
Abstract
The pseudorabies virus (PRV) UL42 protein, known as the DNA polymerase processivity factor, is an essential protein required for viral replication. The in vitro function of UL42 has been characterized; however, there is little information concerning the linear B cell epitopes of UL42 that are recognized during humoral immune responses. We generated and characterized six UL42-reactive monoclonal antibodies (mAbs) from mice that had been immunized with a recombinant form of UL42. Through western blotting analysis, we identified two regions of UL42 (amino acids 39–148 and 302–384) that reacted with these mAbs. We then synthesized a panel of UL42-derived peptides spanning the two regions and screened the six mAbs. We were able to identify three linear epitopes (116SGGVLDALK124, 354KRPAAPR360, and 360RMYTPIAK367) by enzyme-linked immunosorbent assays. The 116SGGVLDALK124 epitope was located at the amino-terminus, while the other two epitopes were at the carboxy-terminus. Using these mAbs, we found that UL42 localized to the nucleus during viral replication and could be immunoprecipitated from PRV-infected PK-15 cells. We also established a UL42 mAb-based immunoperoxidase monolayer assay for the determination of PRV titers. Sequence analysis showed that the linear epitopes of UL42 were highly conserved among PRV strains. Taken together, our results indicate that the six generated mAbs could be useful tools for investigating the structure and function of UL42 during viral replication. In addition, these mAbs could be applied to diagnostic and therapeutic approaches for the effective control of PRV infections.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pseudorabies virus (PRV) is the causative agent of pseudorabies (PR), which is also known as Aujeszky’s disease. PRV is prevalent in most swine herds, with PR responsible for severe economic losses in the pig industry worldwide. PR exhibits a variety of clinical signs and lesions, depending on the age of the infected animals. In general, PR is characterized by neurological signs, severe respiratory syndromes, abortions, reduced litter size, and decreased growth rates of survivors (Muller et al. 2011). It can result in high morbidity and mortality in newborn piglets (Pomeranz et al. 2005; Marcaccini et al. 2008).
PRV belongs to the genus Varicellovirus of the subfamily Alphaherpesvirinae in the family Herpesviridae (Klupp et al. 2004). It has a linear double-stranded DNA (dsDNA) genome that is approximately 150 kb. The organization of genes in the PRV genome is similar to that seen for other alphaherpesviruses, such as herpes simplex virus type 1 (HSV-1), varicella-zoster virus (VZV), and equine herpesvirus (EHV). In addition, the amino acid sequences of gene products share high levels of homology with those from other alphaherpesviruses (Ben-Porat et al. 1983). The replication of PRV DNA occurs via a rolling circle mechanism, yielding concatemers that then become the substrate for genome encapsidation (Mettenleiter 1994b). In HSV-1, seven viral genes are required for its replication. These genes encode an origin-binding protein (UL9), three components of a helicase-primase complex (UL5, UL8, and UL52), DNA polymerase (UL30), an accessory protein for DNA polymerase (UL42), and a single-stranded DNA-binding protein (UL29) (Lundh 1990). These seven genes are also found in PRV and are presumed to function similarly (Pomeranz et al. 2005). The genes encoding UL30 and UL42 are essential for viral DNA replication and are conserved within all members of the Herpesviridae. The UL30 gene encodes the catalytic subunit of the viral DNA-dependent DNA polymerase, while the UL42 gene encodes an accessory subunit and is sometimes referred to as a processivity factor (Romero et al. 1997). UL30 possesses DNA polymerase activity and can be stimulated by the addition of UL42 in vitro (Berthomme et al. 1995). UL42 binds to dsDNA in the absence of other proteins and interacts, either directly or via other proteins that are yet to be identified, with the viral DNA polymerase (Murphy et al. 1989).
The in vitro functions of the PRV UL42 protein have been characterized (Berthomme et al. 1995). However, there is little available information concerning the linear B cell epitopes of UL42 that are recognized during the humoral immune responses. In the current study, we sought to generate six monoclonal antibodies (mAbs) against the UL42 protein and to identify its epitopes. We also attempted to characterize the subcellular localization of UL42 during viral replication and wished to determine whether UL42 could be applied to immunoprecipitation assays. Moreover, we established a UL42 mAb-based immunoperoxidase monolayer assay (IPMA) to detect PRV titers instead of the traditional approach. These data indicate that the mAbs characterized here would become very useful tools to investigate the function of UL42.
Materials and methods
Cell lines and viruses
The myeloma cell line SP2/0 was cultured in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen, Carlsbad, NM, USA) supplemented with 10 % heat-inactivated fetal bovine serum (FBS; Gibco-BRL, Grand Island, NY, USA), 100 μg/mL streptomycin, and 100 IU/mL penicillin at 37 °C/5 % CO2. The JF strain of PRV (PRV-JF) had been previously isolated from a pig farm on mainland China (Zhang et al. 2008) and was maintained in our laboratory in PK-15 cells.
Recombinant protein expression and purification
The pET28a-UL42 plasmid, which we had previously generated, was used to express UL42 (GenBank accession: KP279683) of PRV-JF. We transformed pET28a-UL42 into Escherichia coli BL21 (Novagen, Madison, WI, USA) and induced expression of recombinant UL42 with 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) over 6 h in Luria-Bertani medium containing 100 μg/mL kanamycin. Bacterial cultures were harvested, and crude lysates were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Separated proteins were visualized by soaking polyacrylamide gels in 0.25 M KCl. Bands corresponding to UL42 were excised, homogenized, and then added to an appropriate volume of sterile phosphate-buffered saline (PBS). After several cycles of freeze-thawing, PBS was separated from the solid gel by centrifugation. The purity of proteins was analyzed by SDS-PAGE, and the purified proteins were used to immunize mice. The recombinant UL42 protein was identified using a mouse mAb against the histidine (His) tag (Sigma-Aldrich, St. Louis, MO, USA) that was incorporated into the recombinant protein.
Generation of monoclonal antibodies against UL42
We subcutaneously immunized four female BALB/c mice (6 weeks old) with 50 μg of purified recombinant UL42 that was emulsified in complete Freund’s adjuvant. Booster immunization was performed in the same manner after 3 weeks, except that the protein was emulsified in incomplete Freund’s adjuvant. Following the booster immunization, mice were intraperitoneally administered 100 μg of recombinant UL42 without adjuvant at a 2-week interval. Mice were then euthanized 3 days later, and harvested spleen cells were fused with SP2/0 cells using standard procedures (Galfre and Milstein 1981). Fused cells were cultured in DMEM containing hypoxanthine-aminopterin-thymidine (Sigma-Aldrich) and 20 % FBS. The resulting hybridoma cells were maintained in DMEM containing hypoxanthine-thymidine (Sigma-Aldrich) and 10 % FBS. Hybridoma supernatants were assayed for the presence of UL42-specific antibodies by western blotting and IPMA as described previously (Liu et al. 2004). Briefly, PK-15 cells in 96-well plates were infected with PRV-JF at a multiplicity of infection (MOI) of 1 and allowed to incubate at 37 °C/5 % CO2 for 24 h. Cells were fixed with 33.3 % acetone in PBS for 20 min at room temperature and then dried at 37 °C for 30 min. Cells were then incubated with 100 μL of hybridoma supernatants at 37 °C for 1 h. Following three washes with PBS, 100 μL of horseradish peroxidase (HRP)-conjugated protein A (Invitrogen) diluted 1:3000 was added and allowed to incubate for 30 min at 37 °C. After further washing, color development was carried out with 3-amino-9-ethylcarbazole (AEC) and H2O2 in 0.05 M acetate buffer (pH 5.0) for 30 min at room temperature. The color development reaction was terminated by the removal of the substrate. Cells were washed again with PBS and then examined with an inverted light microscope. Selected positive hybridomas were cloned three times by limiting dilution. Ascites containing UL42 mAbs were prepared from mice injected intraperitoneally with 0.5 mL of sterile paraffin oil and hybridomas (105 cells/mouse) suspended in DMEM. Titers of mAbs were determined using immunofluorescence assays, and antibody subtypes were determined using a Mouse MonoAb-ID Kit (HRP) (Invitrogen) according to the manufacturer’s instructions.
Polypeptide design and expression
To map the epitopes of the generated mAbs, a series of polypeptides were expressed. Three overlapping peptides spanning the UL42 protein (amino acids 1–148, 124–280, and 256–384) were expressed as enhanced green fluorescent protein (EGFP) fusion proteins. Fragments encoding these peptide sequences were amplified from pET28a-UL42 and cloned into the pEGFP-C1 vector for their expression. The resulting constructs were transfected into HEK293T cells for eukaryotic expression using Attractene Transfection Reagent (Qiagen, Dusseldorf, Germany) according to the manufacturer’s instructions. Cells were harvested at 24 h post-transfection. Expression of each of the EGFP-fused recombinant proteins was confirmed by western blotting using a mouse mAb against EGFP (Sigma). Subsequently, two polypeptides spanning amino acids 1–148 (amino acids 1–54 and 39–148) and two polypeptides spanning amino acids 256–384 (amino acids 256–317 and 302–384) were also designed and expressed as EGFP fusion proteins. In the same way, the coding sequences of these four polypeptides were inserted into the pEGFP-C1 vector for eukaryotic expression. These recombinant proteins were also expressed in HEK293T cells and confirmed by western blot as described above. In addition, five peptides spanning amino acids 39–148 and four peptides spanning amino acids 302–384 of PRV UL42 were synthesized by GL Biochem (Shanghai, China) (Table 1(A)). Thirty-six other overlapping peptides spanning amino acids 105–136 and 343–374 of PRV UL42 were synthesized by GL Biochem for further epitope mapping (Table 1(B)).
Western blotting
The reactivity of our mAbs with the EGFP-fused UL42 polypeptides was analyzed by western blotting. Briefly, culture lysates containing EGFP-fused polypeptides or EGFP alone were subjected to SDS-PAGE using 12 % polyacrylamide gels. Proteins were transferred onto nitrocellulose membranes, which were then blocked with 3 % (w/v) skim milk in PBS overnight at 4 °C. Nitrocellulose membranes were incubated with mAbs against UL42 (1:10,000 dilution) for 1 h. After washing three times with PBS containing 0.05 % (v/v) Tween 20 (PBS-T), membranes were incubated with IRDye 800CW goat anti-mouse IgG (H+L) (1:10,000; Li-Cor Biosciences, Lincoln, NE, USA) for 1 h. Membranes were washed with PBS-T and then scanned and analyzed with an Odyssey infrared imaging system (Li-Cor Biosciences).
Enzyme-linked immunosorbent assays
Synthesized peptides were screened by an indirect enzyme-linked immunosorbent assay (ELISA) as described previously (Huang et al. 2012). We used 96-well ELISA plates, and each well was coated with synthesized peptide (30 μg/well) and incubated overnight at 4 °C. Plates were blocked with 1 % bovine serum albumin (BSA) in PBS for 1 h at 37 °C. Plates were washed three times with PBS-T, and then, we added 100 μL of ascites fluid (diluted 1:10,000 in PBS containing 1 % BSA) to each well and incubated plates at 37 °C for 1 h. Following three washes with PBS-T, bound mAbs were detected using HRP-conjugated protein A (1:5000). After three washes with PBS-T, color was developed by adding 100 μL of 2,2-azino-di [3-ethylbenzthiazoline sulfonic acid] (ABTS; 0.21 mg/mL) in 0.1 M citrate buffer (pH 4.2) containing 0.003 % H2O2. Color development reactions were terminated by adding 50 μL of 1 % NaF. The optical density (OD) value at 405 nm for well was measured using a microplate reader. Purified UL42 and an irrelevant peptide (amino acids 1–15 of UL42) served as positive and negative controls, respectively. Relative affinity of each mAb was also assayed by ELISA as described previously (Restle et al. 1992; Sheaffer et al. 1995). Each well of ELISA plates was coated with purified UL42 protein (15 μg/well). MAb IgGs were purified from ascites fluid using HiTrap Protein G HP (GE Healthcare, Fairfield, CT, USA) according to the manufacturer’s instructions and used as primary antibodies with serial dilutions.
Complementary DNA cloning of monoclonal antibody variable region genes
RNA was prepared from about 5 × 106 hybridoma cells that produced mAbs against UL42 using RNeasy Mini Kit (Qiagen) according to the manufacturer’s protocol. Reverse transcription reaction was performed to synthesize first-strand complementary DNA (cDNA) using Transcriptor First-Strand cDNA Synthesis Kit (Roche, Basel, Switzerland) according to the manufacturer’s instructions. cDNA fragments encoding variable regions of mAb heavy and light chains were amplified using primers listed in Table 2. The primers were designed based on the published mouse mAb sequences. PCRs were performed using the Ex Taq DNA polymerase kit (Takara, Dalian, China) according to the manufacturer’s protocols with the cDNA as template. The reactions consisted of initial denaturation at 94 °C for 5 min, 35 cycles of denaturation at 94 °C for 30 s, annealing at 58 °C for 30 s, and an extension at 72 °C for 1 min and a final extension at 72 °C for 7 min. The fragments were then cloned into pMD18-T vectors (Takara) and subject to sequencing analysis by Comate Bioscience Company Limited (Changchun, China).
Indirect immunofluorescence assays
We used immunofluorescence assays (IFAs) to determine the location of UL42 during PRV replication. We conducted IFAs as described previously by Liu et al. (Liu et al. 2007). PK-15 cells were infected with PRV-JF at an MOI of 1 for 24 h. Cells were then fixed with cold acetone for 10 min at −20 °C. Fixed cells were incubated with the six mAbs against UL42 for 1 h at 37 °C in a humidified chamber and were then washed three times with PBS. Cells were incubated with a goat anti-mouse IgG conjugated to DyLight™ 488 (1:500; Pierce, Rockford, IL, USA) for 1 h at 37 °C, followed by three washes with PBS. Cells were also stained with DAPI for 10 min at room temperature to visualize nuclei. Fluorescence was assessed with the assistance of a Leica SP2 confocal system (Leica Microsystems, Wetzlar, Germany).
Immunoprecipitation of UL42
We attempted to immunoprecipitate UL42 from cells infected with PRV-JF. PK-15 cells infected with PRV-JF at an MOI of 3 were harvested at 24 h post-infection, washed three times with cold PBS, and lysed with Triton X-100 buffer (20 mM Tris pH 7.5, 150 mM NaCl, 1 % Triton X-100, and 0.5 mM EDTA) containing 1 mM phenylmethylsulfonyl fluoride for 30 min at 4 °C. Lysates were centrifuged (10,000×g, 10 min, 4 °C), and the resulting supernatants were subjected to immunoprecipitation. We used protein A/G plus agarose immunoprecipitation reagent (Santa Cruz Biotechnology, Dallas, TX, USA) according to the manufacturer’s protocols to precipitate UL42. Lysates were pretreated with 20 μL of protein A/G agarose at 4 °C for 2 h. Supernatants were collected following centrifugation and subjected to immunoprecipitation using the six mAbs against UL42 (1:100). Samples were then incubated with 40 μL of protein A/G agarose at 4 °C overnight. The agarose beads were washed with PBS, centrifuged, and boiled in SDS-PAGE sample buffer (250 mM Tris pH 6.8, 10 % SDS, 0.5 % bromophenol blue, 50 % glycerol, and 5 % β-mercaptoethanol) for 5 min. Samples were separated using 12 % SDS-PAGE and then subjected to western blotting analysis using the six mAbs against UL42 as primary antibodies.
Pseudorabies virus titration
We used traditional method, along with a modified IPMA employing the generated mAbs against UL42, to determine PRV titers. PRV-JF was serially diluted 10-fold (10−1–10−8), and 100 μL aliquots of virus was used to infect confluent PK-15 cultures in 96-well plates. At 24 h post-infection, cultures were examined with an inverted light microscope for the presence of cytopathic effects (CPEs). For the IPMA, at 24 h post-infection, cultures were treated with the mAbs against UL42 (1:10,000). The staining procedure was similar to that described above in “Generation of monoclonal antibodies against UL42” section. PRV titers were determined by as 50 % tissue culture infectious dose (TCID50) and calculated using the Reed-Muench method. Each technique was performed in triplicate, and the resulting titers were averaged.
Computer analysis
To investigate the conservation of identified epitopes among PRV reference strains, the amino acid sequences of the identified epitopes in UL42 were compared with those from 14 other PRV strains, using ClustalW in Lasergene 7.1 (DNASTAR Inc., Madison, WI, USA). We also analyzed defined epitopes and corresponding regions of other alphaherpesviruses. Moreover, the variable region sequences of those specific mAb clones were also compared and analyzed. The spatial distribution of the identified epitopes for UL42 was analyzed by mapping epitope locations onto a 3D model of PRV UL42 using PyMOL software based on results of the PHYRE 2 online server.
Results
Expression of UL42
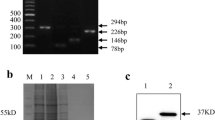
We successfully expressed recombinant PRV UL42 in E. coli (Fig. 1a). The induced and purified UL42 appeared to have a molecular weight of 40–55 kDa (Fig. 1b).

Western blotting analysis of recombinant pseudorabies virus (PRV) UL42 produced in Escherichia coli. a Recombinant UL42 was fused to histidine (His) residues at the N-terminus of the protein, yielding His-UL42. Bacterial lysates were analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). His-UL42 was approximately 46 kDa. Lane Non-induced, lysate from an E. coli culture where isopropyl β-d-1-thiogalactopyranoside was not added; Induced, lysate from an E. coli culture where isopropyl β-d-1-thiogalactopyranoside was added; Purified UL42, recombinant His-UL42 purified from E. coli; M, protein molecular weight marker. b Western blotting analysis of His-UL42. Lane Non-induced, lysate from an E. coli culture where isopropyl β-d-1-thiogalactopyranoside was not added; Induced, lysate from an E. coli culture where isopropyl β-d-1-thiogalactopyranoside was added; Purified UL42, recombinant His-UL42 purified from E. coli; M, protein molecular weight marker
Characterization of mAbs against UL42
We established six hybridoma cell lines producing mAbs that reacted strongly with recombinant UL42 as determined by western blotting (data not shown). These mAb clones were designated as 2D5, 2G11, 4E2, 5F8, 7C11, and 8F9. The heavy and light chains of each mAb were determined using a Mouse Mono Ab-ID (HRP) kit (Table 3). All of the UL42-reactive mAbs contained κ light chains. With the exception of clone 5F8 containing an IgG2b heavy chain, the other mAbs contained IgG1 heavy chains. The titers of antibodies secreted into culture supernatants and ascites fluid were found to range from 64–512 to 64,000–512,000 (Table 3). The resulting titration curves for each mAb displayed a variety of maximum ELISA signals, from which relative affinities for UL42 were ascertained (Table 3). A value of 1.0 was given to the mAb with the highest affinity. MAb 4E2 had the highest affinity with the value of 0.74 while mAb 8F9 had the lowest affinity with the value of 0.10.
Identification of linear B cell epitopes
UL42-derived peptides were expressed with EGFP tags, with EGFP alone and full-length UL42 used as negative and positive controls, respectively. The peptide spanning amino acids 1–148 was recognized by clones 2D5 and 4E2 (Fig. 2). The peptide spanning residues 256–384 was recognized by mAb clones 2G11, 5F8, 7C11, and 8F9. Based on the results, the other four peptides spanning amino acids 1–148 and 256–384 were also expressed as EGFP fusion products. Both the mAb clones 2D5 and 4E2 reacted with amino acids 39–148 of UL42, while clones 2G11, 5F8, 7C11, and 8F9 reacted with residues 302–384 of PRV UL42 (Fig. 2).

Primary identification of PRV UL42 epitopes using monoclonal antibodies (mAbs). The mAb clones 2D5 and 4E2 reacted with amino acids 1–148 and 39–148 of UL42. Clones 2G11, 5F8, 7C11, and 8F9 reacted with amino acids 256–384 and 302–384 of UL42. Lane P, purified UL42 protein; N, enhanced green fluorescent protein; M, protein molecular weight marker
Subsequently, ELISAs were performed using eight synthesized peptides, spanning amino acids 39–148 and 302–384 of UL42, as coating antigens. Full-length UL42 and an irrelevant peptide (amino acids 1–15 of UL42) were used as positive and negative controls, respectively. The peptides spanning amino acids 104–129 were recognized by clones 2D5 and 4E2, while peptides spanning residues 342–367 were recognized by mAb clones 2G11, 5F8, 7C11, and 8F9 (Fig. 3a). To identity the minimum linear epitope recognized by these mAbs, ELISAs were conducted again using 36 synthesized peptides targeting UL42 residues 105–136 and 343–374, as coating antigens (Fig. 3b). Peptides corresponding to residues 110–124, 111–125, 112–126, 113–127, 114–128, 115–129, and 116–130 were recognized by the mAb clones 2D5 and 4E2 (Fig. 3b). The common amino acid sequence among these peptides was 116SGGVLDALK124. Peptides corresponding to amino acids 346–360, 347–361, 348–362, 349–363, 350–364, 351–365, 352–366, 353–367, and 354–368 were recognized by mAb clones 2G11, 7C11, and 8F9 (Fig. 3b). Due to the low solubilities of peptides corresponding to amino acids 348–362, 349–363, 350–364, 351–365, and 352–366 in PBS, the values of OD405 nm of these peptides were lower than other four peptides. But, it still could be seen that clones 2G11, 7C11, and 8F9 produced positive reaction with these peptides. The common amino acid sequence among these peptides was 354KRPAAPR360. Peptides spanning amino acids 353–367, 354–368, 355–369, 356–370, 357–371, 358–372, 359–373, and 360–374 were only recognized by clone 5F8 (Fig. 3b). These peptides contained 360RMYTPIAK367 as the common amino acid sequence. Interestingly, the peptide ELISA results with amino acids 109–123 and 117–131 were negative, while the results with amino acids 110–124 and 116–130 were positive (Fig. 3b). This demonstrated that S116 and K124 were essential to the epitopes of 116–124. Likewise, we could conclude that K354, R360, and R360, K367 were essential to the epitopes of 354–360 and 360–367, respectively.

Fine mapping of the epitopes by peptide enzyme-linked immunosorbent assay (ELISA) with mAbs. a A panel of overlapping peptides spanning amino acids 38–149 and 302–384 of UL42 protein was analyzed for their reactivity with the generated mAbs. b A panel of overlapping peptides spanning UL42 amino acids 105–136 and 343–374 was analyzed for their reactivity with the generated mAbs. Purified UL42 was coated as a positive control, while PEP control (amino acids 1–15 of UL42) severed as a negative control
Comparison of the monoclonal antibody variable region genes
The amplification fragments of light and heavy chains were about 360 and 380 bp, respectively. Amino acid sequences of the mAb variable regions were aligned, and the results are shown in Fig. 4. The light chain amino acid sequences of mAbs that recognized an identical epitope had high identity. There were only two amino acids (positions 4 and 8) different among clones 2G11, 7C11, and 8F9 (Fig. 4a). However, the heavy chain sequences of those clones shared low identity (Fig. 4b). A similar phenomenon occurred in clones 2D5 and 4E2 (Fig. 4). The light and heavy chain sequences of mAb 5F8 shared low identity with other clones (Fig. 4).

Amino acid sequence alignment of the UL42 mAb variable regions. a Sequence alignment of the UL42 mAb light chains. b Sequence alignment of the UL42 mAb heavy chains. Red solid boxes show residues that differ from the consensus. The consensus was used as the majority sequence for this alignment. Blue rectangles indicate high identity among the light chains of the mAbs that recognized the identical epitopes
Subcellular localization of UL42 during pseudorabies virus replication
To investigate whether the mAbs that we generated against UL42 could be used to determine the location of UL42 during replication of PRV, we conducted IFAs on PRV-infected PK-15 cells. We detected UL42 in the nucleus of PK-15 cells during PRV replication, regardless of the mAb clone used (Fig. 5a). We also observed that the shape of the nucleus in PRV-infected PK-15 cells was somewhat altered compared with that in uninfected PK-15 cells.

Application of the generated mAbs to various immunological assays and techniques. a Subcellular localization of UL42 during PRV replication as determined by immunofluorescence assays using the six mAbs (green) generated against UL42. Nuclei were stained with DAPI (blue). Red and yellow arrows indicate normal and pathological nuclei, respectively. b Immunoprecipitation and western blotting analysis of UL42 involving PRV-infected PK-15 cells. Immunoprecipitation was performed using the six mAbs that we generated, which specifically recognized UL42. Precipitates were separated by SDS-PAGE and then subjected to western blotting. The asterisk indicates the heavy chain of IgG. Values to the right indicate the approximate sizes of the protein bands in kilodalton. c Cytopathic effects (CPEs) in PK-15 cell cultures as a result of PRV infection. CPE could be visualized (1) without staining and (3) by staining via an immunoperoxidase monolayer assay (IPMA). Enlarged images from (1) and (3) are shown in (2) and (4) to exhibit the CPE clearly. d PRV titers determined by traditional methods and IPMA
Immunoprecipitation of pseudorabies virus UL42
We were able to detect UL42 among the proteins immunoprecipitated from PRV-infected PK-15 lysates, regardless of the mAb used. As the results were similar, one of the results was shown in case of repetition (Fig. 5b). When normal uninfected PK-15 cells were used, immunoprecipitation of UL42 was unsuccessful.
Pseudorabies virus titers
The extent of CPE as a result of PRV infection was clearly visible when we conducted IPMAs, regardless of the mAb used (Fig. 5c). In addition, it was relatively easy to assess CPE and determine the titer of PRV by IPMA comparing to the traditional methods that did not involve staining. The PRV-JF titer determined by IPMA was 108.47 TCID50/mL, while, using traditional methods, the titer was 107.76 TCID50/mL (Fig. 5d).
Specificity and conservation of identified linear epitopes among alphaherpesviruses
The amino acid sequences of the three identified UL42 epitopes from 14 PRV isolates were analyzed. The three epitopes that we identified were highly conserved among PRV strains (Fig. 6a). These epitopes likely play a pivotal role in the function of UL42 during viral replication. In addition, we analyzed the homologous sequences of the defined epitopes in seven different alphaherpesviruses (HSV-1, VZV, EHV-1, EHV-3, EHV-4, feline herpesvirus 1, and bovine herpesvirus 1.2). The three identified epitopes shared low identity among the seven alphaherpesviruses, indicating that these epitopes were specific for PRV (Fig. 6b).

Amino acid sequence alignment of the identified epitopes in PRV UL42. a The UL42 amino acid sequence for 14 PRV reference strains was aligned. b Amino acid sequences from other alphaherpesviruses that were homologous to PRV UL42 were also aligned. These other alphaherpesviruses were herpes simplex virus type 1 (HSV-1), bovine herpesvirus type 1.2 (BoHV-1.2), feline herpesvirus type 1 (FHV-1), varicella-zoster virus (VZV), and equine herpesvirus (EHV). Black dots indicate residues that are exact matches. Homologous regions in the various alphaherpesviruses that correspond to the identified PRV UL42 epitopes are also indicated by black dots. GenBank accession numbers are shown in parentheses
Protein modeling
Modeling of the PRV UL42 protein was conducted to spatially localize the epitopes recognized by mAb clones 2D5 and 4E2. The complete three-dimensional (3D) structure of the UL42 protein is not currently available; however, we were able to localize one of the identified epitopes (amino acids 116–124) on a predicted 3D structure of UL42. The epitope recognized by clones 2D5 and 4E2 spanned amino acids 116–124 and partially protruded from the surface of the structure (Fig. 7a), forming an alpha helix sheet loop (Fig. 7b).

Relative localization of an identified linear epitope in a partially predicted 3D structure of PRV UL42. The spatial location of the binding epitope (shown in red) (amino acids 116–124) for mAbs 2D5 and 4E2 in PRV UL42 is highlighted. The epitope recognized by 2D5 and 4E2 partially protrudes from the predicted structure (a) and forms an alpha helix sheet loop (b)
Discussion
Apart from its economic importance, PRV has been shown to be an excellent model of alphaherpesvirus biology (Enquist et al. 1998; Mettenleiter 1994a, 2000, 2002) particularly the areas benefiting from the PRV research, including the molecular mechanisms of virus attachment, entry, replication, infection, virion morphogenesis, egress, neuroinvasion, and transneuronal spread. Mapping the epitopes of viral proteins and determining the degree of conservation of identified epitopes could facilitate our understanding of their antigenic structure and function and might also be useful in clinical applications. PRV glycoprotein E (gE) is important for virulence and spread of the virus (Babic et al. 1996; Banfield et al. 1998; Kratchmarov et al. 2013; Kritas et al. 1994). The epitopes of gE have been reported and were shown to play an important role in the diagnosis of PRV and in helping to determine the specific functions of gE (Fuchs et al. 1990; Jacobs and Kimman 1994; Jacobs et al. 1990). The PRV UL42 protein is essential for viral DNA replication (Pomeranz et al. 2005). Mapping the epitopes of UL42 would assist with the functional analysis of UL42 during viral DNA replication.
In the current study, we generated six mAbs that recognized three spatially distinct epitopes of PRV UL42. In epitope mapping through western blotting analysis, the actual molecular weight of EGFP-fused peptides corresponding to amino acids 256–384 and 384–302 is larger than the expected (Fig. 2). We deduced that these two peptides may form glycosylation in eukaryotic expressing system, as it was predicted online that there are glycosylation sites (S345, S346, T363, T371, and S373) on PRV UL42 protein (http://www.cbs.dtu.dk/services/NetOGlyc/). MAbs 2D5 and 4E2 had no reactivity with amino acids 109–123 and 117–131 while they could react with amino acids 110–124 and 116–130 in peptide ELISA analysis (Fig. 3b); this indicates that S116 and K124 were key residues for the activity of the epitopes of 116–124. Similarly, we could infer that K354 and R360, and R360 and K367 were essential to the epitopes of 354–360 and 360–367, respectively. Further research is necessary to identify the specific function of the residue K in those identified epitopes. We were unable to identify an immunodominant epitope, as no more than four antibodies recognized an identical epitope. In contrast, it was previously reported that seven hybridomas from two different spleen fusions reacted with the epitope between amino acids 360 and 369 of HSV-1 UL42 protein (Murphy et al. 1989). One of the three epitopes (116SGGVLDALK124) that we identified in PRV UL42 was located at the amino-terminus, with other two epitopes (354KRPAAPR360 and 360RMYTPIAK367) at the carboxy-terminus. We postulated that the central portion of UL42 was less immunogenic than the amino- or carboxy-terminal regions. A similar finding was observed for HSV-1, which is also an alphaherpesvirus (Sheaffer et al. 1995). In addition, it was demonstrated that five of six epitopes recognized by HSV-1 UL42 were outside the minimal active portion of the protein (Sheaffer et al. 1995). At present, there is no information regarding the minimal active portion of PRV UL42. We also compared the sequences of those specific mAb variable regions and found that light chains of the mAbs that recognized an identical epitope shared high identity while heavy chains shared low identity (Fig. 4). We speculated that the main reason is that UL42 is most likely to bind the light chain of the mAbs more efficiently. This intriguing hypothesis would be a good research orientation in our laboratory.
We found that the six mAbs that we generated against UL42 could be applied to various assays. We used the mAbs to characterize the location of UL42 during viral replication and to immunoprecipitate UL42 from the lysates of PRV-infected PK-15 cells. These mAbs were also helpful in determining PRV titers in infected cell cultures. The subcellular localization of UL42 during PRV replication has not been previously reported. We determined that UL42 strictly localizes to the nucleus during viral DNA replication (Fig. 5a). This result confirms the function of PRV UL42 as a DNA polymerase processivity factor on the other side. The ability of the mAbs to immunoprecipitate UL42 will benefit future investigations into the interactions of UL42 with viral and cellular proteins. Our results suggest that the six mAbs that we generated could be used to immunoprecipitate viral and cellular proteins that interact with PRV UL42 (Fig. 5b). The UL42 mAb-based IPMA that we developed was able to be used to determine PRV titers with greater accuracy than traditional methods. The titer determined by IPMA was higher than that determined by traditional methods possibly because the UL42 protein, which acts as the DNA polymerase processivity factor, was detected earlier during PRV replication (Fig. 5d). Moreover, the gene encoding UL42 is essential to PRV and cannot be deleted without serious adverse consequences to PRV. Therefore, mAbs against UL42 can be used to determine the titers of PRVs where certain genes have been deleted that do not affect virus propagation. Taken together, our findings indicate that the mAbs against UL42 that we generated could become essential tools in the functional analysis of UL42 during viral DNA replication.
Sequence analysis demonstrated that the three identified epitopes in PRV UL42 were highly conserved among different PRV strains, with 100 % identity (Fig. 6a). The highly conserved nature of these epitopes in UL42 results in an advantage in developing technologies for epitope-based diagnoses. Despite the structural and functional similarities of the DNA polymerase processivity proteins among other alphaherpesviruses, they do not share any obvious sequence homology, with low identity (Fig. 6b). Minor differences in the overall structure of DNA polymerase processivity proteins could affect their ability to interact with polymerases (Berthomme et al. 1995). The specificity by which the processivity proteins interact with their respective polymerases and the necessity for this functional interaction in vivo (Digard et al. 1993a, b; Stow 1993) demonstrate the feasibility for developing antiviral compounds that target this interaction. In theory, these compounds would effectively and specifically block viral DNA replication. These compounds could be antibodies that neutralize UL42 protein activity or peptides that bind to the active portions of UL42. This interesting hypothesis may provide new approaches for PRV therapy and control. Protein modeling of UL42 demonstrated that at least one of the identified epitopes was partially exposed and formed an alpha helix sheet loop (Fig. 7). Further research is required to determine the biological function of the epitopes in this region of PRV UL42.
In summary, we isolated six mAbs against PRV UL42 and subsequently identified three PRV-specific and highly conserved B cell epitopes in UL42. The generated mAbs helped us to determine the location of UL42 during PRV replication in porcine kidney cells. The mAbs were also used to immunoprecipitate UL42 from the lysates of PRV-infected PK-15 cells and assisted us in determining the titer of PRV in infected cell cultures. Taken together, our findings provide a solid foundation for further investigations into the antigenic functions of UL42 and for the development of diagnostic and therapeutic approaches to PRV infection.
References
Babic N, Klupp B, Brack A, Mettenleiter TC, Ugolini G, Flamand A (1996) Deletion of glycoprotein gE reduces the propagation of pseudorabies virus in the nervous system of mice after intranasal inoculation. Virology 219:279–284
Banfield BW, Yap GS, Knapp AC, Enquist LW (1998) A chicken embryo eye model for the analysis of alphaherpesvirus neuronal spread and virulence. J Virol 72:4580–4588
Ben-Porat T, Veach RA, Ihara S (1983) Localization of the regions of homology between the genomes of herpes simplex virus, type 1, and pseudorabies virus. Virology 127:194–204
Berthomme H, Monahan SJ, Parris DS, Jacquemont B, Epstein AL (1995) Cloning, sequencing, and functional characterization of the two subunits of the pseudorabies virus DNA polymerase holoenzyme: evidence for specificity of interaction. J Virol 69:2811–2818
Digard P, Bebrin WR, Weisshart K, Coen DM (1993a) The extreme C terminus of herpes simplex virus DNA polymerase is crucial for functional interaction with processivity factor UL42 and for viral replication. J Virol 67:398–406
Digard P, Chow CS, Pirrit L, Coen DM (1993b) Functional analysis of the herpes simplex virus UL42 protein. J Virol 67:1159–1168
Enquist LW, Husak PJ, Banfield BW, Smith GA (1998) Infection and spread of alphaherpesviruses in the nervous system. Adv Virus Res 51:237–347
Fuchs W, Rziha HJ, Lukacs N, Braunschweiger I, Visser N, Lutticken D, Schreurs CS, Thiel HJ, Mettenleiter TC (1990) Pseudorabies virus glycoprotein gI: in vitro and in vivo analysis of immunorelevant epitopes. J Gen Virol 71:1141–1151
Galfre G, Milstein C (1981) Preparation of monoclonal antibodies: strategies and procedures. Methods Enzymol 73:3–46
Huang LP, Lu YH, Wei YW, Guo LJ, Liu CM (2012) Identification of three new type-specific antigen epitopes in the capsid protein of porcine circovirus type 1. Arch Virol 157:1339–1344
Jacobs L, Kimman TG (1994) Epitope-specific antibody response against glycoprotein E of pseudorabies virus. Clin Diagn Lab Immunol 1:500–505
Jacobs L, Meloen RH, Gielkens AL, Van Oirschot JT (1990) Epitope analysis of glycoprotein I of pseudorabies virus. J Gen Virol 71:881–887
Klupp BG, Hengartner CJ, Mettenleiter TC, Enquist LW (2004) Complete, annotated sequence of the pseudorabies virus genome. J Virol 78:424–440
Kratchmarov R, Kramer T, Greco TM, Taylor MP, Ch’ng TH, Cristea IM, Enquist LW (2013) Glycoproteins gE and gI are required for efficient KIF1A-dependent anterograde axonal transport of alphaherpesvirus particles in neurons. J Virol 87:9431–9440
Kritas SK, Pensaert MB, Mettenleiter TC (1994) Role of envelope glycoproteins gI, gp63 and gIII in the invasion and spread of Aujeszky’s disease virus in the olfactory nervous pathway of the pig. J Gen Virol 75:2319–2327
Liu CM, Ihara T, Nunoya T, Ueda S (2004) Development of an ELISA based on the baculovirus-expressed capsid protein of porcine circovirus type 2 as antigen. J Vet Med Sci 66:237–242
Liu CM, Wei YW, Zhang CF, Lu YH, Kong XG (2007) Construction and characterization of porcine circovirus type 2 carrying a genetic marker strain. Virus Res 127:95–99
Lundh B (1990) Spread of vesicular stomatitis virus along the visual pathways after retinal infection in the mouse. Acta Neuropathol 79:395–401
Marcaccini A, Lopez Pena M, Quiroga MI, Bermudez R, Nieto JM, Aleman N (2008) Pseudorabies virus infection in mink: a host-specific pathogenesis. Vet Immunol Immunopathol 124:264–273
Mettenleiter TC (1994a) Initiation and spread of alpha-herpesvirus infections. Trends Microbiol 2:2–4
Mettenleiter TC (1994b) Pseudorabies (Aujeszky’s disease) virus: state of the art. August 1993. Acta Vet Hung 42:153–177
Mettenleiter TC (2000) Aujeszky’s disease (pseudorabies) virus: the virus and molecular pathogenesis—state of the art, June 1999. Vet Res 31:99–115
Mettenleiter TC (2002) Herpesvirus assembly and egress. J Virol 76:1537–1547
Muller T, Hahn EC, Tottewitz F, Kramer M, Klupp BG, Mettenleiter TC, Freuling C (2011) Pseudorabies virus in wild swine: a global perspective. Arch Virol 156:1691–1705
Murphy M, Schenk P, Lankinen HM, Cross AM, Taylor P, Owsianka A, Hope RG, Ludwig H, Marsden HS (1989) Mapping of epitopes on the 65k DNA-binding protein of herpes simplex virus type 1. J Gen Virol 70:2357–2364
Pomeranz LE, Reynolds AE, Hengartner CJ (2005) Molecular biology of pseudorabies virus: impact on neurovirology and veterinary medicine. Microbiol Mol Biol Rev 69:462–500
Restle T, Pawlita M, Sczakiel G, Muller B, Goody RS (1992) Structure-function relationships of HIV-1 reverse transcriptase determined using monoclonal antibodies. J Biol Chem 267:14654–14661
Romero CH, Meade P, Santagata J, Gillis K, Lollis G, Hahn EC, Gibbs EP (1997) Genital infection and transmission of pseudorabies virus in feral swine in Florida, USA. Vet Microbiol 55:131–139
Sheaffer AK, Hurlburt WW, Stevens JT, Bifano M, Hamatake RK, Colonno RJ, Tenney DJ (1995) Characterization of monoclonal antibodies recognizing amino- and carboxy-terminal epitopes of the herpes simplex virus UL42 protein. Virus Res 38:305–314
Stow ND (1993) Sequences at the C-terminus of the herpes simplex virus type 1 UL30 protein are dispensable for DNA polymerase activity but not for viral origin-dependent DNA replication. Nucleic Acids Res 21:87–92
Zhang CF, Liu CM, Wei YW, Liu NH (2008) Isolation and identification of pathogenic procine pseudorabies virus PRV-JF strain. Chinese J Prev Vet Med 30:212–215
Acknowledgments
This study was supported by the Public Welfare Special Funds for Agricultural Scientific Research (no. 201203039) and the National Natural Science Foundation of China (no. 31302110).
Conflict of interest
The authors declare that they have no competing interests.
Ethical statement
This study was approved by the Harbin Veterinary Research Institute of Chinese Academy of Agricultural Sciences (approval number Heilongjiang-SYXK-2006-032) and implemented in accordance with the animal care and ethics guidelines.
Author information
Authors and Affiliations
Corresponding author
Additional information
Wenjuan Du and Yiping Wang contributed equally to this work.
Rights and permissions
About this article

Cite this article
Du, W., Wang, Y., Huang, L. et al. Characterization of monoclonal antibodies that recognize the amino- and carboxy-terminal epitopes of the pseudorabies virus UL42 protein. Appl Microbiol Biotechnol 100, 181–192 (2016). https://doi.org/10.1007/s00253-015-6957-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-015-6957-7