
Abstract
Bacteria represent an underexplored source of diglycosidases. Twenty-five bacterial strains from the genera Actinoplanes, Bacillus, Corynebacterium, Microbacterium, and Streptomyces were selected for their ability to grow in diglycosylated flavonoids-based media. The strains Actinoplanes missouriensis and Actinoplanes liguriae exhibited hesperidin deglycosylation activity (6-O-α-L-rhamnosyl-β-D-glucosidase activity, EC 3.2.1.168), which was 3 to 4 orders of magnitude higher than the corresponding monoglycosidase activities. The diglycosidase production was confirmed in A. missouriensis by zymographic assays and NMR analysis of the released disaccharide, rutinose. The gene encoding the 6-O-α-L-rhamnosyl-β-D-glucosidase was identified in the genome sequence of A. missouriensis 431T (GenBank accession number BAL86042.1) and functionally expressed in Escherichia coli. The recombinant protein hydrolyzed hesperidin and hesperidin methylchalcone, but not rutin, which indicates its specificity for 7-O-rutinosylated flavonoids. The protein was classified into the glycoside hydrolase family 55 (GH55) in contrast to the known eukaryotic diglycosidases, which belong to GH1 and GH5. These findings demonstrate that organisms other than plants and filamentous fungi can contribute to an expansion of the diglycosidase toolbox.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Flavone glycosides are abundant secondary metabolites of plants that are involved in numerous aspects of the manufacturing of products derived from citrus and grapes. Some characteristics like bitter taste or juice clouding are attributed in some extent to these compounds (Manthey and Grohmann 1996). The deglycosylation of different flavone glycosides represents a challenge in food technology for debittering and clarifying of fruit juices (Hemingway et al. 1999; Wang et al. 2001). The deglycosylation processes also find application in plant-based beverages, like wine or tea, to increase the aroma of the final product by releasing volatile terpens (Ma et al. 2001).
The screening of microorganisms that grow on diglycosylated flavonoids usually provides strains that produce monoglycosidases as the main tools for deglycosylation. These strains hydrolyze the disaccharidic flavonoid in a sequential manner, releasing first the exo-glycosidic moiety followed by the action of a β-glucosidase (EC 3.2.1.21), which cleaves the glycosidic bond between the remaining glucose residue and the aglycone (Sarry and Gunata 2004). The two consecutive reactions can be summarized as follows:
-
Reaction 1:
where M is a monosaccharidic moiety and M-hydrolase is a specific monoglycosidase that cleaves the glycosidic bond between M and glucose (Glc) (e.g. EC 3.2.1.40: α-L-rhamnosidase, or EC 3.2.1.37: β-D-xylosidase), and
-
Reaction 2:
Nevertheless, a few fungal strains were reported to produce diglycosidases that perform the deglycosylation of flavonoids and other diglycosylated plant metabolites in a single step (Mazzaferro and Breccia 2011):
Diglycosidases have been predominantly reported in eukaryotes, specifically in filamentous fungi and plants (see Mazzaferro and Breccia 2011 for a review; Šimčíková et al. 2015). Although a few bacterial strains have been identified as diglycosidase producers (Sang Joon et al. 1990; Yamamoto et al. 2006), to the best of our knowledge, no thorough studies about these bacterial enzymes are available. Therefore, bacteria constitute an underexplored source of diglycosidases for biotechnological applications. This work deals with the search for diglycosidase-producing bacterial strains from the genera Actinoplanes, Streptomyces, Microbacterium, Corynebacterium, and Bacillus. The diglycosidase producers were found by zymograms with the fluorescent substrate methylumbelliferyl-rutinoside, which was synthesized by transglycosylation of hesperidin using a fungal diglycosidase (Mazzaferro et al. 2012). The selected diglycosidase (6-O-α-L-rhamnosyl-β-D-glucosidase) from Actinoplanes missouriensis was recombinantly expressed and purified. In addition, the substrate specificity was characterized in view of its potential application for the hydrolysis of 7-O-rutinosylated flavonoids and other rutinose-containing compounds.
Materials and methods
Chemicals
p-Nitrophenyl-β-D-glucopyranoside (pNGlcp), p-nitrophenyl-α-L-rhamnopyranoside (pNRhap), 4-methylumbelliferyl-β-D-glucopyranoside (MU-β-glucoside), 4-methylumbelliferyl-α-L-rhamnopyranoside (MU-α-rhamnoside), 4-methylumbelliferyl-β-D-cellobioside (MU-β-cellobioside), hesperidin, hesperidin methylchalcone, diosmin, rutin, and naringin were purchased from Sigma Chemical (Buenos Aires, Argentina). 4-Methylumbelliferyl-β-D-rutinoside (MU-β-rutinoside) and p-nitrophenyl-β-D-rutinoside (pNRut) were synthesized as described before (Mazzaferro et al. 2012; Šimčíková et al. 2015).
Microbial sources, culture conditions and purification of wild-type enzyme
The microorganisms were isolated from sediment and soil samples collected from Antarctica (Base Marambio S56° 14′ 31″ W56° 37′ 48″), Patagonia Argentina (Rio Turbio S51° 31′ 49″ W72° 21′ 54″; El Calafate S50° 18′ 36″ W72° 14′ 42″), and Shala Lake (Ethiopia N7° 25′ 30″ E38° 23′ 42″) (Minig et al. 2009). Reference strains were kindly provided by the United States Department of Agriculture (USDA). The enrichment medium was adjusted to pH 10 with 100 mM Na2CO3 and contained (g/l) yeast extract 5, meat peptone 10, NaCl 10. The composition of the selection medium was as follows (g/l): hesperidin or rutin 2.5, meat peptone 2, and yeast extract 1. For solid media, 15 g/l of agar were added. Pieces of agar from the clarification zone around the colonies or culture broth from strains cultivated in the selection medium were used as the source of enzymatic activities. The agar samples were processed according to González et al. (1996).
The selected strain (A. missouriensis 431T = NRRL B-3342) was cultured in a benchtop bioreactor (1.0 l working volume) with an agitation rate of 200 rpm and aeration of 0.4 vvm at 30 °C. The culture medium contained (g/l) hesperidin 2.5, meat peptone 2, and yeast extract 1. The pH was maintained at 7.0 with 0.5 M H2SO4 and 1 M NaOH. The bioreactor was inoculated with 10 ml of an overnight culture grown in LB medium, and aliquots of 1 ml were periodically withdrawn for activity measurements. The samples were centrifuged (10 min, 12,000×g), and the pellets were washed twice with 0.16 M NaCl and resuspended in 1 ml of 50 mM Tris–HCl buffer (pH 7.0). Cell suspensions were used to detect cell-bound enzymatic activities. The wild-type protein was purified from the supernatant by anion exchange chromatography (DEAE-Sephadex, 40–120 μm, GE Healthcare Life Sciences), (NH4)2SO4 precipitation, and size exclusion chromatography (Bio-Gel P-60, Bio-Rad).
Trypsin digestion and MALDI-TOF mass spectrometry
The Coomassie brilliant blue-stained protein bands were excised from the gel, cut into small pieces and destained using 50 mM 4-ethylmorpholine acetate (pH 8.1) in 50 % v/v acetonitrile (ACN). The gel was washed with water, dehydrated in ACN, reswelled in water and partly dried in a SpeedVac concentrator. The gel pieces were incubated overnight at 37 °C with 100 ng sequencing grade trypsin (Promega, USA) in cleavage solution (25 mM 4-ethylmorpholine acetate, 5 % v/v ACN). The resulting peptides were transferred to 40 % v/v ACN/0.1 % v/v TFA. An aqueous 50 % v/v ACN/0.1 % v/v TFA solution of 5 mg/ml α-cyano-4-hydroxycinnamic acid (Sigma) was used as the MALDI matrix. One microliter of the peptide mixture was deposited on the MALDI plate and overlaid with 0.4 μl of the matrix. MALDI mass spectra were recorded on an Ultraflex III instrument (Bruker Daltonics, Germany) in the mass range of 700–4000 Da calibrated with PepMix II (Bruker Daltonics). Both MS and MS/MS data sets were searched against the NCBInr20130322 database subset of bacterial proteins using the in-house MASCOT search engine.
Cloning
Genomic DNA from A. missouriensis was isolated according to Sambrook and Russell (2001). The secretory signal sequence in the 6-O-α-L-rhamnosyl-β-D-glucosidase-encoding gene was predicted using SignalP (Petersen et al. 2011). The gene encoding the putative protein without its native signal peptide sequence was PCR amplified using the Q5 DNA polymerase (New England Biolabs), the isolated genomic DNA as a template and the primers Act-101F (5′-CACCATGGCGACCACTCCGGA) and Act-101R (5′-TCAGCGGGGGTGGGCA). The PCR product was used for cloning into the expression vector pET101/D-TOPO (Invitrogen). Replacement of the stop codon located upstream of the pET101/D-based His-tag sequence by a Ser-encoding codon was accomplished using the Phusion site-directed mutagenesis kit (Thermo Scientific) and the primers Act-His-101F (5′-CCACCCCCGCTCAAAGGGCGAGCT) and Act-His-101R (5′-GCAGCCAGCCGGGCAGAT). The PCR product, which comprised the entire insert-containing plasmid, was gel purified, phosphorylated using T4 polynucleotide kinase (New England Biolabs), and circularized. The resulting construct pET101/D-TOPO::rgh was used to transform E. coli TOP10 cells.
Bioinformatics
ExPASy software tools were used to predict the molecular mass and pI of the protein (Gasteiger et al. 2005). Protein sequence alignments and unrooted phylogenetic tree were generated using MUltiple Sequence Comparison by Log-Expectation (MUSCLE).
Heterologous expression and purification of 6-O-α-L-rhamnosyl-β-D-glucosidase
The pET101-based constructs (encoding the protein with and without a C-terminal His-tag) were transformed into E. coli BL21 Star (DE3) cells (Life Technologies) harboring the plasmid pGro7 (TaKaRa Bio Inc.) for co-expression of the cloned protein with the chaperones GroES and GroEL. Following overnight growth of a selected transformant at 28 °C in LB medium containing ampicillin (100 μg/ml) and chloramphenicol (34 μg/ml), the cultivation temperature was decreased to 22 °C and L-arabinose was added (to a final concentration of 1.7 g/l). After 1 h, IPTG was added (final concentration of 450 μM), and the cultivation continued overnight. The biomass was harvested by centrifugation (3600×g at 8 °C, 15 min), and the cell pellet was washed twice in 25 mM Tris–HCl buffer (pH 7.0). The cells were resuspended in binding buffer (see below), and cell lysis was accomplished by lysozyme and DNase treatment (0.2 mg/ml and 20 μ/ml, respectively) for 1 h, followed by sonication on ice for 15 min (pause of 1 min between pulses of 1 min; 80 % amplitude; UP50H sonicator, Hielscher Ultrasonics GmbH, Germany). The soluble fraction of the lysate, which was obtained by centrifugation (15,500×g at 4 °C, 15 min), was loaded on a Ni Sepharose column (HisTrap HP, 5 ml, GE Healthcare Life Sciences) and equilibrated with binding buffer (20 mM sodium phosphate, 500 mM NaCl, 30 mM imidazole, pH 7.4). The column was washed with binding buffer, and the bound protein was eluted with elution buffer (20 mM sodium phosphate, 500 mM NaCl, 500 mM imidazole, pH 7.4) after 24 min using a flow rate of 3 ml/min and a gradient time of 40 min.
Zymograms and agar-based activity determination
Native polyacrylamide gel electrophoresis (PAGE) was carried out according to Laemmli (1970). Enzymatic activities were detected by submerging the gel in 50 mM sodium phosphate buffer (pH 8.0) containing MU-β-glucoside, MU-α-rhamnoside, MU-β-cellobioside, or MU-β-rutinoside (Mazzaferro et al. 2012). After 10 min at 37 °C, the bands were visualized by UV illumination (312 nm). The enzymatic activity was also detected by overlaying 2 mm thick agar layer (1.5 % w/v) containing hesperidin or rutin (0.25 % w/v) and incubating them at 37 °C. Transparent halos in a clouded light yellow background indicated deglycosylation activity. The enzyme 6-O-α-L-rhamnosyl-β-D-glucosidase from Acremonium sp. DSM24697 was used as a positive control.
Enzyme assays
The total amount of reducing sugars produced during enzymatic deglycosylation of hesperidin was determined using 2,4-dinitrosalicylic acid, and the substrate specificity of the 6-O-α-L-rhamnosyl-β-D-glucosidase was assessed as described by Mazzaferro et al. (2010). Diglycosidase (6-O-α-L-rhamnosyl-β-D-glucosidase) and monoglycosidase (α-L-rhamnosidase and β-D-glucosidase) activities were determined according to Orrillo et al. (2007) using the corresponding substrates (pNRut, pNRhap, or pNGlcp). Activity measurements at different temperatures (50 mM Tris–HCl buffer pH 7.0) and at different pH values (50 °C) were performed with the following buffers (50 mM): sodium citrate (pH 4.0–6.0), sodium phosphate (pH 6.0–7.0), Tris–HCl (pH 7.0–8.9), and Na2CO3 (9.2–10.8). The products of the enzymatic reactions were analyzed by thin layer chromatography (silica gel 60 W) using ethyl acetate/2-propanol/water (3:2:2) as a mobile phase and the anthrone reagent for staining. Rutinose, which was generated by enzymatic hesperidin hydrolysis, was purified for NMR structure determination as follows: the reaction mixture was extracted twice with ethyl acetate (1:1), and the aqueous phase was concentrated and loaded on a size-exclusion chromatography column (Bio-Gel P-2, Bio-Rad). The fractions containing rutinose were pooled and lyophilized.
Results
Search for flavonoid glycoside-deglycosylating bacteria
Twenty-five bacterial strains were selected based on their ability to grow on rutin or hesperidin as the sole carbon source. Four strains were able to form a clarification zone in the solid culture medium, which was considered as indication of flavonoid glycoside degradation (Table S1, Supplementary material). The strains of the genera Bacillus, Corynebacterium, and Microbacterium increased their microbial biomass in the hesperidin- or rutin-based media, but a halo around the colonies was not observed. On the other hand, four out of 11 strains of the genera Actinoplanes and Streptomyces generated a clarification zone in the solid culture media. The extracellular extracts of these four strains were tested for hesperidin deglycosylation, α-L-rhamnosidase, and β-D-glucosidase activities. All of them were active against hesperidin and the monoglycosylated artificial substrates. The strains with the highest extracellular hesperidin deglycosidase activities (>5 U/l), namely Streptomyces sp. SES405, Actinoplanes liguriae, and A. missouriensis, were selected for further studies (Table 1). Remarkably, the strain Streptomyces sp. SES405 showed comparable values of hesperidin deglycosylation and monoglycosidase activities, while the strains A. liguriae and A. missouriensis produced only traces of monoglycosidase activities. These data suggest that the deglycosylation activities of the latter strains are likely based on the action of a diglycosidase enzyme.
Screening for diglycosidase activities
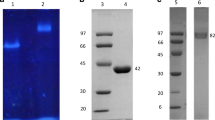
Enzyme activities were detected in native PAGE by zymography using the corresponding 4-methylumbelliferyl glycosides as substrates. The fluorogenic compounds MU-α-rhamnoside and MU-β-glucoside were used for the detection of monoglycoside-specific glycosidase activities, while MU-β-cellobioside and MU-β-rutinoside were used for diglycosidase activities. A. missouriensis and A. liguriae displayed a positive band of activity with MU-β-rutinoside; on the other hand, extracellular protein extracts from Streptomyces sp. SES405 did not exhibit activity (Fig. 1). None of the screened strains showed activity towards MU-β-cellobioside. When hesperidin was used as a substrate, the diglycosidase activities present in the supernatants of A. missouriensis and A. liguriae resulted in rutinose formation as revealed by thin-layer chromatography, whereas glucose and rhamnose were not detected (Fig. 2 and Figure S1, Supplementary material). These data, together with the fact that the α-L-rhamnosidase activities were 3–4 orders of magnitude lower than the total hesperidin deglycosylation activity (Table 1), strongly suggest the presence of an extracellular diglycosidase in these two strains. The enzyme A. missouriensis was selected for further studies because the degradation of the flavone ring by this strain is well documented and its genome publically available (GenBank: AP012319.1) (Rose and Fetzner 2006, Yamamura et al. 2012).

Zymographic analysis of extracellular extracts using MU-β-rutinoside as the substrate. Positive control: 6-O-α-L-rhamnosyl-β-D-glucosidase from Acremonium sp. DSM24697. A. missouriensis and Streptomyces sp. SES405 were grown in the presence of hesperidin as the sole carbon source, whereas A. liguriae was grown with rutin as the carbon source

Reaction scheme of the hesperidin hydrolysis catalyzed by 6-O-α-L-rhamnosyl-β-D-glucosidase (EC 3.2.1.168) from A. missouriensis
Diglycosidase production, cellular location, and hydrolysis of other substrates by A. missouriensis
The strain A. missouriensis was able to grow on hesperidin as the sole carbon source, and the diglycosidase activity was detected only in media containing the flavonoid. The maximum enzyme activity was determined at 55 °C with an apparent optimum pH value at 7.0. The strain, when cultivated in a 1-l bioreactor, produced maximum diglycosidase activity after 4 days (27.7 U/l). Minor monoglycosidase activities were also detected in the growth medium (0.11 U/l of α-L-rhamnosidase and 0.42 U/l of β-D-glucosidase activities). After 72 h of cultivation, 80 % of the enzyme activity was found in the extracellular space and 20 % associated with the cells. The enzyme, which was responsible for hesperidin deglycosylation, was purified from A. missouriensis using a three-step purification protocol. The zymographic assay revealed a positive band (R f of 0.45) for both substrates, MU-β-glucoside and MU-β-rutinoside. The main product of hesperidin hydrolysis was confirmed by 1H and 13C NMR to be rutinose (Table S2, Supplementary material; Schubert et al. 2010).
Cloning and expression of 6-O-α-L-rhamnosyl-β-D-glucosidase from A. missouriensis
The purified 6-O-α-L-rhamnosyl-β-D-glucosidase was subjected to in-gel trypsin digestion followed by MALDI-TOF mass spectrometry. Besides peptide fingerprint data, the sequences of eight peptides (SLDNLSINPIQRPVGADAER, TNASGVNWATDSTAGTSIPISR, NQVIWNGER, HLILTPGIYR, VILDDNWLWR, ISGTVNSGGDAGPGQAQYYVR, IGTGTANNIGDATNPTTLSDLFVR, NIVGTTGAAVDATQPNQVVPGFTASAR) were obtained. The MS data unambiguously identified a hypothetical protein sequence (GenBank accession number BAL86042.1) from A. missouriensis 431. An open reading frame consisting of 1902 bp was identified, starting with the codon GTG. The encoded protein was predicted to be composed of 633 amino acids, including a 34-residue long secretory signal peptide at its N-terminus. A molecular mass of 62303 Da and a pI of 6.89 was predicted for the secreted enzyme. The highest sequence identity (52 %) with experimentally verified glycoside hydrolases was determined for an endo-β-1,3-glucanase from Arthrobacter sp. NHB-10 (GenBank: BAF52916) (Okazaki et al., 2007). A CAZy database search revealed that the putative protein BAL86042.1 has been previously classified as a member of the glycoside hydrolase family 55 (GH55) (Lombard et al. 2014). A phylogenetic tree was generated to assess the sequence relationships between known diglycosidases, the 6-O-α-L-rhamnosyl-β-D-glucosidase from A. missouriensis, and closely related sequences within GH55 (Fig. 3).

Phylogenetic relationships among known β-1,3-glucanases and diglycosidases. The data set included 16 protein sequences from plantae, fungi, and bacteria. The clades are colored according to the CAZy classification of glycoside hydrolases: GH1 (green), GH5 (red), and GH55 (blue). The accession numbers or names for each protein sequence are as follows: A2SY66.1, BAC78656.1, BAD14925.1, A3RF67.1, BAG70961.1, CAK39791.1, PcLam55A, AAW47927.1, CAF32160.1, P49426.1, AJ002397.1, BAF52916.1, BAL86042.1, BAL88862.1, BAL87999.1 and SacteLam55A (from the top to the bottom). The bar represents 0.3 amino acid substitutions per site
E. coli BL21 Star (DE3) cells harboring the construct pET101/D-TOPO::rgh were cultivated for heterologous production of the C-terminal His-tagged 6-O-α-L-rhamnosyl-β-D-glucosidase (Rgh) from A. missouriensis. The enzyme Rgh was expressed in the cytosolic fraction in a soluble and active form and purified to homogeneity by metal affinity chromatography. The molecular mass estimated by SDS-PAGE (∼60 kDa) was in agreement with the predicted molecular mass of 62 kDa (Fig. 4, lane 3). Rgh showed a specific hydrolytic activity of 1.8 U/mg with hesperidin as the substrate.

SDS–PAGE analysis of the His-tagged 6-O-α-rhamnosyl-β-glucosidase during its purification. Line 1, molecular mass marker; line 2, unbound fraction of the crude extract loaded onto the Ni Sepharose column; line 3, purified 6-O-α-rhamnosyl-β-glucosidase
Substrate specificity of Rgh
The substrate specificity was tested with different mono- and diglycosides (Table 2). Two flavonoid 7-O-rutinosides, hesperidin and hesperidin methylchalcone, were shown to be substrates of the enzyme. By contrast, the hydrolysis of diosmin (diosmetin 7-O-rutinoside), rutin (quercetin 3-O-rutinoside), and naringin (naringenin-7-O-neohesperidoside) was not detected. Since Rgh exhibited the highest protein sequence identity with a known endo-1,3-β-glucanase from Arthrobacter sp., activity against laminarin, which is a predominantly linear β(1→3)-glucan with some β(1→6)-branches, was also tested. However, a detectable amount of reducing sugars was not released from laminarin when incubated with the cloned enzyme. Regarding the artificial p-nitrophenyl monoglycosides, the enzyme was not able to hydrolyze pNRhap and showed only traces of activity towards pNGlcp. By contrast, 26 % relative activity was detected for the diglycoside pNRut. The Rgh was stable in the temperature range of 25–50 °C for at least 1 h (Fig. 5). Moreover, >90 % of the initial activity was determined after incubation of the enzyme at 8 °C for 10 days (data not shown). The presence of the metal ions Mn2+, Ca2+, and Na+ (1 mM) did not affect the enzyme activity, while Zn2+, Mg2+, and the chelating agent EDTA reduced the activity by 20–50 %. The activity was only increased (25 %) by 10 mM K+.

Effect of temperature on hesperidin hydrolysis (red circles) and stability (black circles) of the recombinant 6-O-α-L-rhamnosyl-β-D-glucosidase. Enzyme stability was assessed by determining the residual activity at 50 °C after incubation at different temperatures for 60 min. The 100 % activity corresponded to 0.24 U/ml for hydrolysis and 0.48 U/ml for stability assays
Discussion
Diglycosidases have been reported to be chiefly produced by eukaryotic organisms, specifically by filamentous fungi and plants (Mazzaferro and Breccia 2011). Only a few bacterial diglycosidase producers and one archaebacterium have been identified (Sang Joon et al. 1990; Yamamoto et al. 2006; Nam et al. 2012). In this work, we screened a panel of bacterial strains for diglycosidase activities. Two strains, A. missouriensis and A. liguriae, out of 25 tested, were able to grow on hesperidin and rutin, respectively, as the sole carbon source and at the same time exhibited extracellular diglycosidase activities. In contrast to monoglycosidase activities such as α-L-rhamnosidase and β-D-glucosidase, which seem to be widespread, diglycosidase activities are rather scarce. The bacterial degradation of flavonoids has been reported for some strains within the order Rhizobiales and the genera Streptomyces, Actinoplanes, Bacillus, Corynebacterium, Pseudomonas, and Xanthomonas (Merkens et al. 2008; Rao and Cooper 1994; Rose and Fetzner 2006; Westlake et al. 1959). These studies focused on the capability of bacteria to oxidize the flavonoid aromatic rings. In the case of glycosylated flavonoids, a deglycosylation step preceding oxidation is usually required. Streptomyces sp. FLA is able to utilize quercetin as the sole carbon source; however, the growth is poor (Merkens et al. 2008). Our results with Streptomyces sp. SES405 indicate that this strain deglycosylates hesperidin via a sequential mechanism since it displayed monoglycosidase activities while diglycosidase activity was not detected. A work published by Rose and Fetzner (2006) describes the identification of a linear plasmid in A. missouriensis, which is involved in the degradation of the flavone ring. This strain was able to degrade not only the non-glycosylated flavonoid quercetin but also the glycosylated flavonoids rutin and hesperidin. The 6-O-α-L-rhamnosyl-β-D-glucosidase is likely responsible for the deglycosylation of the latter flavonoid in A. missouriensis.
The genome of A. missouriensis (strain 431T) consists of 8204 predicted genes; out of these, 8125 encode proteins and 79 encode RNAs. A putative function was assigned to 55.9 % of the protein-encoding genes (Yamamura et al. 2012). In this work, the putative protein BAL86042.1 was experimentally linked to a catalytic function, specifically 6-O-α-L-rhamnosyl-β-D-glucosidase activity. The enzyme was found to be a member of GH55. This family comprised until now only exo-β-1,3-glucanases (EC 3.2.1.58) and endo-β-1,3-glucanases (EC 3.2.1.39). A phylogenetic analysis with the known diglycosidases, GH55 exo-β-1,3-glucanases and GH55 endo-β-1,3-glucanases revealed that the diglycosidases from plants clustered in a separate clade as GH1 family members, whereas the fungal diglycosidases gathered in GH5; enzymes in both glycoside hydrolase families exhibit a retaining catalytic mechanism.
Recently, the glutamate at the position 502 of the β-1,3-exo-glucanase SacteLam55A from Streptomyces sp. SirexAA-E was identified by site-directed mutagenesis and structural studies as the catalytic acid (Bianchetti et al. 2015). This catalytic glutamate is conserved throughout the GH55 family in both prokaryotic and eukaryotic microorganisms (Ishida et al. 2009, Bianchetti et al. 2015). The sequence alignment of the 6-O-α-L-rhamnosyl-β-D-glucosidase from A. missouriensis with SacteLam55A, the β-1,3-exo-glucanase PcLam55Afrom Phanaerochaete chrysosporium (Ishida et al. 2009), and other GH55 β-1,3-glucanases (Okazaki et al. 2007) allowed the identification of Glu517 as the presumable catalytic acid (Table S3, Supplementary material).
Several glycosidases were reported to recognize both mono- and diglycosides with different degrees of specificity. The Acremonium sp. DSM24697 6-O-α-L-rhamnosyl-β-D-glucosidase and the Aspergillus niger K2 rutinosidase are highly specific for diglycosylated substrates and exhibit rather low or null activities against monoglycosylated compounds (Mazzaferro et al. 2010; Šimčíková et al. 2015). The ratio of β-D-glucosidase activity to 6-O-α-L-rhamnosyl-β-D-glucosidase activity (<0.04) for the A. missouriensis enzyme clearly shows its preference for the diglycosylated substrate, exhibiting only traces of β-D-glucosidase activity. By contrast, the Pyrococcus furiosus β-D-glucosidase is promiscuous enough to hydrolyze diglycosylated compounds, albeit with a low ratio of β-D-glucosidase activity to β-D-rutinosidase activity (>7000) (Nam et al. 2012). The 6-O-α-L-rhamnosyl-β-D-glucosidases from A. missouriensis and Acremonium sp. DSM24697 exhibited both a preference for 7-O-rutinosylated flavonoids (hesperidin and hesperidin methylchalcone) (Mazzaferro et al. 2010). The hydrolysis of rutin, a 3-O-rutinosylated flavonoid, was not detected. Among diglycosidases, the reports on enzymes that preferentially hydrolyze hesperidin are circumscribed to these two proteins, while there are at least six enzymes that hydrolyze rutin (Sang Joon et al. 1990, Narikawa et al. 2000, Nam et al. 2012, Šimčíková et al. 2015).
Until now, two activities have been described in the GH55 family: β-1,3-exo-glucanases and β-1,3-endo-glucanases. Recently, 51 members of the GH55 were tested with a panel of substrates, from which 34 proteins were found to be active (Bianchetti et al. 2015). Laminarin was the only substrate that gave a positive assay response (Bianchetti et al. 2015). The authors suggested that the enzymes whose activities could not be experimentally verified might be specific for other substrates. The enzyme described in this work represents the first member of the family with an enzymatic activity other than β-1,3-glucanase.
Eukaryotic diglycosidases are effective catalysts when applications such as the release of volatile compounds during aroma modulation of plant-based foods are envisioned (Mizutani et al. 2002; Minig et al. 2011). These enzymes group under GH1 and GH5, respectively. Because of their retaining mechanism, transglycosylation side activity is to be expected. Indeed, the synthetic potential of diglycosidases from fungal origin has been demonstrated: they in vitro glycosylate alkylic and phenolic alcohols (Mazzaferro et al. 2012; Šimčíková et al. 2015). However, transglycosylation reactions might be disadvantageous for industrial biotransformations where solely hydrolysis is expected. The bacterial diglycosidase herein described belongs to GH55 and, consistently with the inverting mechanism of the family, did not show transglycosylation activity. Therefore, our results open up the possibility to carry out predictable biotransformations of complex mixtures. We demonstrated as well that bacteria contribute to an expansion of the diglycosidase toolbox.
References
Bianchetti CM, Takasuka TE, Deutsch S, Udell HS, Yik EJ, Bergeman LF, Fox BG (2015) Active site and laminarin binding in glycoside hydrolase family 55. J Biol Chem 290:11819–11832. doi:10.1074/jbc.M114.623579
Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A (2005) Protein identification and analysis tools on the ExPASy server. In: John M. Walker (ed): The Proteomics Protocols Handbook, Humana Press, pp 571–607
González C, Martínez A, Vázquez F, Baigori M, Figueroa LIC (1996) New method of screening and differentiation of exoenzymes from industrial strains. Biotechnol Tech 10:519–522. doi:10.1007/BF00159517
Hemingway KM, Alston MJ, Chappell CG, Taylor AJ (1999) Carbohydrate-flavour conjugates in wine. Carbohydr Polymer 38:283–286. doi:10.1016/S0144-8617(98)00103-9
Ishida T, Fushinobu S, Kawai R, Kitaoka M, Igarashi K, Samejima M (2009) Crystal structure of glycoside hydrolase family 55 β-1,3-glucanase from the basidiomycete Phanerochaete chrysosporium. J Biol Chem 284:10100–10109. doi:10.1074/jbc.M808122200
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. doi:10.1038/227680a0
Lombard V, Golaconda RH, Drula E, Coutinho PM, Henrissat B (2014) The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res 42:D490–D495
Ma SJ, Mizutani M, Hiratake J, Hayashi K, Yagi K, Watanabe N, Sakata K (2001) Substrates specificity of β-primeverosidase, a key enzyme in aroma formation during oolong tea and black tea manufacturing. Biosci Biotechnol Biochem 65:2719–2729
Manthey JA, Grohmann K (1996) Concentrations of hesperidin and other orange peel flavonoids in citrus processing by products. J Agric Food Chem 44:811–814. doi:10.1021/jf950572g
Mazzaferro L, Piñuel L, Minig M, Breccia JD (2010) Extracellular monoenzyme deglycosylation system of 7-O-linked flavonoid β-rutinosides and its disaccharide transglycosylation activity from Stilbella fimetaria. Arch Microbiol 2010;192:383–393 Erratum in: Arch Microbiol (2011) 193:461. doi:10.1007/s00203-010-0567-7
Mazzaferro L, Breccia JD (2011) Functional and biotechnological insights into diglycosidases. Biocat Biotrans 29:103–112. doi:10.3109/10242422.2011.594882
Mazzaferro L, Piñuel L, Erra-Balsells R, Giudicessi SL, Breccia JD (2012) Transglycosylation specificity of Acremonium sp. α-rhamnosyl-β-glucosidase and its application to the synthesis of the new fluorogenic substrate 4-methylumbelliferyl-rutinoside. Carbohydr Res 347:69–75. doi:10.1016/j.carres.2011.11.008
Merkens H, Kappl R, Jakob RP, Schmid FX, Fetzner S (2008) Quercetinase QueD of Streptomyces sp. FLA, a monocupin dioxygenase with a preference for nickel and cobalt. Biochemistry 47:12185–12196. doi:10.1021/bi801398x
Minig M, Walker D, Ledesma P, Martínez MA, Breccia JD (2009) Bacterial isolates from Ethiopian soda lake producers of alkaline-active β-glucanases resistant to chelating and surfactant compounds. Res J Microbiol 4:194–201. doi:10.3923/jm.2009.194.201
Minig M, Mazzaferro LS, Erra-Balsells R, Petroselli G, Breccia JD (2011) α-Rhamnosyl-β-glucosidase-catalyzed reactions for analysis and biotransformations of plant-based foods. J Agr Food Chem 59:11238–11243
Mizutani M, Nakanishi H, Ema J, Ma SJ, Noguchi E, Inohara-Ochiai M, Fukuchi-Mizutani M, Nakao M, Sakata K (2002) Cloning of β-primeverosidase from tea leaves, a key enzyme in tea aroma formation. Plant Physiol 130:2164–2176
Nam HK, Hong SY, Shin KC, Oh DK (2012) Quercetin production from rutin by a thermostable β-rutinosidase from Pyrococcus furiosus. Biotechnol Lett 34:483–489. doi:10.1007/s10529-011-0786-2
Narikawa T, Shinoyama H, Fujii T (2000) A β-rutinosidase from Penicillium rugulosum IFO 7242 that is a peculiar flavonoid glycosidase. Biosci Biotechnol Biochem 64:1317–1319. doi:10.1271/bbb.64.1317
Okazaki K, Nishimura N, Matsuoka F, Hayakawa S (2007) Cloning and characterization of the gene encoding endo-β-1,3-glucanase from Arthrobacter sp. NHB-10. Biosci Biotechnol Biochem 71(6):1568–1571. doi:10.1271/bbb.70030
Orrillo AG, Ledesma P, Delgado OD, Spagna G, Breccia JD (2007) Cold-active α-L-rhamnosidase from psychrotolerant bacteria isolated from a sub-Antarctic ecosystem. Enz Microb Technol 40:236–241. doi:10.1016/j.enzmictec.2006.04.002
Petersen TN, Brunak S, Heijne G, Nielsen H (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8:785–786. doi:10.1038/nmeth.1701
Rao JR, Cooper JE (1994) Rhizobia catabolize nod gene-inducing flavonoids via C-ring fission mechanisms. J Bacteriol 176:5409–5413. doi:0021–9193/94/$04.00+0
Rose K, Fetzner S (2006) Identification of linear plasmid pAM1 in the flavonoid degrading strain Actinoplanes missouriensis (DSM 43046). Plasmid 55:249–254. doi:10.1016/j.plasmid.2005.10.003
Sambrook J, Russell D (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY
Sang-Joon L, Omorl T, Kodama T (1990) Purification and some properties of rutinosidase from Arthrobacter sp. Kor J Appl Microbiol Biotech 18:360–367
Sarry JE, Gunata Z (2004) Plant and microbial glycoside hydrolases: volatile release from glycosidic aroma precursors. Food Chem 87:509–521. doi:10.1016/j.foodchem.2004.01.003
Schubert M, Melnikova AN, Mesecke N, Zubkova EK, Fortte R, Batashev DR, Barth I, Sauer N, Gamalei YV, Mamushina NS, Tietze LF, Voitsekhovskaja OV, Pawlowski K (2010) Two novel disaccharides, rutinose and methylrutinose, are involved in carbon metabolism in Datisca glomerata. Planta 231:507–521. doi:10.1007/s00425-009-1085-1
Šimčíková D, Kotik M, Weignerová L, Halada P, Pelantová H, Adamcová K, Křen V (2015) α-L-Rhamnosyl-β-D-glucosidase (rutinosidase) from Aspergillus niger: characterization and synthetic potential of a novel diglycosidase. Adv Synth Catal 357(1):107–117. doi:10.1002/adsc.201400566
Wang D, Kurasawa E, Yamaguchi Y, Kubota K, Kobayashi A (2001) Analysis of glycosidically bound aroma precursors in tea leaves. 2. Changes in glycoside contents and glycosidase activities in tea leaves during the black tea manufacturing process. J Agric Food Chem 49:1900–1903. doi:10.1021/jf001077+
Westlake DWS, Talbot G, Blakley ER, Simpson FJ (1959) Microbial decomposition of rutin. Can J Microbiol 5:621–629. doi:10.1139/m59-076
Yamamoto S, Okada M, Usui T, Sakata K, Toumoto A, Tsuruhami K (2006) Diglycosidase isolated from microorganisms. United States Patent 71090142006 Sep 19.
Yamamura H, Ohnishi Y, Ishikawa J, Ichikawa N, Ikeda H, Sekine M, Harada T, Horinouchi S, Otoguro M, Tamura T, Suzuki K, Hoshino Y, Arisawa A, Nakagawa Y, Fujita N, Hayakawa M (2012) Complete genome sequence of the motile actinomycete Actinoplanes missouriensis 431T (= NBRC 102363T). SIGS 7:294–303. doi:10.4056/sigs.3196539
Acknowledgments
This work was supported by the National University of La Pampa (UNLPam), the National Council of Scientific and Technical Research (CONICET), and The National Agency for Science and Technology Promotion (ANPCyT) of Argentina. Funding of the bilateral cooperation project 7AMB13AR005 (ARC/12/04) by the Ministry of Education, Youth and Sports (MEYS) of the Czech Republic and the Ministry of Science, Technology and Innovation (MINCYT) of Argentina is acknowledged. This work was also supported by the MEYS projects LD13042 and LD15085. Further, the authors also thank the support for the networking by the European COST Chemistry project MultiGlycoNano CM1102.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no competing interests.
Electronic supplementary material
ESM 1
(PDF 546 kb)
Rights and permissions
About this article

Cite this article
Neher, B.D., Mazzaferro, L.S., Kotik, M. et al. Bacteria as source of diglycosidase activity: Actinoplanes missouriensis produces 6-O-α-l-rhamnosyl-β-d-glucosidase active on flavonoids. Appl Microbiol Biotechnol 100, 3061–3070 (2016). https://doi.org/10.1007/s00253-015-7088-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-015-7088-x