
Abstract
Bacillus fastidious uricase (BF uricase) containing 322 amino acid residues exhibited high stability under physiological conditions. Its crystal structure was solved to 1.4-Å resolution, showing homotetramer containing two homodimers. After the intersubunit antiparallel β-sheet in its homodimer, each subunit had a total of 18 C-terminus residues forming an α-helix (Q305-A313) and random coil (S314-L322) on surface to bury other two α-helices (I227-T238 and I244-R258). In comparison, reported crystal structures of Arthrobacter globiformis and Aspergillus flavus uricases had atomic coordinates of only some C-terminus residues, while the crystal structures of all the other uricases accessible before September 2014 missed atomic coordinates of all their C-terminus residues, after the intersubunit antiparallel β-sheets. In each homodimer of BF uricase, H-bonds were found between E311 and Y249 and between Y319 and D257; electrostatic interaction networks were found to surround D307 plus R310 and intersubunit R3, K312 plus D257, E318 plus K242, and L322 plus R258. Amino acid mutations that disrupted those interactions when R3 and D307 were reserved caused moderate decreases of activity at pH 9.2 while negligible decreases of activity at pH 7.4, but destroyed stability at pH 7.4 while slightly decreased stability at pH 9.2. Such structural information guided the fusion of 6His-tag to the C-terminus of the mutant L322D with SNSNSN as a linker to reserve the activity and stability. Hence, the folding of the C-terminus residues is crucial for thermal stability of BF uricase under physiological conditions; these new structural insights are valuable for molecular engineering of uricases.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Uricase (E.C. 3.1.2.4) is currently used as an important tool for serum uric acid assay in clinical laboratories (Zhao et al. 2009a; Huang et al. 2015), a biodrug for therapeutic treatment of diseases associated with acute hyperuricemia and the sole drug to handle refractory gout associated with persistent hyperuricemia (Crittenden and Pillinger 2013; Ea et al. 2011; Schlesinger et al. 2011; Yang et al. 2012). To facilitate large-scale production via recombinant expression, microbial uricases are preferable over mammalian ones (Alvares et al. 1992). For applications of any uricase, it is highly desirable to have an activity and stability as high as possible (Feng et al. 2010; Sarkissian et al. 2008). However, most microbial uricases reported to date exhibit limited enzyme activities/stability under physiological conditions (Huang et al. 2015; Liu et al. 2009; Liu et al. 2011). For example, Aspergillus flavus uricase as a biodrug to treat acute hyperuricemia has favorable thermal stability at 4 °C, but unsatisfactory stability under physiological conditions besides a low activity (Liu et al. 2009). Arthrobacter globiformis uricase has significantly higher thermal stability under physiological conditions, but still a low activity (Huang et al. 2015). Hence, it is necessary to improve enzyme activities and thermal stability of uricases through molecular engineering via directed evolution and/or rational design of mutants (Chica et al. 2005; Turner 2009). In practice, the lack of suitable uricases as starting materials has limited directed evolution of uricases. Three-dimensional crystal structures are available for quite a few uricases (Colloc’h et al. 1997; Juan et al. 2008; Oksanen et al. 2014), but the insufficient understanding of the action mechanism of uricases and/or relationship between sequences and activities/stability has limited rational design of uricase mutants (Colloc’h et al. 1997; Gabison et al. 2008, 2010; Kahn and Tipton 1997; Retailleau et al. 2004, 2005). Therefore, it is pivotal to discover uricases of high activity/stability and reveal mechanistic and structural determinants of their enzyme activities/stability.
The putative active form of uricase is homotetramer that contains two homodimers; each homodimer is a closed tunnel bearing 2-fold rotation symmetry. There is an intersubunit antiparallel β-sheet between residues near the N-terminus of one subunit and those near the C-terminus of the other subunit of a homodimer (Colloc’h et al. 1997; Colloc’h et al. 2008; Gabison et al. 2008; Juan et al. 2008; Oksanen et al. 2014; Retailleau et al. 2004). Interestingly, truncation of C-terminus decreases stability of Bacillus sp. TB-90 uricase (Hibi et al. 2014; Yamamoto et al. 1996); uricase stability is associated with the number of C-terminus residues after each intersubunit antiparallel β-sheet in homodimer (Hibi et al. 2014; Liu et al. 2009; Suzuki et al. 2004; Yamamoto et al. 1996). Folding of C-terminus may thus play some roles in thermal stability of uricases. According to crystal structures of all microbial uricases accessible in the Protein Data Bank before September 2014, there are some C-terminus residues in each subunit after the intersubunit antiparallel β-sheet. In crystal structures, however, only some of the C-terminus residues in subunits of Arthrobacter globiformis and Aspergillus flavus uricases have coordinates (Colloc’h et al. 1997; Colloc’h et al. 2008; Gabison et al. 2008; Juan et al. 2008; Oksanen et al. 2014), while no coordinates are available for any of these C-terminus residues in the other microbial uricases (Kahn and Tipton 1997; Retailleau et al. 2004). These facts prevent analysis of the possible roles of C-terminus folding in thermal stability of uricases. On the other hand, fusion of a six-histidine (6His)-tag to N- or C-terminus is preferred for purification of recombinant proteins, but few uricases are expressed via fusion of 6His-tag (Pfrimer et al. 2010; Wu et al. 2009; Zhang et al. 2012), potentially due to negative impacts of 6His-tag on activities/stability of uricases. Structure-guided fusion of 6His-tag to a protein is promising to alleviate negative impact of 6His-tag on the activity/stability of the 6His-tagged protein but requires coordinates of all residues at one terminus. Hence, for elucidation of potential roles of C-terminus residues folding in stability/activity and structure-guided fusion of 6His-tag to the C-terminus, the atomic coordinates of all residues in the C-terminus of uricases are needed.
The pharmaceutical value of a uricase as a biodrug depends on its activity and thermal stability under physiological conditions. The activities of most microbial uricases show strong susceptibility to the reaction pH as their activities at pH 7.4 are usually less than 50 % of the corresponding maximum activities that usually occur at pH ~9.2. In addition, microbial uricases usually have a lower thermal stability at pH 7.4 compared to those at pH 9.2 (Huang et al. 2015). An intracellular uricase from Bacillus fastidious A.T.C.C. 29604 (BF uricase) is a promising candidate of biodrug for the treatment of hyperuricemia (Feng et al. 2010; Schiavon et al. 2000; Tan et al. 2012; Zhang et al. 2010; Zhang et al. 2014) and a practical tool for serum uric acid assay (Huang et al. 2015; Liao et al. 2006; Zhao et al. 2006). Predominantly, the half-life of BF uricase under physiological conditions reached 3 weeks (Huang et al. 2015). Crystal structure of BF uricase was thus determined by X-ray diffraction. Surprisingly, our newly solved crystal structure contained the atomic coordinates for all of a total of 18 C-terminus residues in each subunit after the intersubunit antiparallel β-sheet. This report thus presented for the first time a crystal structure of uricase containing the coordinates for all of the C-terminus residues after each intersubunit antiparallel β-sheet. More importantly, these 18 C-terminus residues and their folding were found to be main determinants of its thermal stability, but minor factors of its enzyme activity, under physiological conditions; such structural information has guided successful fusion of 6His-tag to the C-terminus of the mutant L322D.
Materials and methods
Chemicals and materials
DEAE-cellulose was from Whatman. Diethyltraminepentaacetic acid and uric acid were from Alfa Aesar. Poly-(ethylene glycol) (PEG)-3350 was from Sigma-Aldrich. Bacillus fastidious A.T.C.C 29604 was that we reserved (Zhao et al. 2006). Superdex 200 16/60 size exclusion column was from GE Healthcare. Ni2+-NTA superflow sepharose was purchased from QIAGEN. Restriction enzymes on DNA, competent cells of Escherichia coli DH5a and BL21 (DE3), and isopropyl-β-D-thiogalactoside (IPTG) were from Takara Ltd. (Dalian, China) or Sangon Biotechnology (Shanghai, China). Syntheses of primers and sequencing of DNA fragments were performed by these two companies. Sodium borate, boric acid, and other chemicals were domestic products of analytical or better grade.
Recombinant expression of BF uricase and mutants
The coding sequence of BF uricase as recorded in GenBank (accession no. FJ393559.1) was cloned from the genomic DNA of Bacillus fastidious A.T.C.C 29604 via PCR (Huang et al. 2015; Zhang et al. 2010; Zhao et al. 2006). To have the N-terminus of AERTMFYGKGDV as suggested by Edman degradation of the corresponding wild-type uricase (Zhao et al. 2006), the forward primer of 5′-CAGGATGGTAAAGGCGACGTATA-3′ and the reversed primer of 5′-GTCAACGCCATAGTCGATTTTAAC-3′ were used. Mutants were generated via site-directed mutagenesis or replacement of DNA fragments with reagent kits from Taihe Biotechnology, Co., Ltd. (Beijing, China). An engineered pET28a vector was used to construct expression vectors of mutants with carefully designed primers carrying on sites for digestion by NcoI and Hind III. Competent Escherichia coli DH5α cells were transformed to amplify each vector, and then E. coli BL21 cells (DE3) were transformed for induced expression. Each active uricase in cell lysates was purified as described previously (Huang et al. 2015; Zhang et al. 2010; Zhang et al. 2014). To estimate the highest specific activity and reflect catalytic capacity for each uricase, a small amount of enzyme after purification at 4 °C was further purified by using polyacrylamide gel electrophoresis (PAGE) (Zhao et al. 2006). To confirm sequences of resulting uricase/mutant, molecular weights of uricase/mutant and fragments after digestion with trypsin and V8 protease were determined by matrix-assisted-laser-desorption-ionization time-of-flight mass-spectrometry and tandem mass spectrometry (Analytical Center of Proteomics, Shanghai Institution of Biological Sciences, Chinese Academy of Sciences, Shanghai, China). Results confirmed that the first residue methionine was eliminated in BF uricase and tested mutants. Hence, positions of residues in polypeptides were counted after the elimination of the first residue methionine.
Analysis of kinetic properties of uricase
To measure initial rates as index of catalytic capacity, final uric acid was 75.0 μM in 200-mM sodium borate buffer plus 0.10 mM diethylenetriaminepentaacetic acid at pH 9.2 or 7.4 (Feng et al. 2013; Liao et al. 2005). The buffer was preheated to 25 °C by incubation for no less than 10 min prior to use. Unless stated otherwise, reaction was initiated by the addition of uricase sample. Absorbance of reaction mixture at 293 nm was recorded on MAPADA UV 1600 PC spectrophotometer linked to a computer in an isolated small room air-conditioned at 25 °C. The Bradford method was used to quantify proteins with BSA as the reference (Bradford 1976). As usual, one unit of uricase oxidizes one micromole uric acid per min; the absorptivity of uric acid at 293 nm was fixed at 11.5 mM−1 cm−1 for calculation of substrate concentrations. Michaelis-Menten constant (K m) was determined by Lineweaver-Burk plot analysis of initial reaction rates at uric acid levels from 80 to 400 μM in borate buffers; reaction was monitored by absorbance at 308 nm (Feng et al. 2013; Zhao et al. 2006).
Analysis of homotetramer structure and activity of resolved component
Purified BF uricase and mutants were analyzed by both nondenatured PAGE to resolve oligomers and sodium dodecyl sulfate-PAGE (SDS-PAGE) to detect potential contaminated polypeptides and/or degradation. BF uricase had molecular weight of about 36 kDa for each polypeptide and an isoelectric point of about 4.7, close to those of serum albumins. For PAGE, bovine serum albumin (BSA, molecular weight of about 68 kDa) and ovarian serum albumin (OVA, molecular weight of about 45 kDa) after purification via preparative PAGE were used as references to differentiate homodimers from homotetramers and subunits. To check activities of oligomers, uricase in gel after PAGE was stained with uric acid plus peroxidase and its chromogenic substrate (Zhao et al. 2009b).
Characterization of thermo-inactivation of BF uricase and mutants
Sodium borate and boric acid buffer at 200 mM for pH at 7.4 or 9.2 were used. Final concentrations of kanamycin C at 1.25 g/L, ampicillin at 1.25 g/L, 4-aminobenzamidine at 2.0 mM, and EDTA at 0.1 mM were added to prevent protein degradation. For thermo-inactivation, solutions of uricases in 5.0-mL Ependorf tubes sealed with Parafilm were incubated in an incubator thermo-stated at 37 °C (Shanghai Yuejin Medical Instruments, Shanghai, China). After being kept for an indicated time, aliquots were withdrawn under sterile environment to measure the activity with 75.0 μM uric acid in 200-mM sodium borate buffer at pH 9.2 as described above. Half-life was estimated for 50 % residual activity according to single-component exponential decrease model. Meanwhile, amino groups were determined by 2,4,6-trinitrobenzenesulfonic acid (TNBS) assay with glycine as the reference, following those described previously (Zhang et al. 2010; Zhang et al. 2014).
Crystallization of two uricases, collection, and analysis of X-ray diffraction data
Crystal structures of BF uricase, and a fusion expression of six-histidine (6His) via a linker (KLAAALK) to the C-terminus of BF uricase (BFU-6His), were determined as described below. To obtain protein material of the highest possible purity, BF uricase in cell lysates after induced expression as described above was purified over two consecutive DEAE-cellulose chromatography via elution with 0.10 M Tris-HCl at pH 8.0 plus a linear gradient of NaCl from 0 to 0.40 M (Huang et al. 2015; Zhang et al. 2010; Zhang et al. 2014; Zhao et al. 2006). Only fractions with specific enzyme activity over 11 kU/g were pooled for further processing. BFU-6His was purified using Ni2+-NTA superflow sepharose, with 20 mM Tris-HCl at pH 8.0 plus 150 mM NaCl and 30 mM imidazole as the buffer for binding of tagged proteins and washing of untagged proteins; elution buffer was 300 mM imidazole in the same binding/washing buffer. Pooled protein of BF uricase or BFU-6His was concentrated via ultrafiltration and further purified through a Superdex 200 16/60 size exclusion column in 20 mM Tris-HCl at pH 8.0 plus 150 mM NaCl; the resulted preparation of either uricase had negligible contaminated proteins.
BF uricase and BFU-6His were further concentrated via ultrafiltration to 20 g/L. Crystals of each uricase were grown with sitting drop vapor diffusion method by mixing 1 μL of protein sample with 1-μL well solution at 4 °C. BF uricase was crystallized in 0.2 M sodium citrate plus 20 % (w/v) PEG-3350 (final pH was about 7.0), and BFU-6His was crystallized in 0.1 M Tris-HCl at pH 8.5 plus 0.2 M ammonium sulfate and 25 % (w/v) PEG-3350 (final pH was about 7.5). Crystals appeared and grew to full sizes in 2 weeks and were flash-frozen with liquid nitrogen in mother liquid after the addition of final 15 % (w/v) glycerol. All diffraction data sets were collected at Shanghai Synchrotron Radiation Facility (SSRF) beamline BL17U at 100 K. Data were indexed, integrated, and processed with DENZO and SCALEPACK as implemented in HKL2000 (Adams et al. 2010). BF uricase structure was solved by molecular replacement with the program PHASER (McCoy et al. 2007), using PDB accession ID 1j2g as the search model. Then, BFU-6His structure was solved similarly using BF uricase as the search model.
Structure refinement was done with PHENIX (Zwart et al. 2008). The overall quality of each model was assessed by MolProbity (Chen et al. 2010; Davis et al. 2007). Results showed that S243 in chain A, V174 in chain B and D of BF uricase, while S177 in chain A, V174 in chain B, and T194 and D195 in chain D of BFU-6His, had disallowed phi/psi angles. Electron densities of those residues bearing disallowed phi/psi angles were thus carefully examined one-by-one and compared against those of the nearby well-defined residues. In fact, as for BFU-6His, except for V174 in chain B and D195 in chain D, there were insufficient reflections of both S177 of chain A and T194 of chain D in comparison to nearby residues in the same chains. As a result, the coordinates of those two residues in BFU-6His were deleted to avoid misleading information (Supplementary material Fig. S1). After deletions of the coordinates of those two residues, the structure of BFU-6His was refined again with PHENIX (Zwart et al. 2008). As for BF uricase, however, electron densities of those three residues indicated that there were sufficient reflections in comparison to nearby residues in the same chains, and thus, their coordinates were reserved (Supplementary material Fig. S2). Data collection and structure refinement statistics are given in Table 1. The structures of BF uricase and BFU-6His had been submitted to Protein Data Bank with IDs of 4r8X and 4r99.
Analysis of conformation of uricase
PyMol (v11.0) (http://www.pymol.org/) was used for conformational analysis. For simplicity to analyze interactions, only residues within 1.0 nm for electrostatic interactions and within 0.60 nm for other noncovalent interactions were considered. Unless otherwise stated, labeled distances were measured for carbon atoms in carboxyl groups, carbon atoms in guanidine groups, and nitrogen atoms in side chains of lysine and histidine. Hydrophobicity and charges were calculated with DNAMAN (v5.0, Lynnon Corp., Quebec, Canada). Folding of the C-terminus of BF uricase was compared with those of Arthrobacter globiformis uricase (AG uricase) and Aspergillus flavus uricase (AF uricase).
Results
Overall structure of BF uricase
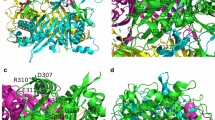
Crystal structure of BF uricase had a resolution of 1.4 Å (pdb ID 4r8x). For each subunit of the BF uricase, β-sheets clustered on one side while α-helices clustered on the other side; both sides were mainly hydrophilic (Fig. 1a, supplementary material Fig. S3); M5-G10 of subunit A interacted with F296-H304 of subunit C via an intersubunit antiparallel β-sheet to form a homodimer (Fig. 1b, c). Moreover, in a homodimer as a tightly closed tunnel, residues Y180–T194 on surface from subunit C formed a large loop to contact three α-helices of subunit A (E52-E60, T69-A81, and L88-K102) (Supplementary material Fig. S4). In each homodimer, the other intersubunit antiparallel β-sheet and the other large contacting loop on surface were located according to the 2-fold rotation symmetry. Putatively, in the bottom of a homodimer, there was a large cleft as the active site, involving residues near K9 of subunit A in the intersubunit antiparallel β-sheet, residues nearby T69 in the α-helix of A68-S82 and most residues in the medium linking loop of G61-A68 of subunit A, residues nearby I244 in the α-helix of I244-R258, and some residues in the large contacting loop of Y180-T194 of subunit C on surface of the homodimer. The other active site was located according to the 2-fold rotation symmetry. For making homotetramer, Y116-E142 and T273-P295 in each subunit of a homodimer formed two large extending loops to interact with the counterparts of the other homodimer (Fig. 1c, d; Supplementary material Fig. S4).

Overall structure of BF uricase. Cartoon in blue represented the 18 C-terminus residues after an intersubunit antiparallel β-sheet. a Side view of subunit inside a closed tunnel of BF uricase; b one intersubunit antiparallel β-sheet in a closed tunnel of BF uricase; c side view of a closed tunnel of BF uricase, those in cyan represented the extending arms; d top view of BF uricase homotetramer; e structural alignment of subunits of BF uricase (cyan), AF uricase (green), and AG uricase (magenta) to demonstrate differences in folds of the C-terminus residues after their intersubunit antiparallel β-sheets
Folding of the C-terminus residues of BF uricase and the associated interactions
In AG uricase (pdb ID 2yzb), only the first 12 residues (E286-N297) in a total of 17 C-terminus residues after each intersubunit antiparallel β-sheet had accessible coordinates; I294-N297 formed an α-helix while E286-P293 folded as a loop to drive the α-helix into the closed tunnel (Supplementary material Fig. S5). In AF uricase (pdb ID 1ws3), only six residues in subunit C among a total of eight C-terminus residues after the intersubunit antiparallel β-sheet had accessible coordinates while coordinates of the last five C-terminus residues after the intersubunit antiparallel β-sheets in other subunits were missed; those six C-terminus residues of subunit C formed random coli and stretched away from the closed tunnel (Supplementary material Fig. S6). Fortunately, in BF uricase, all of the 18 C-terminus residues (Q305 to L322) after each intersubunit antiparallel β-sheet had accessible coordinates. Much unexpectedly, Q305-A313 in the C-terminus of BF uricase folded into an α-helix while S314-L322 was random coil, and they turned away from the closed tunnel to spread out on homodimer surface and contact spatially near residues (Fig. 1c, d). Structural alignment of subunit C of those three uricases depicted the differences in the folding of their C-terminus residues after the intersubunit antiparallel β-sheets (Fig. 1e).
To simplify analysis of the conformation of the 18 C-terminus residues of BF uricase, the α-helix (Q305-A313) is denoted as the first region, and the random coil (S314-L322) is designated as the second region (Fig. 2a). To elaborate interactions associated with those 18 C-terminus residues (Q305 to L322) after each intersubunit antiparallel β-sheet in homodimer of BF uricase, residues in subunit A were considered, unless otherwise stated.

Interactions associated with C-terminus residues after each intersubunit antiparallel β-sheet. Residues within 1.0 nm from the two regions were displayed. Cartoons in magentas, green, and cyan indicated residues from different subunits. Distances were measured for carbon atom in carboxyl group, carbon atom in guanidine group, and nitrogen atom in amino group. Residues for involved interactions were labeled. a Two regions in blue of the last 18 residues in the C-terminus of a subunit; b electrostatic interactions and H-bonding with residues in the first region; c electrostatic interaction and H-bonding with residues in the second region; d hydrophobicity of the 18 C-terminus residues predicted with DNAMAN 5.0
The α-helix (Q305-A313) as the first region had two interaction centers. One center was located on D307 plus R310 of subunit A while R3 of subunit C, both of which were within 0.35 nm from D307. This center was surrounded by E306, E311, and E318 of subunit A and E2, E52, E60, and D62 of subunit C (Fig. 2b). These residues thus formed a network that was connected to the second region via electrostatic interactions with E60 and D62 of subunit C. Additionally, E311 in this center formed H-bond with Y249. The other center was located on K312 and D257 at a distance within 0.34 nm and connected with the second region via a charge-relay network (Fig. 2b). Those two interaction centers fixed the α-helix (Q305-A313) on the surface to bury the long α-helix (I244-R258) of subunit A (Fig. 2b). In the buried long α-helix, I244 was included in active site, and only L253 and L256 had hydrophobic side chains that were away from the side chain of L308 by about 0.60 nm.
The random coil (S314-L322) of those 18 C-terminus residues as the second region had three interaction centers. The first center was located on E318 and K242 at a distance within 0.50 nm and included H246 and H250 within 0.55 nm from E318, D240 within 0.80 m from K242 of subunit A, and E60 and D62 of subunit C to form a network. The second center was located on L322 and R258 at a distance within 0.33 nm (Fig. 2c), involving D257 and K312 in subunit A while D126 and K127 in subunit B of the other homodimer to exert interhomodimer electrostatic interactions. Via interactions with K312 and D257 of subunit A, and interactions with E60 and D62 of subunit C, there was an integrated interaction network involving residues in the first and second regions of those 18 C-terminus after each intersubunit antiparallel β-sheet. The third center involved a hydrophobic surface formed by side chains of the last four residues and H-bond between Y319 and D257 (Fig. 2d); the main chains of the last four residues isolated solvent water from a large hydrophobic groove generated by two buried α-helices of subunit A (Q227-T238 and I244-R258).
Roles of folding of those C-terminus residues in thermal stability and catalytic activity
During storage at 37 °C and pH 9.2 or 7.4, there were no significant changes of total amino groups in solution detected with TNBS before the loss of more than 90 % activity. Activities of BF uricase and mutants followed single-component exponential decreases (Fig. 3). During storage at 37 °C, half-lives of the BF uricase were about 3 weeks at pH 7.4 and 6 weeks at pH 9.2 (Huang et al. 2015). At 65 °C, the half-lives of the BF uricase were about 8.0 min at pH 9.2 (Supplementary material Fig. S7), but too short to be quantified at pH 7.4.

Characterization of some mutants. a SDS-PAGE (the left part) and PAGE (the right part) analysis of BF uricase (lane A) and mutant (lane B) bearing the N-terminus of AEDTMFYGKGDV (mutant R3D); b stability of mutant E318R at pH 7.4 and 9.2; c stability of mutant Y249F at pH 7.4 and 9.2; d stability of mutant R310E at pH 7.4 and 9.2
When R3 was reserved, mutations of the second residue slightly reduced mutant activities at pH 9.2 while hardly affected the stability at pH 7.4 or 9.2 (Table 2). When positive charge on R3 was eliminated, the mutants became inactive dimers (Fig. 3a). However, the ratios of activities at pH 9.2 to those at pH 7.4 remained consistent after such mutations (Table 2).
Mutant D307R or the deletion of residues after D307 gave an inactive homotetramer of lower solubility (Table 2). The deletion of residues after S314 gave a homotetramer bearing about 25 % activity, 5 % half-life at pH 7.4 but 100 % half-life at pH 9.2 (Table 2). Mutant E318R exhibited enzyme activity and thermal stability comparable to the mutant after the deletion of residues since S314 (Fig. 3b). Thermal stability of mutant Y319F was reduced by one magnitude at pH 7.4, but by just 2-fold at pH 9.2 (Fig. 3c). Interestingly, the mutant Y249F showed the reduction of thermal stability by more than 50-fold at pH 7.4 while by just about 7-fold at pH 9.2. More importantly, double mutant Y319F/Y249F was an inactive homotetramer. Disruption of interactions with E311 also greatly destabilized the mutant at pH 7.4, but slightly weakened its thermal stability at pH 9.2. Most notably, mutant R310E exhibited reasonable activity, but the lowest stability at pH 7.4 or 9.2 among tested mutants and the strongest effects of pH values on thermal stability (Fig. 3d). The mutant L322D had an activity and thermal stability at pH 9.2 or 7.4 comparable to BF uricase (Table 2). The electrostatic repulsion interaction between side chains of R258 and R322 in the mutant L322R reduced activity and thermal stability at pH 7.4, but enhanced stability at pH 9.2.
The amino acid mutations that disrupted the interactions associated with those 18 C-terminus residues observed in crystal structure of BF uricase reduced susceptibility of enzyme activity to variations of reaction pH while produced consistent affinity for the substrate. In particular, enzyme activities of all such mutants at pH 7.4 were comparable to those of BF uricase at pH 7.4, but their activities at pH 9.2 were sometimes reduced by more than 3-fold in comparison with that of BF uricase at pH 9.2. For the mutant R310E bearing a half-life of only about 0.4 % of that of BF uricase at pH 7.4, its activity was still >70 % of that of BF uricase at pH 7.4. Interestingly, within all of the mutants tested, the mutant L322D exhibited the highest activity at pH 9.2 (about 80 % of BF uricase), and its activity exhibited the susceptibility to reaction pH comparable to BF uricase (Table 2).
Structure-guided fusion of 6His-tag to the C-terminus of BF uricase
The mutant after the direct fusion of 6His-tag to the C-terminus of BF uricase was unable to be effectively bound by Ni2+-NTA at pH 8.0. The use of KLAAALK as a linker for fusion of 6His-tag to the C-terminus of BF uricase gave BFU-6His that was successfully purified with Ni2+-NTA and had moderate activity at pH 9.2. Moreover, it exhibited stability at pH 7.4 or 9.2 comparable to BF uricase under the same conditions (Table 3). The use of EAAAR as a linker for the fusion of 6His-tag to the C-terminus of BF uricase produced comparable alterations of stability and activity at pH 7.4 or 9.2.
In crystal structure of BFU-6His (pdb ID 4r99), atomic coordinates of most residues in the linker (KLAAALK) and all those of 6His-tag were missed, but those 18 C-terminus residues (Q305-L322) had their atomic coordinates and the unchanged folding and interaction networks (Supplementary material Fig. S8). For convenience, D62 and T69 of subunit C, and I244 and Y180 of subunit A in the active sites of BF uricase and BFU-6His were selected as points to reflect the size of the active site. By structural alignment of BF uricase and BFU-6His, there were no significant differences in the folding of main chains of residues in ordered secondary structures within 1.0 nm from those four residues (Fig. 4a), and in the conformation of side chains of residues within 1.0 nm from those four residues (Fig. 4b).

Conformation of residues around active site of BF uricase and BFU-6His. a Structural alignment of main chains of ordered secondary structures within 1.0 nm from residues involved in catalysis of those two uricases. b Structural alignment of side chains of residues within 1.0 nm from four indicated residues (D62 and T69 of subunit C, and I244 and Y180 of subunit A) in active sites of those two uricases
Fusion of the neutral peptide SNSNSN alone to the C-terminus of BF uricase gave a mutant bearing a ~2-fold improved activity at pH 9.2 over BFU-6His while thermal stability at pH 7.4 or 9.2 comparable to BF uricase (Table 3). The mutant after fusion of 6His-tag to the C-terminus of mutant L322D via linker SNSNSN was denoted as L322D-SN-6His, for convenience. The mutant L322D showed the closest activity at pH 9.2 to that of BF uricase. In comparison to the mutant after the fusion of SNSNSN alone to the C-terminus of BF uricase, the mutant after the fusion of SNSNSN alone to the C-terminus of mutant L322D showed even higher activity to account for nearly 85 % activity of the mutant L322D. Interestingly, L322D-SN-6His had activity for about 70 % of BF uricase, while the stability was close to that of BF uricase at pH 7.4 or 9.2 (Table 3). Moreover, BFU-6His and L322D-SN-6His after purification via affinity to Ni2+-NTA had specific activities comparable to those after DEAE-cellulose chromatography, but the purification with once Ni2+-NTA needed only 10 % of the time required for single DEAE-cellulose chromatography.
The fusion of any of such peptides directly to the C-terminus of BF uricase did not significantly alter susceptibility of the enzyme activity to reaction pH; the ratios of activities of such fusion mutants at pH 7.4 were comparable to those at pH 9.2. More importantly, by the same site-directed mutagenesis, relationship of sequence and activity or stability, at pH 7.4 or 9.2, of L322D-SN-6His was consistent with that of BF uricase (Table 3).
Discussion
Distinctive folding of the C-terminus residues of BF uricase
Apparently, overall structure of homotetramer of BF uricase had few differences from those of other uricases that had lower stability. For the first time, however, the atomic coordinates were available for all of the 18 C-terminus residues (Q305 to L322) after the intersubunit antiparallel β-sheet in each subunit of BF uricase homotetramer. As for subunit C of AG uricase at 1.9-Å resolution in pdb ID 2yzb, and AF uricase at 3.2-Å resolution in pdb ID 1ws3, there were coordinates for only some C-terminus residues after the intersubunit antiparallel β-sheets. AG uricase had much more atomic coordinates of the C-terminus residues after the intersubunit antiparallel β-sheet in each subunit than AF uricase; the C-terminus residues of AG uricase turned into the closed tunnel of a homodimer (Fig. 1e). Differently, the 18 C-terminus residues after the intersubunit antiparallel β-sheet in each subunit of BF uricase had an unexpected folding (Fig. 1e). For the first time, thus, potential roles of the unexpected folding of all those 18 C-terminus residues of BF uricase and their associated interactions in thermal stability and enzyme activity were investigated.
Roles of folding and associated interactions in stability and activity of BF uricase
No degradation of proteins under stated conditions, the single-component exponential decreases of activities and significant susceptibility of thermal stability to temperatures suggested that the inactivation of BF uricase and its mutants was attributed to noncovalent structural change of the homotetramer, rather than chemical degradation.
The change in the charge on the second residue slightly decreased the enzyme activity, but did not significantly change thermal stability of mutants at pH 7.4 or 9.2 (Table 2). The elimination of positive charge on the third residue essentially inactivated the mutants and sometimes generated inactive homodimers. Hence, the intersubunit electrostatic attraction between side chains of R3 and D307 in a homodimer was essential for homotetramer integrity and the activity of BF uricase. K9 was essential for activity of BF uricase (data unpublished); it was included in the nearby active site and the intersubunit antiparallel β-sheet. Side chain of R3 easily exerted long-distance electrostatic interaction with side chain of K9; altered electrostatic interaction with R3 was prone to distort conformation of side chain of K9 in the active site and thus decreased enzyme activity of mutants. It was unknown why and how the mutation disrupting the interaction networks involving R3 produced the inactive homodimer. R3 of subunit C and D307 of subunit A were far away from interfaces of homodimers in homotetramer (Fig. 1c); R3 and D307 should be connected via networks with interactions essential for the association of homodimers into homotetramer.
Two H-bonds with tyrosine residues were found to be associated with the unexpected folding of those 18 C-terminus residues of BF uricase (Fig. 2b, c). Mutation of tyrosine into phenylalanine eliminated such H-bonds and caused significant decreases of thermal stability at pH 7.4, but resulted in slight decreases of thermal stability at pH 9.2. Meanwhile, such mutations decreased enzyme activities at pH 9.2; the double mutant Y319F/Y249F was even inactive. Hence, H-bonds from E311-Y249 and Y319-D257 are determinants of thermal stability of BF uricase under physiological conditions and positive factors of BF uricase activity at pH 9.2. There may be the disruption of the localization of the α-helix (Q305-A313) to alter the localization of the last four residues in mutants Y249F and Y319F. Such domino effects of mutation may account primarily for the significant reduction of thermal stability of mutants at pH 7.4 after the loss of any of those two H-bonds.
The decrease of thermal stability of the mutant R310E at pH 7.4 was the most manifest; mutants E311R and E318R exhibited moderate decreases of thermal stability at pH 7.4, but consistent stability at pH 9.2. R310 participated in electrostatic interaction centers involving R3, D307, and E311 (Fig. 2b, c). Significant alterations of electrostatic interactions with such central residues may cause reformation of electrostatic interaction networks on homodimer surface and alteration of localization of residues in the first and second regions of the C-terminus to destabilize the mutants; such effects of mutations may account for the lowered thermal stability of R310E and Y249F (Table 2). On the other hand, electrostatic attraction between α-carboxylic group of L322 and side chain of R258 was a determinant of the stability at pH 7.4, but was a minor factor of both the activity and thermal stability at pH 9.2. However, it was unusual that the reversal of the electrostatic attraction between side chain of R258 and α-carboxylic group of L322 into electrostatic repulsion enhanced thermal stability of the mutant L322R at pH 9.2 (Table 2). Taken together, interactions associated with those 18 C-terminus residues were minor factors of both the stability at pH 9.2 and the activity at pH 9.2 or 7.4, but were crucial determinants of thermal stability at pH 7.4.
As discussed above, the disruption of the interactions associated with the 18 C-terminus residues of BF uricase dramatically decreased the thermal stability at pH 7.4 and moderately decreased catalytic activity at pH 9.2 but did not significantly change catalytic activity at pH 7.4 and thermal stability at pH 9.2. These differential effects of the mutations on catalytic activity and thermal stability of BF uricase at pH 7.4 and 9.2 are interesting and support that structural factors crucial for thermal stability are not necessarily the same as those crucial for the catalytic activity of BF uricase. BF uricase had pI of about 4.8, and all of the mutants had pI below 6.0. BF uricase, and the mutants are all expected to have more net negative charges on surface at pH 9.2 compared to those at pH 7.4 according to the ionization properties of side chains of amino acid residues in proteins. With the increase of the net negative charges on surface, the conformation of BF uricase or mutant is expected to be more dynamic at pH 9.2 due to electrostatic repulsion. Engineering of optimally distributed residues bearing charged side chains on surface is recognized as a plausible way to enhance thermal stability of proteins (Chan et al. 2012; Der et al. 2013; Raghunathan et al. 2013). Thus, the stability of BF uricase and mutants at pH 9.2 might be at least partially associated with the increase in the net negative charges on surface. Further efforts are needed to clarify the actual mechanism of the effects of pH on thermal stability and catalytic activity of BF uricase.
Structure-guided fusion of 6His tag to the C-terminus of BF uricase
For molecular engineering via rational design of mutants, in principle, many mutants may be generated and characterized at considerable cost. The use of 6His-tag for fusion expression and affinity purification will greatly facilitate characterization of mutants. On the other hand, if 6His-tag has negligible or small negative impact on enzyme activity/stability, or such impacts are independent of the designed mutations for molecular engineering, a new technique based on affinity of a fusion tag to an immobilized adsorbent may be applicable for rapid comparison of specific activities of tagged mutants in cell lysates, which thus may greatly accelerate the screening of mutant libraries during directed evolution and the elucidation of relationship between sequences and activity/stability of tagged enzymes (Li et al. 2014; Li et al. 2015). When the fused 6His-tag has a sufficiently high affinity to Ni2+-NTA but negligible negative impact on the enzyme activity/stability, a 6His-tagged fusion mutant bearing favorable properties may be utilized directly for large-scale recombinant production. The 6His-tag and its linker may be removed in coding sequence of the vector before large-scale recombinant production if they have significant negative impact(s) on the activity and/or thermal stability of the enzyme, or be removed after affinity purification at some cost. Thus, 6His-tag fused to either terminus of an enzyme should have sufficient affinity for Ni2+-NTA to facilitate purification and negligible negative impacts on thermal stability and activity of the enzyme. Namely, the fused 6His-tag at either terminus should cause negligible alterations of interaction networks in space of the untagged protein.
In BF uricase, residues of the N-terminus were involved in active site via the intersubunit antiparallel β-sheet; the end of the C-terminus was much farther away from active site than the end of the N-terminus as revealed by the high-resolution crystal structure (Fig. 1c). Indeed, the fusion of 6His-tag to the N-terminus of BF uricase decreased both the stability and activity at pH 7.4 (data not shown), but the fusion of 6His-tag to the C-terminus of BF uricase only decreased enzyme activity (Table 3). Low affinity of the mutant after direct fusion of 6His-tag to the C-terminus may be due to attraction of 6His-tag by spatially near residues bearing negatively charged side chains. The last four residues of BF uricase formed a hydrophobic region to bury a large hydrophobic groove inside (Fig. 2c); alteration of localization of the last four residues may cause partial exposure of the buried large hydrophobic groove to destabilize mutants. As a result, KLAAALK was tested as a linker. In this linker, two lysine residues were included to alleviate attraction of 6His-tag by negatively charged side chains of spatially near residues; the sequence of LAAAL was used to drive the linker to the vicinity of the last four residues via hydrophobic interactions to mask the large hydrophobic groove. Indeed, BFU-6His had reasonable affinity to Ni2+-NTA and exhibited thermal stability at pH 7.4 or 9.2 comparable to BF uricase, but a lower activity.
Clearly, the reconstruction of all complicated interaction networks with those C-terminus residues may restore effectively the activity of BF uricase. To consider potential roles of the lost negative charge of the α-carboxyl group of L322 after the fusion of KLAAALK, EAAAR was tested as another linker, but the use of this linker produced no improvement on the catalytic activity of the fusion mutant even its thermal stability at both pH 7.4 and 9.2 was reserved. There were no significant differences in conformation of residues involved in catalysis between BF uricase and BFU-6His (Fig. 4a, b). Electrostatic interactions are effective at long distances. At pH 9.2, side chain of lysine is still ionized even residues in the 6His-tag are not. To alleviate long-distance interactions with residues involved in catalysis, a neutral hydrophilic linker was tested. Indeed, the fusion of SNSNSN to the C-terminus of BF uricase gave a fusion mutant bearing ~2-fold improved activity compared to BFU-6His and thermal stability comparable to BF uricase.
Residues near I244 in the long α-helix of I244-R258 were included in the nearby active site. Distortion of electrostatic interactions with R258 was prone to alter conformation of residues near I244 and thus the enzyme activity of the fusion mutant. To further restore the enzyme activity of fusion mutant of BF uricase, reconstruction of electrostatic interaction between α-carboxylic group of L322 and side chain of R258 was elaborated based on the unexpected folding of those C-terminus residues. According to the crystal structure of BFU-6His, there may be improper orientation in space of the δ-carboxylic group of E323 from the linker EAAAR in the obtained 6His-tagged mutant to exert electrostatic interaction with side chain of R258. The size of side chain of aspartic acid was close to that of α-carboxylic group of L322, in comparison to that of glutamic acid; the substitution of L322 with aspartic acid may reconstruct electrostatic interaction with side chain of R258. Indeed, L322D-SN-6His after the fusion of 6His-tag to the mutant L322D via the linker SNSNSN showed an activity much higher than that of BFU-6His while thermal stability comparable to BF uricase. Interestingly, L322D-SN-6His reserved nearly 85 % activity of that after the fusion of SNSNSN alone to the mutant L322D. More importantly, by site-directed mutagenesis, the similar relationships between the sequences and activity/stability, as well as the effects of reaction pH on catalytic activities, were observed with L322D-SN-6His and BF uricase. Hence, L322D-SN-6His may be an alternative for molecular engineering of BF uricase.
The pivotal roles of enzyme activity and thermal stability of uricases under physiological conditions in its pharmaceutical value as biodrugs to handle hyperuricemia support structural insights of the C-terminus residues of BF uricase revealed in this report should be valuable for molecular engineering of uricases. Clearly, folding of the C-terminus residues after each intersubunit antiparallel β-sheet of BF uricase is crucial for the design of L322D-SN-6His, which is an active example of molecular engineering of BF uricase. Engineering of C-terminus on surface to enhance interactions with spatially near residues may improve thermal stability of other microbial uricases under physiological conditions.
References
Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221
Alvares K, Widrow RJ, Abu-Jawdeh GM, Schmidt JV, Yeldandi AV, Rao MS, Reddy JK (1992) Rat urate oxidase produced by recombinant baculovirus expression: formation of peroxisome crystalloid core-like structures. Proc Natl Acad Sci U S A 89:4908–4912
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Chan CH, Wilbanks CC, Makhatadze GI, Wong KB (2012) Electrostatic contribution of surface charge residues to the stability of a thermophilic protein: benchmarking experimental and predicted pKa values. PLoS One 7(1):e30296
Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66:12–21
Chica RA, Doucet N, Pelletier JN (2005) Semi-rational approaches to engineering enzyme activity: combining the benefits of directed evolution and rational design. Curr Opin Biotechnol 16:378–384
Colloc’h N, El Hajji M, Bachet B, L’Hermite G, Schiltz M, Prangé T, Castro B, Mornon JP (1997) Crystal structure of the protein drug urate oxidase-inhibitor complex at 2.05 Å resolution. Nat Struct Biol 4:947–952
Colloc’h N, Gabison L, Monard G, Altarsha M, Chiadmi M, Marassio G, Sopkova-de Oliveira Santos J, El Hajji M, Castro B, Abraini JH, Prangé T (2008) Oxygen pressurized X-ray crystallography: probing the dioxygen binding site in cofactorless urate oxidase and implications for its catalytic mechanism. Biophys J 95:2415–2422
Crittenden DB, Pillinger MH (2013) New therapies for gout. Annu Rev Med 64:325–337
Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB 3rd, Snoeyink J, Richardson JS, Richardson DC (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 35:W375–W383
Der BS, Kluwe C, Miklos AE, Jacak R, Lyskov S, Gray JJ, Georgiou G, Ellington AD, Kuhlman B (2013) Alternative computational protocols for supercharging protein surfaces for reversible unfolding and retention of stability. PLoS One 8(5):e64363
Ea HK, Chales G, Lioté F (2011) Pegloticase and chronic gout. JAMA 306:1979
Feng J, Li X, Yang X, Zhang C, Yuan Y, Pu J, Zhao Y, Xie Y, Yuan H, Bu Y, Liao F (2010) A new practical system for evaluating the pharmacological properties of uricase as a potential drug for hyperuricemia. Arch Pharm Res 33:1761–1769
Feng J, Liu H, Yang X, Gao A, Liao J, Feng L, Pu J, Xie Y, Long G, Li Y, Liao F (2013) Comparison of activity indexes for recognizing enzyme mutants of higher activity with uricase as model. Chem Cent J 7:69
Gabison L, Prangé T, El Colloc’h N, Hajji M, Castro B, Chiadmi M (2008) Structural analysis of urate oxidase in complex with its natural substrate inhibited by cyanide: mechanistic implications. BMC Struct Biol 8:32
Gabison L, Chiadmi M, El Hajji M, Castro B, Colloc’h N, Prangé T (2010) Near-atomic resolution structures of urate oxidase complexed with its substrate and analogues: the protonation state of the ligand. Acta Crystallogr D Biol Crystallogr 66:714–724
Hibi T, Hayashi Y, Fukada H, Itoh T, Nago T, Nishiya Y (2014) Intersubunit salt bridges with a sulfate anion control subunit dissociation and thermal stabilization of Bacillus sp. TB-90 urate oxidase. Biochemistry 53:3879–3888
Huang Y, Chen Y, Yang X, Zhao H, Hu X, Pu J, Liao J, Long G, Liao F (2015) Optimization of pH values to formulate the bireagent kit for serum uric acid assay. Biotechnol Appl Biochem 62:137–144
Juan EC, Hoque MM, Shimizu S, Hossain MT, Yamamoto T, Imamura S, Suzuki K, Tsunoda M, Amano H, Sekiguchi T, Takénaka A (2008) Structures of Arthrobacter globiformis urate oxidase-ligand complexes. Acta Crystallogr D Biol Crystallogr D64:815–822
Kahn K, Tipton PA (1997) Kinetic mechanism and cofactor content of soybean root nodule urate oxidase. Biochemistry 36:4731–4738
Li Y, Yang X, He C, Hu X, Pu J, Liu L, Long G, Liao F (2014) Facile quantitative comparison of specific activities of fusion-tagged enzyme/mutants in cell lysates via prediction of their maximum adsorption by anti-tag antibody immobilized in microplate wells. RSC Adv 4:29925–29932
Li Y, Long G, Yang X, Hu X, Feng Y, Tan D, Xie Y, Pu J, Liao F (2015) Approximated maximum adsorption of His-tagged enzyme/mutants on Ni2+-NTA for comparison of specific activities. Int J Biol Macromol 74C:211–217
Liao F, Zhu XY, Wang YM, Zuo YP (2005) The comparison of the estimation of enzyme kinetic parameters by fitting reaction curve to the integrated Michaelis-Menten rate equations of different predictor variables. J Biochem Biophys Methods 62:13–24
Liao F, Zhao YS, Zhao LN, Tao J, Zhu XY, Liu L (2006) Evaluation of a kinetic uricase method for serum uric acid assay by predicting background absorbance of uricase reaction solution with an integrated method. J Zhejiang Univ Sci B 7:497–502
Liu Z, Lu D, Li J, Chen W, Liu Z (2009) Strengthening intersubunit hydrogen bonds for enhanced stability of recombinant urate oxidase from Aspergillus flavus: molecular simulations and experimental validation. Phys Chem Chem Phys 11:333–340
Liu X, Wen M, Li J, Zhai F, Ruan J, Zhang L, Li S (2011) High-yield expression, purification, characterization and structure determination of tag-free Candida utilis uricase. Appl Microbiol Biotechnol 92:529–537
McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007) Phaser crystallographic software. J Appl Crystallogr 40:658–674
Oksanen E, Blakeley MP, El-Hajji M, Ryde U, Budayova-Spano M (2014) The neutron structure of urate oxidase resolves a long-standing mechanistic conundrum and reveals unexpected changes in protonation. PLoS One 9:e86651
Pfrimer P, de Moraes LM, Galdino AS, Salles LP, Reis VC, De Marco JL, Prates MV Jr, Bloch C, Torres FA (2010) Cloning, purification, and partial characterization of Bacillus subtilis urate oxidase expressed in Escherichia coli. J Biomed Biotechnol 2010:674908. doi:10.1155/2010/674908
Raghunathan G, Sokalingam S, Soundrarajan N, Madan B, Munussami G, Lee SG (2013) Modulation of protein stability and aggregation properties by surface charge engineering. Mol Biosyst 9(9):2379–2389
Retailleau P, Colloc’h N, Vivarès D, Bonneté F, Castro B, El-Hajji M, Mornon JP, Monard G, Prangé T (2004) Complexed and ligand-free high-resolution structures of urate oxidase (Uox) from Aspergillus flavus: a reassignment of the active-site binding mode. Acta Crystallogr D Biol Crystallogr 60:453–462
Retailleau P, Colloc’h N, Vivarès D, Bonneté F, Castro B, El Hajji M, Prangé T (2005) Urate oxidase from Aspergillus flavus: new crystal-packing contacts in relation to the content of the active site. Acta Crystallogr D Biol Crystallogr 61:218–229
Sarkissian CN, Gámez A, Wang L, Charbonneau M, Fitzpatrick P, Lemontt JF, Zhao B, Vellard M, Bell SM, Henschell C, Lambert A, Tsuruda L, Stevens RC, Scriver CR (2008) Preclinical evaluation of multiple species of PEGylated recombinant phenylalanine ammonia lyase for the treatment of phenylketonuria. Proc Natl Acad Sci U S A 105:20894–20899
Schiavon O, Caliceti P, Ferruti P, Veronese FM (2000) Therapeutic proteins: a comparison of chemical and biological properties of uricase conjugated to linear or branched poly(ethylene glycol) and poly(N-acryloylmorpholine). Farmaco 55:264–269
Schlesinger N, Yasothan U, Kirkpatrick P (2011) Pegloticase. Nat Rev Drug Discov 10:17–18
Suzuki K, Sakasegawa S, Misaki H, Sugiyama M (2004) Molecular cloning and expression of uricase gene from Arthrobacter globiformis in Escherichia coli and characterization of the gene product. J Biosci Bioeng 98:153–158
Tan Q, Zhang J, Wang N, Li X, Xiong H, Teng Y, He D, Wu J, Zhao C, Yin H, Zhang L (2012) Uricase from Bacillus fastidious loaded in alkaline enzymosomes: enhanced biochemical and pharmacological characteristics in hypouricemic rats. Eur J Pharm Biopharm 82:43–48
Turner NJ (2009) Directed evolution drives the next generation of biocatalysts. Nat Chem Biol 5:567–573
Wu S, Chen B, Liu C, Ou Y, Yi J (2009) Expression in Escherichia coli, purification and enzymatic properties of porcine urate oxidase. Sheng Wu Gong Cheng Xue Bao 25:1664–1670
Yamamoto K, Kojima Y, Kikuchi T, Shigyo T, Sugihara K, Takashio M, Emi S (1996) Nucleotide sequence of the uricase gene from Bacillus sp.TB-90. J Biochem 119:80–84
Yang X, Yuan Y, Zhan CG, Liao F (2012) Uricases as therapeutic agents to treat refractory gout: current states and future directions. Drug Dev Res 73:66–72
Zhang C, Yang X, Feng J, Yuan Y, Li X, Bu Y, Xie Y, Yuan H, Liao F (2010) Effects of modification of amino groups with poly(ethylene glycol) on a recombinant uricase from Bacillus fastidiosus. Biosci Biotechnol Biochem 74:1298–1301
Zhang P, Xu L, Li Q, Lin X, Liu H, Ma X (2012) Cloning and characterization of a thermostable urate oxidase from Microbacterium sp. strain ZZJ4-1. Sheng Wu Gong Cheng Xue Bao 28:813–822
Zhang C, Yang X, Gao A, Hu X, Pu J, Liu H, Feng J, Liao J, Li Y, Liao F (2014) Comparison of modification of a bacterial uricase with N-hydroxysuccinimide esters of succinate and carbonate of monomethoxyl poly(ethylene glycol). Biotechnol Appl Biochem 61:683-690
Zhao Y, Zhao L, Yang G, Tao J, Bu Y, Liao F (2006) Characterization of a uricase from Bacillus fastidious ATCC26904 and its application to serum uric acid assay by a patented kinetic uricase method. Biotechnol Appl Biochem 45:75–80
Zhao Y, Yang X, Lu W, Liao H, Liao F (2009a) Uricase based method for determination of uric acid in serum. Microchim Acta 164:1–6
Zhao Y, Yang X, Li X, Bu Y, Deng P, Zhang C, Feng J, Xie Y, Zhu S, Yuan H, Yu M, Liao F (2009b) Reversible inactivation of an intracellular uricase from Bacillus fastidiosus via dissociation of homotetramer into homodimers in solutions of low ionic strength. Biosci Biotechnol Biochem 73:2141–2144
Zwart PH, Afonine PV, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, McKee E, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Storoni LC, Terwilliger TC, Adams PD (2008) Automated structure solution with the PHENIX suite. Methods Mol Biol 426:419–435
Acknowledgments
This work was supported by the National Natural Science Foundation of China (no.30672139), Natural Sciences Foundation of CQ (nos. CSTC2011BA5039 and CSTC2012JJA0057), the Education Ministry of China (grant no.20125503110007), and the National Science Foundation of USA (grant CHE-1111761). We thank scientists in Shanghai Synchrotron Radiation Facility for technical support during collection of data.
Author information
Authors and Affiliations
Corresponding author
Additional information
Juan Feng, Lu Wang and Hongbo Liu contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 3360 kb)
Rights and permissions
About this article

Cite this article
Feng, J., Wang, L., Liu, H. et al. Crystal structure of Bacillus fastidious uricase reveals an unexpected folding of the C-terminus residues crucial for thermostability under physiological conditions. Appl Microbiol Biotechnol 99, 7973–7986 (2015). https://doi.org/10.1007/s00253-015-6520-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-015-6520-6