamine synthetase and glutamate dehydrogenase in nitrogen assimilation and possibilities for improvement in the nitrogen utilization of crops. J Exp Botany. 53(370):979–987
Minambres B, Olivera ER, Jensen RA, Luengo JM (2000) A new class of glutamate dehydrogenases (GDH). Biochemical and genetic characterization of the first member, the AMP-requiring NAD-specific GDH of Streptomyces clavuligerus. J Biol Chem. 275(50):39529–39542. doi:10.1074/jbc.M005136200
Nieselt K, Battke F, Herbig A, Bruheim P, Wentzel A, Jakobsen OM, Sletta H, Alam MT, Merlo ME, Moore J, Omara WA, Morrissey ER, Juarez-Hermosillo MA, Rodriguez-Garcia A, Nentwich M, Thomas L, Iqbal M, Legaie R, Gaze WH, Challis GL, Jansen RC, Dijkhuizen L, Rand DA, Wild DL, Bonin M, Reuther J, Wohlleben W, Smith MC, Burroughs NJ, Martin JF, Hodgson DA, Takano E, Breitling R, Ellingsen TE, Wellington EM (2010) The dynamic architecture of the metabolic switch in Streptomyces coelicolor. BMC genom 11:10. doi:10.1186/1471-2164-11-10
Ninfa AJ, Jiang P (2005) PII signal transduction proteins: sensors of alpha-ketoglutarate that regulate nitrogen metabolism. Curr Opin Microbiol 8(2):168–173. doi:10.1016/j.mib.2005.02.011
Penyige A, Keseru J, Fazakas F, Schmelczer I, Szirak K, Barabas G, Biro S (2009) Analysis and identification of ADP-ribosylated proteins of Streptomyces coelicolor M145. J Microbiol 47(5):549–556. doi:10.1007/s12275-009-0032-y
Ratcliffe PJ (2013) Oxygen sensing and hypoxia signalling pathways in animals: the implications of physiology for cancer. J Physiol 591(Pt 8):2027–2042. doi:10.1113/jphysiol.2013.251470
Rexer HU, Schaberle T, Wohlleben W, Engels A (2006) Investigation of the functional properties and regulation of three glutamine synthetase-like genes in Streptomyces coelicolor A3(2). Arch Microbiol 186(6):447–458. doi:10.1007/s00203-006-0159-8
Rigali S, Titgemeyer F, Barends S, Mulder S, Thomae AW, Hopwood DA, van Wezel GP (2008) Feast or famine: the global regulator DasR links nutrient stress to antibiotic production by Streptomyces. EMBO Rep 9(7):670–675. doi:10.1038/embor.2008.83
Ryu YG, Butler MJ, Chater KF, Lee KJ (2006) Engineering of primary carbohydrate metabolism for increased production of actinorhodin in Streptomyces coelicolor. App Environ Microbiol. 72(11):7132–7139. doi:10.1128/AEM.01308-06
Sola-Landa A, Rodriguez-Garcia A, Amin R, Wohlleben W, Martin JF (2013) Competition between the GlnR and PhoP regulators for the glnA and amtB promoters in Streptomyces coelicolor. NA Research 41(3):1767–1782. doi:10.1093/nar/gks1203
Sprusansky O, Stirrett K, Skinner D, Denoya C, Westpheling J (2005) The bkdR gene of Streptomyces coelicolor is required for morphogenesis and antibiotic production and encodes a transcriptional regulator of a branched-chain amino acid dehydrogenase complex. J Bacteriol 187(2):664–671. doi:10.1128/JB.187.2.664-671.2005
Strauch E, Takano E, Baylis HA, Bibb MJ (1991) The stringent response in Streptomyces coelicolor A3(2). Mol Microbiol 5(2):289–298
Swiatek MA, Gubbens J, Bucca G, Song E, Yang YH, Laing E, Kim BG, Smith CP, van Wezel GP (2013) The ROK family regulator Rok7B7 pleiotropically affects xylose utilization, carbon catabolite repression, and antibiotic production in Streptomyces coelicolor. J Bacteriol 195(6):1236–1248. doi:10.1128/JB.02191-12
Vancurova I, Vancura A, Volc J, Kopecky J, Neuzil J, Basarova G, Behal V (1989) Purification and properties of NADP-dependent glutamate dehydrogenase from Streptomyces fradiae. J Gen Microbiol 135(12):3311–3318
Velvizhi S, Nagalashmi T, Essa MM, Dakshayani KB, Subramanian P (2002) Effects of alpha-ketoglutarate on lipid peroxidation and antioxidant status during chronic ethanol administration in Wistar rats. Polish J Pharmacol. 54(3):231–236
Wentzel A, S. H, Consortium SE, Bruheim P (2012) Intracellular metabolite pool changes in response to nutrient depletion induced metabolic switching in Streptomyces coelicolor. Metabolites 2(1):178–194. doi:10.3390/metabo2010178
Acknowledgments
Authors would like to appreciate the scientific comments from Prof. Gilles van Wezel, Leiden University, the Netherlands.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Ethical approval
This article does not contain any studies with human participants or animals formed by any of the authors.
Funding
This research was supported by the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT, and Future Planning (NRF-2013R1A2A2A01069197).
Conflict of interest
Byung-Gee Kim declares that he has no conflict of interest. Songhee Kim declares that she has no conflict of interest.
Electronic supplementary materials
ESM 1
(PDF 107 kb)
Rights and permissions
About this article

Cite this article
Kim, S.H., Kim, BG. NAD+-specific glutamate dehydrogenase (EC.1.4.1.2) in Streptomyces coelicolor; in vivo characterization and the implication for nutrient-dependent secondary metabolism. Appl Microbiol Biotechnol 100, 5527–5536 (2016). https://doi.org/10.1007/s00253-016-7433-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-016-7433-8