
Abstract
Intestinal microbes are part of a complex ecosystem. They have a mutual relationship with the host and play an essential role in maintaining the host’s health. To optimize the feeding strategies and improve the health status of the dhole, which is an endangered species, we analyzed the structure of fecal microbes in four captive dholes using high-throughput Illumina sequencing targeting the V3–V4 region of the 16S rRNA gene. The diversity indexes and rarefaction curves indicated high microbial diversity in the intestines of the four dholes. The average number of operational taxonomical units (OTUs) in the four samples was 1196, but the number of OTUs common to all libraries was 126, suggesting only a few dominant species. Phylogenetic analysis identified 19 prokaryotic phyla from the 16S rRNA gene sequences, of which only 5 phyla were core microbiota: Bacteroidetes (21.63–38.97 %), Firmicutes (20.97–44.01 %), Proteobacteria (9.33–17.60 %), Fusobacteria (9.11–17.90 %), and Actinobacteria (1.22–2.87 %). These five phyla accounted for 97 % of the bacteria in all the dholes apart from one, in which 78 % of the bacteria were from these phyla. The results of our study provide an effective theoretical basis from which to reach an understanding of the biological mechanisms relevant to the protection of this endangered species.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The dhole (Cuon alpinus) is one of six canid species in China, the others being the wolf (Canis lupus), Tibetan fox (Vulpes ferrilata), red fox (Vulpes vulpes), orsac fox (Vulpes corsac), and racoon dog (Nyctereutes procyonoides). The dhole is on the endangered species list of the International Union for Conservation of Nature and Natural Resources (IUCN) and is also under state protection (category ii) in China. The population has fallen to less than 2500 individuals across its native territories (Srivathsa et al. 2014). Wild dholes are vigilant hunters with a rich sense of smell. As typical mountain animals, they engage in frequent and diverse hunting activities, preying on hare, wild boar, and other small animals (Karanth and Sunquist 1995; Thinley et al. 2011). A comparison with the other five canids showed that dholes’ digestive system is similar to that of wolves (Leonard et al. 2002; Savolainen et al. 2002). The most abundant phylum in the feces of healthy wolves was found to be Firmicutes (60 %), followed by Bacteroidetes (16.9 %), Proteobacteria (9.2 %), Fusobacteria (9.2 %), and Actinobacteria (4.6 %) (Zhang and Chen 2010). Unlike wolves, dholes are not exclusively carnivorous and they are known to graze in protected areas. We therefore expect the intestinal microbiota of dholes to be similar to that of other omnivorous mammals. Previous reports have shown that Firmicutes and Bacteroidetes bacteria are predominant in the feline intestine (Tun et al. 2012) and in horses (Costa et al. 2012). However, to the best of our knowledge, molecular techniques, particularly high-throughput Illumina sequencing, have not been used to assess the diversity of the bacterial community in the distal intestinal tract of dholes. With recent advances in next-generation sequencing (NGS) technologies, high-throughput sequence information can help in environment and ecosystem studies (Liu et al. 2012). The Illumina MiSeq genome analyzer (Illumina, USA) is a representative NGS system. Comprehensive use has shown these technologies to be reliable. The aim of this study was to characterize the bacterial microbial community in the gastrointestinal tract in a group of dholes via next-generation sequencing targeting the V3–V4 region of the bacterial 16S rRNA gene. Our result indicated that diet and sex might be associated with the composition of the dholes’ intestinal microbiota. These results were compared with previously reported gut microbial communities of other mammals.
Materials and methods
Sample collection
Four adult dholes with a mean live weight of 18.3 kg were captured in Jiangxi in 2010 and fed in a wild animal rescue station. Captive dholes are usually fed commercially prepared diets containing moderate quantities of carbohydrates and plant-derived soluble fibers. We gave each dhole a unique number: sample 3 (female), sample 36 (male), sample 5 (female), and sample 6 (male). They were fed a regular canine maintenance diet once a week and were not given any drugs that might affect the composition of their intestinal microbial community before the research began. All but sample 36 had no clinical signs of gastrointestinal disease. The feces of sample 36 were soft, and we speculated that the animal had loose bowels. Fecal collections were made within 30 min of defecation during the late morning of December 2014. Fecal samples were immediately placed into sterile plastic tubes. The collected samples were frozen in sample containers and carried to the lab and stored at −80 °C until DNA extraction.
DNA extraction
Total community genomic DNA extraction was performed using a QIAamp DNA Stool Mini Kit (Qiagen, Germany), following the manufacturer’s instructions. We measured the concentration of the DNA using a UV-vis spectrophotometer (NanoDrop 2000c, USA) to ensure that adequate amounts of high-quality genomic DNA had been extracted (>300 μg/μl).
16S rRNA gene amplification by PCR
Our target was the V3–V4 hyper-variable region of the bacterial 16S rRNA gene. PCR was started immediately after the DNA was extracted. The 16S rRNA V3–V4 amplicon was amplified using KAPA HiFi Hot Start Ready Mix (2×) (TaKaRa Bio Inc., Japan). Two universal bacterial 16S rRNA gene amplicon PCR primers (PAGE purified) were used: the amplicon PCR forward primer (CTACGGGNGGCWGCAG) and amplicon PCR reverse primer (GACTACHVGGGTATCTAATCC). The reaction was set up as follows: microbial DNA (5 ng/μl) 2.5 μl; amplicon PCR forward primer (1 μM) 5 μl; amplicon PCR reverse primer (1 μM) 5 μl; 2× KAPA HiFi Hot Start Ready Mix 12.5 μl (total 25 μl). The plate was sealed and PCR performed in a thermal instrument (Applied Biosystems 9700, USA) using the following program: 1 cycle of denaturing at 95 °C for 3 min, followed by 25 cycles of denaturing at 95 °C for 30 s, annealing at 55 °C for 30 s, elongation at 72 °C for 30 s, and a final extension at 72 °C for 5 min. The PCR products were checked using electrophoresis in 1 % (w/v) agarose gels in TBE buffer (Tris, boric acid, EDTA) stained with ethidium bromide (EB) and visualized under UV light. A bioanalyzer (Agilent 2100, USA) with DNA 1000 chip was used to verify the size of the PCR product.
16S gene library construction, quantification, and sequencing
We used AMPure XP beads to purify the free primers and primer dimer species in the amplicon product. To sequence our amplicon, we attached dual indices and Illumina sequencing adapters (Kozich et al. 2013) using the Nextera XT Index Kit and purified the amplicon again using AMPure XP beads. Before sequencing, the DNA concentration of each PCR product was determined using a Qubit® 2.0 Green double-stranded DNA assay and it was quality controlled using a bioanalyzer (Agilent 2100, USA). Depending on coverage needs, all libraries can be pooled for one run. The amplicons from each reaction mixture were pooled in equimolar ratios based on their concentration. Sequencing was performed using the Illumina MiSeq system (Illumina MiSeq, USA), according to the manufacturer’s instructions.
Sequence processing
After sequencing, data were collected as follows:
-
(1)
The two short Illumina readings were assembled according to the overlap and fastq files were processed to generate individual fasta and qual files, which could then be analyzed by standard methods.
-
(2)
Sequences containing ambiguous bases and any longer than 480 base pairs (bp) were dislodged and those with a maximum homopolymer length of 6 bp were allowed (Köchling et al. 2015).
-
(3)
All identical sequences were merged into one.
-
(4)
Sequences were aligned according to a customized reference database.
-
(5)
The completeness of the index and the adaptor was checked and removed all of the index and the adaptor sequence.
-
(6)
Noise was removed using the Pre.cluster tool.
Chimeras were detected by using Chimera UCHIME, and the reads flagged as chimeras were submitted to the Ribosomal Database Project (RDP) Classifier (Yost et al. 2012).We did not remove all the sequences flagged as chimeras because sequences may get flagged as chimeras in one sample but be the most abundant sequence in another sample (Edgar et al. 2011; Kozich et al. 2013). All the software was in the mothur package. Other genetic material, such as 18S rRNA gene fragments or 16S rRNA from Archaea, chloroplasts, and mitochondria, may be present at this point in the process and must be removed. We submitted the effective sequences of each sample to the RDP Classifier again to identify archaeal and bacterial sequences. Species richness and diversity statistics including coverage, chao1, ace, simpson, and shannonever were also calculated using mothur. The modified pipeline is described on the mothur website. Finally, all effective bacterial sequences without primers were submitted for downstream analysis (Kozich et al. 2013).
Nucleotide sequence accession numbers
The raw Illumina sequencing reads were submitted to the Sequencing Read Archive (SRA) database under the accession ID SRA278746.
Results
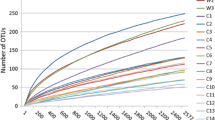
After making reads of contigs, all chimers and sequences of Archaea, chloroplasts, mitochondria and other “undesirables” were checked and filtered out by chimera UCHIME in mothur. We obtained 140,333 high-quality bacterial sequences for further analysis: 37,165 for sample 3; 30,302 for sample 36; 36,411 for sample 5; and 36,455 for sample 6. The average length of high-quality sequence was 451.3 bp. We calculated the number of operational taxonomical units (OTUs), and they were analyzed for each sample with a 97 % sequence similarity cutoff value. Figure 1 shows the rarefaction curve at an OTU definition of 97 % identity.

Rarefaction analysis of V3/V4 MiSeq sequencing reads of the 16S rRNA gene in different fecal ecosystems from four dhole samples. Rarefaction curves at a cutoff level of 3 % were constructed at a 97 % sequence similarity cutoff value in mothur
The Good’s coverage of the four samples ranged from 96.60 to 97.77 %. Each individual sample was used to estimate the completeness of sampling using a probability calculation based on a randomly selected amplicon sequence (Nam et al. 2011). We also analyzed the alpha diversity using chao1, ace, simpson, shannonever, and sobs indexes and generated heatmaps to assess beta diversity. The index of diversity at a genetic distance of 3 % is listed in Table 1.
Taxonomic composition
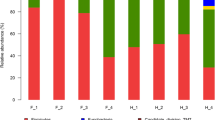
We found 19 prokaryotic phyla in the four dhole samples (Fig. 2). Only five of these were core microbiota: Bacteroidetes (21.63–38.97 %), Firmicutes (20.97–44.01 %), Proteobacteria (9.33–17.60 %), Fusobacteria (9.11–17.90 %), and Actinobacteria (1.22–2.87 %). These five phyla accounted for 97 % of the total phyla in three of the four dholes but 78 % of the phyla in sample 36. There was some variation in microbial composition among the dholes. Sample 36 had a lower abundance of Bacteroidetes (21.63 %) in its feces than did the other animals (38.97, 36.47, 29.42 %) and sample 5 had a lower abundance of Fusobacteria (9.11 %) in its feces than did the other animals (17.90, 16.63, 13.77 %).

The relative abundance of total sequences of bacterial 16S rRNA genes from fecal samples by mothur at the phylum level with a modified 16S rRNA database from the Ribosomal Database Project (RDP)
As shown in Fig. 3, a large of number of sequences could be classified at the family level (93.20, 70.62, 93.39, and 96.17 % of samples 3, 36, 5, and 6, respectively). In sample 3, Bacteroidaceae was the predominant family (20.65 %), followed by Fusobacteriaceae, Prevotellaceae, Erysipelotrichaceae, Helicobacteraceae, and Lachnospiraceae. Bacteroidaceae was also the predominant family in sample 5 (18.66 %), followed by Helicobacteraceae, Lachnospiraceae, Porphyromonadaceae, Fusobacteriaceae, Prevotellaceae, and Erysipelotrichaceae. Sample 6 and sample 36 each contained a unique family. The abundance of Moraxellaceae in sample 36 was 6.43 %, but other samples had hardly any of this family. Clostridiaceae was present in sample 6 (20.47 %), the other three samples’ abundance was averagely 0.45 %. Helicobacteraceae and Campylobacteraceae were found in all samples except sample 36. We also found many unclassified sequences at the family level: Firmicutes, Bacteroidetes, Tenericutes, Fusobacteria, Lentisphaerae, Proteobacteria, Actinobacteria, OD1_family_incertae_sedis, and TM7_family_incertae_sedis. These comprised 10.72, 34.01, 10.75, and 6.14 % of samples 3, 36, 5, and 6, respectively.

The relative abundance of total sequences of bacterial 16S rRNA genes from fecal samples by mothur at the family level with a modified 16S rRNA database from the RDP
At the genus level, unclassified bacteria in the four samples accounted for 39.36, 52.29, 37.34, and 25.66 % of the total reads for samples 3, 36, 5, and 6, respectively(Fig. 4). Bacteroides was the most abundant genus in samples 3 and 5 (20.65 and 18.66 %). Fusobacterium was most abundant in sample 36. There were large differences in the abundance of Helicobacter, Clostridium_XI, Campylobacter, Sutterella, Roseburia, Anaeroplasma, Collinsella, Parabacteroides, and Phascolarctobacterium. We were surprised to find that Helicobacter, Campylobacter, and Roseburia bacteria were common to samples 3, 5, and 6 but not present in sample 36.

The relative abundance of total sequences of bacterial 16S rRNA genes from fecal samples by mothur at the genus level with a modified 16S rRNA database from the RDP
We also compared the microbial community across the different samples. We plotted a heatmap of the jaccard and thetayc coefficients (Fig. 5) to estimate the similarities of the four dholes. Red indicates communities that are more similar than those colored in black. The heatmap shows that the highest degree of similarity was between samples 3 and 5, followed by samples 3 and 6 and then samples 5 and 6. Sample 36 was different from the other three samples. We plotted another heatmap (Fig. 6) to compare the membership and structure of the samples at the genus level. An inspection of the tree reveals that samples 3, 5, and 6 cluster together and sample 36 is excluded.

Heatmap of the jaccard and thetayc coefficients. The outlying mark number was the fourth sample. The color code represents the similarly of the four sample gut microbe communities, where black (value = 0.0) represents the lowest and red (value = 1) the highest level of abundance

Dendrogram showing the bacterial distribution among the four samples. The bacterial phylogenetic tree was calculated using the neighbor-joining method, and the relationship among samples was determined by thetayc distance and the complete clustering method. The heatmap plot depicts the percentage of each bacterial genus (variables clustering on the Y-axis) within each sample (X-axis clustering). The relative values for bacterial genus are depicted by color intensity with the legend indicated at the right of the figure. Clusters based on the distance of the four samples along the X-axis and the bacterial genus along the Y-axis are indicated in the upper part and right of the figure, respectively. Black indicates that no representative bacteria were found
Core fecal microbiota
The core microbiota in the feces of dholes is our major interest, but further research is needed to confirm its existence.
In our study, OTUs were analyzed for each sample at distance levels of 3 %. We obtained 1180, 1280, 1075, and 1248 OTUs from samples 3, 36, 5, and 6, respectively. The number of OTUs common to all libraries was 126, which comprised 12.26, 9.84, 11.72, and 11.10 % of the reads in the libraries of samples 3, 36, 5, and 6, respectively (Table 2). Firmicutes and Bacteroidetes dominated the core microbiota of the dholes. We found Clostridium, Blautia, Lachnospiraceae, Roseburia, Phascolarctobacterium, Oscillibacter, Erysipelotrichaceae, Erysipelothrix, Eubacterium, Butyricicoccus, Peptococcus, Flavonifractor in the Firmicutes; Bacteroides, Parabacteroides, Odoribacter, and Provotella in the Bacteroidetes; Sutterella, Campylobacter, Helicobacter, Anaerobiospirillum, and Parasutterella in the Proteobacteria; Fusobacterium in the Fusobacteria; and Collinsella in the Actinobacteria.
Discussion
The fecal microbiota plays an important role in nutrient pre-processing, assimilation and energy harvest from food (Ghosh et al. 2014). Carnivorous animals in particular rely on the functional bacterial population to digest fiber foods, and intestinal microbes play an essential role in the conversion of fibrous feeds into volatile fatty acids (Liang et al. 2014). However, the fecal microbiota of dholes has not been well studied at present.
We used high-throughput Illumina sequencing technology for the first time to characterize dholes’ fecal microbes. This technology allowed us to gain a deeper insight into the bacterial diversity of our samples. However, high-throughput sequencing technology is imperfect and is subject to a multitude of errors. Errors can originate in sample and library preparation and also in the bioinformatic analysis (Lawrence et al. 2013). We can try to reduce experimental error by improving the methods (Akbari et al. 2005; Lawrence et al. 2013; Williams et al. 1999; Yost et al. 2012), but little work has been done on the classification of fecal microbes, and the database of 16S RNA gene sequences is very limited. Our sequences therefore include a large number of unclassified genera. We may have had many PCR or sequencing errors. Although we tried our best to remove the “noise” (Quince et al. 2011), short-read high-throughput sequencing has weaknesses. Nevertheless, our results describe dholes’ specific fecal microbiota. However, we must make efforts to identify these unclassified bacteria and their specific roles.
The dhole is the only extant species in the genus Cuon (Carnivora: Canidae) (Zhang and Chen 2011). Genetic information shows that the species is closely related to wolves. Like wolves (Andersone and Ozolins 2004; Capitani et al. 2004), dholes eat a low-fiber diet. In recent years, many scientists have paid attention to the fecal microbes of mammals and humans. Nam et al. investigated the overall composition of the Korean human fecal microbiota. They compared microbes from individuals from the USA, China, and Japan by high-throughput sequencing and found Actinobacteria, Firmicutes, Bacteroidetes, Fusobacteria, and Proteobacteria predominated (Nam et al. 2011). Zhang et al. found the same five major phylogenetic lineages in the fecal microbiota of wolves. As in humans and most mammals (Gu et al. 2013), Bacteroidetes and Firmicutes were found to be the dominant phyla in the feces of the dholes. Within these phyla, we found that Bacteroidaceae, Porphyromonadaceae, Prevotellaceae, Erysipelotrichaceae, Lachnospiraceae, Ruminococcaceae, and Peptostreptococcaceae were the most common families. However, we also found evidence of bacteria from Lachnospiraceae and Ruminococcaceae, which are regarded as fiber-degraders in the rumen and hindgut of herbivores (Daly et al. 2012; Gu et al. 2013; Jami and Mizrahi 2012), in the feces of the four dholes. Research has shown that intestinal bacterial communities are similar to fecal bacterial communities (Gu et al. 2013). Animals have a large number of different bacteria living in the intestine. Dominant bacteria continually compete to maintain a well-balanced intestinal flora. Firmicutes is regarded as the most abundant phylum in the feces of wolves, other carnivorous animals, humans, and herbivores (Becker et al. 2014; Bian et al. 2013; Guard et al. 2015; Swanson et al. 2011; Zhang and Chen 2010). Our results indicate that Bacteroidetes and Firmicutes are abundant in the fecal microbial community of all four dholes, which differs from other carnivorous animals. However, a recent study using 454 pyrosequencing reported Bacteroidetes and Firmicutes to be the most abundant bacterial phyla in the feline intestinal microbiota. The dholes’ intestinal microbiota is more similar to that of felines than other carnivores. Felines usually eat carbohydrates and plant-derived soluble fiber, and Bacteroidetes are important degraders of complex and otherwise indigestible dietary polysaccharides in the large intestine into short-chain fatty acids that are reabsorbed by the host as an energy source (Becker et al. 2014). Interestingly, Bacteroidetes were abundant in the feces of dholes. This result may be due to the dhole’s diet. The dhole is well known to be strictly carnivorous, but captive dholes are usually fed commercially prepared diets containing moderate quantities of carbohydrates and plant-derived soluble fibers. If they are not fed this diet, dholes will sometimes get sick and experience diarrhea. We suggest that the dholes’ fecal microbiota might be influenced by the changes in their diet.
Notably, we found that Bacteroides and Fusobacterium were predominant in the four libraries, and Helicobacteraceae was found in the feces of samples 3, 5, and 6 with a relative abundance of 7.07, 11.33, and 5.05 %, respectively, but was absent from sample 36. Some strains of Helicobacteraceae, which are able to thrive in the very acidic mammalian stomach by producing large quantities of the enzyme urease, are pathogenic to humans and strongly associated with peptic ulcers, chronic gastritis, duodenitis, and stomach cancer. Moreover, Helicobacteraceae has been found to be susceptible to antibiotic drugs, including penicillin, which leads us to know that sample 36 had been treated with antibiotics because of constipation. In addition, Moraxellaceae were only found in sample 36. Information about sample 36 can be considered inconclusive.
To describe the similarity of the samples, we generated a dendrogram using jclass and thetayc coefficients. Samples 3, 5, and 6 had highly similar microbiota and samples 3 and 5 were grouped together. There was a clear difference between the clustering of the female and male samples. In addition, we found that the females had more Bacteroidetes than the males. We therefore have reason to believe that the animal’s sex can influence the fecal microbiota. Our findings are consistent with a previous study which reported lower abundance of Bacteroidetes in females than males in humans and other mammals (Dominianni et al. 2015). However, the physiological mechanism of the sexual influence is unknown.
More than 5000 bacterial species have been found in the human intestine (Dethlefsen et al. 2008). Most of the fecal microbiota is present at birth and, once established, host-specific microbial communities remain stable throughout life (Bian et al. 2013), so we can characterize the relationship between core stable microbiota and the body and diseases. This phenomenon is also relevant to the protection of wildlife. However, few previous studies have investigated the fecal microbial composition of dholes. We found that the core microbial population shared between the four individual dholes in our study comprises only 126 OTUs (Fig. 7).

Venn diagram at distance 0.03. There were 1004 species in sample 3; 1280 species in sample 36; 929 species in sample 5; and 1077 species in sample 6. Samples 3 and 36 shared 157 species; samples 3 and 5 shared 236 species; samples 3 and 6 shared 234 species; samples 5 and 36 shared 144 species; samples 6 and 36 shared 150 species; samples 5 and 6 shared 221 species; samples 3, 36 and 5 shared 135 species; samples 3, 36 and 6 shared 139 species; samples 3, 5 and 6 shared 193 species; and samples 5, 6 and 36 shared 129 species. The total richness of all the samples was 3618
In addition, we found that the core Firmicutes and Bacteroidetes bacteria in the four dholes were the same as the core fecal-associated phylotypes of omnivores such as humans and mice but not strictly carnivorous animals such as cheetahs (Becker et al. 2014; Wen et al. 2008). A possible reason for a higher diversity of omnivore-like core bacteria in the dholes is diversification of the diet. To adapt to the destruction of their environment, the dholes must be able to eat different food in order to get more energy, including some toxic phytochemicals.
In conclusion, we first described four captive dhole’s predominant fecal bacterial populations using a 16S rRNA clone library and high-throughput Illumina sequencing technology. Microbial taxon composition and relative abundance has been shown to vary dramatically among host individuals (Bolnick et al. 2014), and our study also found differences between the individual dholes. Although many factors affecting the subsequent bacterial community using high-throughput sequencing, our results reveal the complex bacterial community in the feces of four dholes living in captivity. The dholes’ microbiota, predominantly composed of Firmicutes and Bacteroidetes, resembles the fecal microbiota of omnivorous animals and is unlike that of carnivores. To increase our understanding of the microbial ecosystem diversity in endangered animals, we also provide a first taxonomic baseline for further research into the dhole intestinal ecosystem. Ultimately, understanding the associations between the structure of intestinal bacterial communities and body ecosystem parameters is important for determining animals’ physical condition, and such understanding will also contribute to the optimization of breeding regimes and improvements in the health of endangered species in captivity.
References
Akbari M, Hansen MD, Halgunset J, Skorpen F, Krokan HE (2005) Low copy number DNA template can render polymerase chain reaction error prone in a sequence-dependent manner. JMD 7(1):36–39
Andersone Z, Ozolins J (2004) Food habits of wolves (Canis lupus) in Latvia. Acta Theriol 49(3):357–367
Becker AA, Hesta M, Hollants J, Janssens GP, Huys G (2014) Phylogenetic analysis of faecal microbiota from captive cheetahs reveals underrepresentation of Bacteroidetes and Bifidobacteriaceae. BMC Microbiol 14:43. doi:10.1186/1471-2180-14-43
Bian G, Ma L, Su Y, Zhu W (2013) The microbial community in the feces of the white rhinoceros (Ceratotherium simum) as determined by barcoded pyrosequencing analysis. PLoS One 8(7):e70103. doi:10.1371/journal.pone.0070103
Bolnick DI, Snowberg LK, Hirsch PE, Lauber CL, Org E, Parks B, Lusis AJ, Knight R, Caporaso JG, Svanback R (2014) Individual diet has sex-dependent effects on vertebrate gut microbiota. Nat Commun 5:4500. doi:10.1038/ncomms5500
Capitani C, Bertelli I, Varuzza P, Scandura M, Apollonio M (2004) A comparative analysis of wolf (Canis lupus) diet in three different Italian ecosystems. Mammalian Biology - Zeitschrift für S01ugetierkunde 69:1–10
Costa MC, Arroyo LG, Allen-Vercoe E, Stӓmpfli HR, Kim PT, Sturgeon A, Weese JS (2012) Comparison of the fecal microbiota of healthy horses and horses with colitis by high throughput sequencing of the V3-V5 region of the 16S rRNA gene. PLoS One 7(7):267–278
Daly K, Proudman CJ, Duncan SH, Flint HJ, Dyer J, Shirazi-Beechey SP (2012) Alterations in microbiota and fermentation products in equine large intestine in response to dietary variation and intestinal disease. The British Journal of Nutrition 107(7):989–995. doi:10.1017/s0007114511003825
Dethlefsen L, Huse S, Sogin ML, Relman DA (2008) The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol 6(11):e280. doi:10.1371/journal.pbio.0060280
Dominianni C, Sinha R, Goedert JJ, Pei Z, Yang L, Hayes RB, Ahn J (2015) Sex, body mass index, and dietary fiber intake influence the human gut microbiome. PLoS One 10(4):e0124599. doi:10.1371/journal.pone.0124599
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics (Oxford, England) 27(16):2194–2200. doi:10.1093/bioinformatics/btr381
Ghosh TS, Gupta SS, Bhattacharya T, Yadav D, Barik A, Chowdhury A, Das B, Mande SS, Nair GB (2014) Gut microbiomes of Indian children of varying nutritional status. PLoS One 9(4):e95547. doi:10.1371/journal.pone.0095547
Gu S, Chen D, Zhang JN, Lv X, Wang K, Duan LP, Nie Y, Wu XL (2013) Bacterial community mapping of the mouse gastrointestinal tract. PLoS One 8(10):e74957. doi:10.1371/journal.pone.0074957
Guard BC, Barr JW, Reddivari L, Klemashevich C, Jayaraman A, Steiner JM, Vanamala J, Suchodolski JS (2015) Characterization of microbial dysbiosis and metabolomic changes in dogs with acute diarrhea. PLoS One 10(5):e0127259. doi:10.1371/journal.pone.0127259
Jami E, Mizrahi I (2012) Composition and similarity of bovine rumen microbiota across individual animals. PLoS One 7(3):e33306. doi:10.1371/journal.pone.0033306
Köchling T, Sanz JL, Gavazza S, Florencio L (2015) Analysis of microbial community structure and composition in leachates from a young landfill by 454 pyrosequencing. Appl Microbiol Biotechnol 99:5657–5668
Karanth KU, Sunquist ME (1995) Prey selection by tiger, leopard and dhole in tropical forests. J Anim Ecol 64(4):439–450
Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD (2013) Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol 79(17):5112–5120
Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, Kiezun A, Hammerman PS, McKenna A, Drier Y, Zou L, Ramos AH, Pugh TJ, Stransky N, Helman E, Kim J, Sougnez C, Ambrogio L, Nickerson E, Shefler E, Cortes ML, Auclair D, Saksena G, Voet D, Noble M, DiCara D, Lin P, Lichtenstein L, Heiman DI, Fennell T, Imielinski M, Hernandez B, Hodis E, Baca S, Dulak AM, Lohr J, Landau DA, Wu CJ, Melendez-Zajgla J, Hidalgo-Miranda A, Koren A, McCarroll SA, Mora J, Lee RS, Crompton B, Onofrio R, Parkin M, Winckler W, Ardlie K, Gabriel SB, Roberts CW, Biegel JA, Stegmaier K, Bass AJ, Garraway LA, Meyerson M, Golub TR, Gordenin DA, Sunyaev S, Lander ES, Getz G (2013) Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499(7457):214–218. doi:10.1038/nature12213
Leonard JA, Wayne RK, Wheeler J, Valadez R, Guillen S, Vila C (2002) Ancient DNA evidence for old world origin of new world dogs. Science (New York, NY) 298(5598):1613–1616. doi:10.1126/science.1076980
Liang J, Sha SM, Wu KC (2014) Role of the intestinal microbiota and fecal transplantation in inflammatory bowel diseases. J Dig Dis 15(12):641–646
Liu L, Li Y, Li S, Hu N, He Y, Pong R, Lin D, Lu L, Law M (2012) Comparison of next-generation sequencing systems. Biomed Research International 2012(3):355–355
Nam YD, Jung MJ, Roh SW, Kim MS, Bae JW (2011) Comparative analysis of Korean human gut microbiota by barcoded pyrosequencing. PLoS One 6(7):65–65
Quince C, Lanzen A, Davenport RJ, Turnbaugh PJ (2011) Removing noise from pyrosequenced amplicons. BMC Bioinformatics 12:38. doi:10.1186/1471-2105-12-38
Savolainen P, Zhang YP, Luo J, Lundeberg J, Leitner T (2002) Genetic evidence for an East Asian origin of domestic dogs. Science 298(5598):1610–1613. doi:10.1126/science.1073906
Srivathsa A, Karanth KK, Jathanna D, Kumar NS, Karanth KU (2014) On a dhole trail: examining ecological and anthropogenic correlates of dhole habitat occupancy in the Western ghats of India. PLoS One 9(6):e98803. doi:10.1371/journal.pone.0098803
Swanson KS, Dowd SE, Suchodolski JS, Middelbos IS, Vester BM, Barry KA, Nelson KE, Torralba M, Henrissat B, Coutinho PM, Cann IK, White BA, Fahey Jr GC (2011) Phylogenetic and gene-centric metagenomics of the canine intestinal microbiome reveals similarities with humans and mice. The ISME Journal 5(4):639–649. doi:10.1038/ismej.2010.162
Thinley P, Kamler JF, Wang SW, Lham K, Stenkewitz U, Macdonald DW (2011) Seasonal diet of dholes (Cuon alpinus) in northwestern Bhutan. Mammalian Biology - Zeitschrift für S01ugetierkunde 76(4):518–520
Tun HM, Brar MS, Khin N, Li J, Hui KH, Dowd SE, Leung CC (2012) Gene-centric metagenomics analysis of feline intestinal microbiome using 454 junior pyrosequencing. J Microbiol Methods 88(3):369–376
Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, Hu C, Wong FS, Szot GL, Bluestone JA, Gordon JI, Chervonsky AV (2008) Innate immunity and intestinal microbiota in the development of type 1 diabetes. Nature 455(7216):1109–1113. doi:10.1038/nature07336
Williams C, Ponten F, Moberg C, Soderkvist P, Uhlen M, Ponten J, Sitbon G, Lundeberg J (1999) A high frequency of sequence alterations is due to formalin fixation of archival specimens. Am J Pathol 155(5):1467–1471. doi:10.1016/s0002-9440(10)65461-2
Yost SE, Smith EN, Schwab RB, Bao L, Jung H, Wang X, Voest E, Pierce JP, Messer K, Parker BA, Harismendy O, Frazer KA (2012) Identification of high-confidence somatic mutations in whole genome sequence of formalin-fixed breast cancer specimens. Nucleic Acids Res 40(14):e107. doi:10.1093/nar/gks299
Zhang H, Chen L (2010) Phylogenetic analysis of 16S rRNA gene sequences reveals distal gut bacterial diversity in wild wolves (Canis lupus). Mol Biol Rep 37(8):4013–4022. doi:10.1007/s11033-010-0060-z
Zhang H, Chen L (2011) The complete mitochondrial genome of dhole Cuon alpinus: phylogenetic analysis and dating evolutionary divergence within canidae. Mol Biol Rep 38(3):1651–1660. doi:10.1007/s11033-010-0276-y
Acknowledgments
This research was supported by the Special Fund for Forest Scientific Research in the Public Welfare (201404420), the National Natural Science Fund of China (31372220, 31172119), the Natural Science Fund of Shandong Province of China (ZR2011CM009), and the Ph.D. Programs Foundation of Ministry of Education of China (20113705110001). The authors thank all the supports.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by the Special Fund for Forest Scientific Research in the Public Welfare (201404420), the National Natural Science Fund of China (31372220, 31172119), the Natural Science Fund of Shandong Province of China (ZR2011CM009), and the Ph.D. Programs Foundation of Ministry of Education of China (20113705110001).
Conflict of interest
All of the authors declare that they have no conflict of interest.
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Rights and permissions
About this article

Cite this article
Wu, X., Zhang, H., Chen, J. et al. Comparison of the fecal microbiota of dholes high-throughput Illumina sequencing of the V3–V4 region of the 16S rRNA gene. Appl Microbiol Biotechnol 100, 3577–3586 (2016). https://doi.org/10.1007/s00253-015-7257-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-015-7257-y