
Abstract
Bacteriophage lytic enzymes, either endolysins or virion-associated lysins, have been receiving considerable attention as potential antibacterial agents, particularly for the combat of antibiotic-resistant Gram-positive pathogens. A conclusion that easily emerges from the careful analysis of a great number of reports on the field is that the activity of phage lytic enzymes is rarely studied in conditions that support robust growth of the target bacteria. Here, we report the construction and study of a chimerical lysin, EC300, which was designed to target and kill Enterococcus faecalis in conditions supporting vigorous bacterial growth. EC300 resulted from the fusion of a predicted M23 endopeptidase domain of a virion-associated lysin to the putative cell wall binding domain of a previously characterized amidase endolysin, both produced by the E. faecalis phage F170/08. This bacteriolysin-like protein exhibited a clear enhanced lytic activity over the parental endolysin when both were assayed in a rich bacterial growth medium. We demonstrate the killing efficacy of EC300 against growing cells of a panel of typed E. faecalis clinical strains with high level of antibiotic resistance. The possible reasons for the marked difference between the lytic performance of EC300 and that of the amidase are discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Enterococci are commensal bacteria in the intestines of humans and several animals and can also be found in soil, water, and plants (Klein 2003). Yet, the species Enterococcus faecalis and Enterococcus faecium have also emerged as important opportunistic pathogens worldwide, especially because of life-threatening nosocomial infections (Gilmore et al. 2013). They have been associated to several human infections, such as neonatal sepsis, peritonitis, device-related infections, and infective endocarditis, being also described as leading causes of wound and urinary tract infections and bacteraemias (Emori and Gaynes 1993; Fisher and Phillips 2009; Gilmore et al. 2013; Poh et al. 2006; Sava et al. 2010; Schaberg et al. 1991). Enterococci exhibit intrinsic resistance to several first-line antimicrobial agents (Hammerum 2012) and high propensity to acquire resistance to others, including quinolones, macrolides, tetracyclines, streptogramins, and glycopeptides (Arias and Murray 2008; French 2010; Murray 1990). The reduced susceptibility to antibiotics can make extremely difficult the treatment of an enterococcal infection, and the therapeutic options can be very limited (Theuretzbacher 2012; Werner et al. 2013). Therefore, there is a growing need to find therapeutic alternatives to fight infections caused by these multidrug resistant enterococci.
Virion-associated lysins (VALs) and endolysins are enzymes produced by bacteriophages (phages) that degrade the peptidoglycan (PG) moiety of the bacterial cell wall. VALs are typically carried in the virus particle and are thought to promote a local cleavage of PG bonds to facilitate phage genome transference into the host bacterial cell. Endolysins act at the end of the phage reproductive cycle to destroy the cell wall PG mesh, leading to cell burst and to the consequent release of the virion progeny. The potential of endolysins and more recently of VALs as antibacterial agents toward Gram-positive bacterial pathogens has been intensively studied (for recent reviews, see Fenton et al. 2010; Nelson et al. 2012; Rodríguez-Rubio et al. 2013; Schmelcher et al. 2012a). Endolysins from phages infecting Gram-positive bacteria typically display a modular architecture where one or more catalytic domains (CDs) responsible for PG cleavage are connected by a flexible linker to a cell wall binding (CWB) domain (Schmelcher et al. 2012a). As components of phage virus particles, VALs are frequently multifunctional proteins, playing essential roles during virion morphogenesis and sometimes participating in initial steps of phage infection other than just the degradation of the cell wall PG (Boulanger et al. 2008; Piuri and Hatfull 2006). VALs from phages infecting Gram-positive bacteria are usually larger than cognate endolysins, and a single VAL may also display multiple CDs for PG hydrolysis, while generally no CWB domains are identifiable (Rodríguez-Rubio et al. 2013).
In the vast majority of the studies reporting the antibacterial activity of phage PG hydrolases, the lytic enzymes are tested in conditions that do not support robust bacterial growth; most commonly, in vitro experiments are performed with target cells washed and suspended in buffered solutions (Gu et al. 2011; Proença et al. 2012). Often, the high lytic activity observed in these conditions does not translate to the expected results when assays are transposed to animal infection models, and in some cases, satisfactory levels of animal survival are only obtained when lytic enzymes are administrated to animals soon after the injection of the deadly bacterial inoculum (Gu et al. 2011; Loeffler et al. 2003; Oechslin et al. 2013). The idea that actively growing bacteria are able to mount at least a certain level of resistance to endolysin lytic action is somewhat expected, if we consider that in the context of phage infection, endolysins always act after cells had been killed by another phage-encoded protein, the holin, even when the lytic enzymes are released to the cell wall compartment by a holin-independent mechanism (Catalão et al. 2013). Regarding this aspect, VALs might be viewed as having the advantage of being naturally “designed” to act on dividing bacteria. Another group of proteins sharing this feature are bacteriolysins (formerly class III bacteriocins, Cotter et al. 2005), such as the M23-like endopeptidases lysostaphin and enterolysin A, which are known to display potent lytic activity in growth-promoting conditions (Khan et al. 2013; Kumar 2008).
The goal of this work was to design an enzyme with effective anti-E. faecalis activity in cell growth supporting conditions. For that, we have assumed the theoretical advantages of VALs and bacteriolysins referred to above and have generated an artificial, bacteriolysin-like enzyme (EC300) by fusing the M23 endopeptidase CD of the VAL Orf73 to the CWB domain of the previously characterized endolysin Lys170 (Proença et al. 2012, 2014), both produced by the E. faecalis phage F170/08. The results show the superior lytic activity of EC300 when compared to the endolysin Lys170.
Materials and methods
Bacteria, phage, and growth conditions
Escherichia coli strain XL1-Blue MRF’ (Stratagene) was used for plasmid isolation and propagation, whereas E. coli strain CG61 (São-José et al. 2000) was used for protein production. E. coli strains were routinely grown in LB medium (Sambrook and Russell 2001) in an orbital incubator (200 rpm, SM 30 control, Edmund Bühler GmbH), either at 37 °C (XL1-Blue MRF’) or at 28 °C (CG61 and its derivatives). When necessary, LB was supplemented with ampicillin (100 μg/ml), kanamycin (30 μg/ml), and/or tetracycline (10 μg/ml). The antibacterial activity of EC300 was tested against a panel of typed, multiresistant enterococcal clinical strains (Table 1), which was composed of 28 E. faecalis and 21 E. faecium isolates from patients of a Portuguese hospital between 2004 and 2006 (Mato et al. 2009), plus the two model E. faecalis clinical strains MMH594 (NR-31975, BEI Resources, Manassas, USA) and V583 (ATCC 700802, ATCC/LGC Standards, Teddington, UK) (Huycke et al. 1991; Paulsen et al. 2003; Sahm et al. 1989; Shankar et al. 2002). These strains and the E. faecalis clinical isolates 1518/05 and 926/05 from Technophage collection (Lisbon, Portugal) were grown in Tryptic Soy Broth (TSB) at 37 °C. When required, media were supplemented with 1.4 or 0.7 % agar to obtain solid or soft-agar plates, respectively. All culture media components were purchased from Biokar Diagnostics. E. faecalis phage F170/08, which was isolated from a sewage water sample of the Lisbon area after its incorporation in a soft-agar brain heart infusion (BHI) lawn inoculated with the host strain E. faecalis 926/05 (classic double agar overlay method, Kropinski et al. 2009), was propagated as described previously (Proença et al. 2012).
General DNA techniques
Phage F170/08 DNA was extracted from CsCl-purified lysates as described previously (Vinga et al. 2012). Preparation of E. coli plasmid DNA and purification of polymerase chain reaction (PCR) products were performed with the commercial kits QIAprep Spin Miniprep kit (QIAGEN) and High Pure PCR Product Amplification kit (Roche Applied Science), respectively, following manufacturer’s instructions. Restriction enzymes and T4 DNA ligase were purchased from Fermentas (Thermo Scientific). Recombinant plasmids were confirmed by DNA sequencing (Macrogen, Seoul, Korea). Restriction endonuclease digestions, DNA ligations, and conventional agarose gel electrophoresis were carried out essentially as described by Sambrook and Russell (2001). Development of competence and transformation of E. coli strains by heat shock (45 s in a water bath set to 42 °C) was performed according to the method of Chung et al. (1989).
General protein techniques
The Bradford reagent (Bio-Rad Laboratories) was used for protein quantification using bovine serum albumin as standard. After sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE), gels were either stained with Coomassie blue or transferred to 0.45-μm nitrocellulose membranes (Bio-Rad Laboratories) for Western blotting analyses. EC300 polypeptides were immunodetected using a horseradish peroxidase-conjugated anti-His6 monoclonal antibody (Roche Applied Science). PageRuler Prestained Protein Ladder (Thermo Scientific) was used as protein marker in SDS-PAGE.
Construction and cloning of EC300 chimeric gene and its derivative mEC300
The nucleotide sequence of the primers used to generate the EC300 and mEC300 genes is indicated in Table S1. The coding sequence encompassing the peptidase M23 CD of the VAL Orf73 from the enterococcal phage F170/08 was PCR amplified with the primer pair 73Fw2/Rv_M23_CWB170, using phage DNA as template and the polymerase KOD hot start master mix (Novagen). The PCR conditions were set according to the recommendations of the polymerase supplier. The sequence encoding the C-terminal region of the cognate endolysin Lys170, harboring its CWB domain (CWB170), was similarly amplified in a separate reaction using the primer pair CWB_Fw_170/F170_Xma. Orf73 (VAL) and Lys170 (endolysin) of phage F170/08 are virtually identical (over 99 % amino acid sequence identity) to Orfs 30 and 9 of E. faecalis phage ϕEF24C, respectively (GenBank BAF81298 and BAF81277; Uchiyama et al. 2008). The 3′ and 5′ ends of the M23- and CWB170-containing PCR products, respectively, carried a 28-bp complementary segment that allowed fusing both fragments by overlap extension PCR (OE-PCR, Ho et al. 1989), using the 73Fw2 and the F170_Xma primers. These primers added NdeI and XmaI restriction sites for cloning of the chimerical gene in the expression vector pIVEX2.3d (Roche Applied Science), generating the recombinant plasmid pDPEC300. Gene EC300 in pDPEC300 was subjected to site-directed mutagenesis by using the Quick Change II Site directed mutagenesis kit (Stratagene Agilent Technologies) and the primer pair Fw_BD_M170/ Rv_BD_M170, resulting in plasmid pDPmEC300 carrying mEC300 gene. The introduced nucleotide substitutions eliminated the internal translation start site known to drive the independent synthesis of the CWB170 domain (Proença et al. 2014, see text also). The pIVEX vectors allow the expression of genes under the control of the phage T7 ϕ10 promoter, and the production of the corresponding proteins C-terminally fused to a hexahistidine tag. Details of the primary sequence of EC300, mEC300, and of the parental lytic enzymes are presented in Fig. S1. The pIVEX2.3d derivatives were used to transform E. coli strain CG61, which produces the phage T7 RNA polymerase upon temperate upshift. The production of active EC300 by CG61 clones was confirmed by growing them over a dense lawn of autoclaved enterococcal cells and by checking the development of lysis halos around E. coli colonies (Proença et al. 2012, Fig. S2).
Protein production and purification
Induction of EC300 and mEC300 synthesis by E. coli CG61 carrying pDPEC300 or pDPmEC300, respectively, production of total protein extracts, and subsequent purification by metal chelate affinity chromatography (AFC) were carried out as described by Proença et al. (2012), except for the following changes: induction of protein production by heat shock occurred in a shaking water bath set to 42 °C (100 rpm, model BSH, Raypa—R. Espinar, SL) for 30 min, followed by a 3-h incubation at 37 °C (200 rpm, model SM 30 control, Edmund Bühler GmbH), and removal of insoluble material from total protein extracts was by centrifugation at 35,000 g, 25 min, 4 °C in a High-Performance Centrifuge Avanti J-25I (rotor JA 25-50, Beckman Coulter). Fractions eluted from the AFC step (HisTrap HP columns, GE Healthcare) were analyzed by SDS-PAGE, and those containing the partially purified enzymes were subjected to size-exclusion chromatography (SEC) using a Hi-load 16/600 superdex 75 prep grade column (GE Healthcare), equilibrated and run in protein buffer (20 mM HEPES-Na, 500 mM NaCl, 1 % glycerol, and 1 mM DTT, pH8.0) at a flow rate of 1 ml/min. Purified enzymes were divided in small aliquots and stored at −80 °C until use. Partition coefficients (K av ) extracted from SEC were used to estimate protein masses by extrapolation from a plot of Stokes radii of standard proteins versus (-logK av )1/2 (Cabré et al. 1989). The column void volume (V 0) was determined with blue dextran 2000 (GE Healthcare Life Sciences). The standard proteins (Bio-Rad Laboratories) were thyroglobulin (molecular mass = 670 kDa, Stokes radius = 8.6 nm), γ-globulin (158 kDa, 4.8 nm), ovalbumin (44 kDa, 2.73 nm), myoglobin (17 kDa, 2.08 nm), and vitamin B12 (1.35 kDa, 0.85 nm). Proteins Lys170 and CWB170, also used in this work, were produced from pIVEX2.3 derivatives pDP2 (Proença et al. 2012) and pDP4 (Proença et al. 2014) and purified as described above.
Lytic activity in liquid media
The lytic activity of EC300 and Lys170 was studied against selected E. faecalis strains cultured in TSB medium as described above. Cells from cultures grown to an optical density at 600 nm (OD600) of 0.3–0.4 were collected by centrifugation at 8000 g for 10 min (model 5804R, Eppendorf) and resuspended in ½ volume of fresh TSB. Cell suspensions were challenged with the indicated concentrations of EC300, Lys170 and/or nisin (Sigma Aldrich), and OD600 variations followed over time. Lytic activity was also tested with E. faecalis cells recovered in ½ volume of protein buffer. Negative controls were similarly prepared, except that protein buffer was added instead of the lytic proteins.
Evaluation of EC300 antibacterial activity in solid medium
The bacterial growth inhibition potential of EC300 and Lys170 was evaluated against the panel of typed E. faecalis and E. faecium clinical strains (Table 1) on double-layer agar TSA plates as follows. A 200-μl sample of each target bacteria in exponential growth phase (OD600 = 0.3–0.4) was incorporated in 5-ml of TSA soft-agar and poured over a TSA solid bottom. Plates were allowed to dry for 30 min in a laminar flow cabinet, and then four different amounts of purified EC300 (10, 3.3, 1.1, and 0.37 μg in a final volume of 10 μl) were spotted on each strain lawn. The plates were incubated overnight at 37 °C, and the anti-enterococcal activity was evaluated and scored (− to +++) according to relative diameter and transparency of the growth inhibition halos. Lys170 endolysin was only tested at the maximum amount (10 μg). EC300, mEC300, Lys170, and CWB170, alone or in combination, were also tested on dense lawns of viable E. faecalis strain 1518/05 prepared in agarized protein buffer (0.7 % agar) as described previously (Proença et al. 2014). Negative controls were prepared by spotting 10 μl of protein buffer.
Bioinformatics tools
Phage F170/08 putative genes were recognized by integrating results obtained with GeneMark.hmm and MetaGeneAnnotator web software (Besemer and Borodovsky 2005; Noguchi et al. 2008). Identification of phage F170/08 putative VALs was based on BLASTP homology searches (Altschul et al. 1997) and on the prediction of protein functional domains using NCBI’s CDD (Marchler-Bauer et al. 2011) and Pfam (http://pfam.xfam.org/). Putative linkers connecting protein functional domains were assigned with SVM (Ebina et al. 2009), using the SVM-joint output.
Results
Rationale for the construction of the chimeric lysin EC300
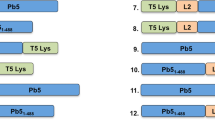
We studied previously the lytic activity of Lys170, an endolysin recently shown to correspond to a heterooligomer (see below). Lys170 displayed a broad spectrum of lytic activity against E. faecalis clinical strains when these were collected from exponentially growing cultures and resuspended in a physiologic buffer before enzyme addition (Proença et al. 2012). However, we observed that the endolysin exhibited very poor or no lytic activity when added directly to logarithmic phase cultures in rich media like TSB (see below) or BHI (Proença et al. unpublished). As mentioned above, the capacity of “healthy” growing bacteria to offer some resistance to endolysin attack from the cell exterior may simply reflect the fact that during phage infection, endolysins always act in cells previously killed by the holin function. In contrast, other PG hydrolases such as VALs and bacteriolysins are meant to act against bacteria in active growth phase. Curiously, some PG hydrolase CDs seem to be shared by VALs and bacteriolysins, like for example the endopeptidase CD of the M23 family (Fig. 1a), which is found in several VALs and in the bacteriolysins lysostaphin and enterolysin A (Khan et al. 2013; Rodríguez-Rubio et al. 2012a; Stockdale et al. 2013; Sudiarta et al. 2010; Thumm and Götz 1997). In silico analysis of phage F170/08 genome sequence allowed the identification of two VAL putative genes, orf72 and orf73. Their deduced products are large proteins bearing putative C-terminal CDs of different PG cleavage specificities but none carrying any detectable CWB domain (not shown). We focused on Orf73 (1061 amino acid residues, 118 kDa) that harbors two putative PG hydrolase CDs, a peptidase M23 (residues 687 to 787, Pfam database entry PF01551) and a NLPC/P60 (residues 926 to 1058, Pfam database entry PF00877; Anantharaman and Aravind 2003) (Figs. 1b and S 1A). We have reasoned that by fusing the peptidase M23 CD of Orf73 to the CWB domain of the cognate endolysin Lys170 (CWB170), we would generate a bacteriolysin-like chimera with the capacity of inducing lysis of actively growing E. faecalis, thus overcoming the limitation described for Lys170. The resulting anti-E nterococcus faecalis chimera of 300 amino acids (EC300) is schematically represented in Fig. 1b, and details of its primary sequence are presented in Fig. S1.

Rationale behind the construction of the lytic chimera EC300. a Examples of bacteriolysins and virion-associated lysins (VALs) harboring the endopeptidase catalytic domain (CD) of the M23 family (M23). Note that the bacteriolysin schemes represent the direct, non-processed products of translation. b Domain architecture of EC300 and of the parental proteins Orf73 and Lys170 of phage F170/08. CWB170 is the CWB domain of endolysin Lys170. CD families (Pfam database entries): M23, peptidase family (PF01551); SLT, soluble lytic transglycosylase (PF01464); NLPC/P60, peptidase family (PF00877); SH3b, cell wall binding domain of the SH3_5 family (PF08460); Amidase_2, amidase family (PF01510)
Purification and features of EC300
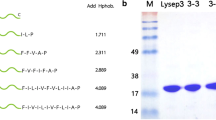
EC300 was produced in E. coli C-terminally fused to a hexahistidine tag (His6), which allowed a first purification step by affinity chromatography, followed by a second step of size-exclusion chromatography (SEC). During protein production, we systematically detected in Coomassie-stained SDS-PAGE gels and in anti-His6 Western blots a C-terminal fragment of EC300 of about 12 kDa, in addition to the expected full length protein (Fig. 2). The same fragment is also produced during synthesis of the parental Lys170, and we showed recently that it is essential for robust lytic activity of the endolysin (Proença et al. 2014). In fact, the C-terminal product (CWB170) results from an in frame, secondary translational start site lying at the beginning of the CWB170 coding sequence; this secondary start site is present both in Lys170 and EC300 sequences (Fig. S1). We showed also that CWB170 oligomerizes and associates with the full length Lys170 via CWB170-CWB170 interactions to form a heterooligomeric complex (Proença et al. 2014). Analysis of the major size-exclusion chromatography (SEC) peak of EC300 (SE1 peak in Fig. 2) indicates that the chimeric protein is an analogous heterooligomer, which also results from the association of the full length EC300 with the CWB170 subunit.

EC300 and mEC300 purification. a EC300 and mEC300 fractions from the corresponding affinity chromatography (AFC) purification steps were subjected to size-exclusion chromatography (SEC). The eluting profile of the proteins was monitored by taking absorbance measurements at 280 nm (A280 nm). Representative UV curves were combined in a single graph. Note the two-peak elution profile of EC300 corresponding to the full length EC300/CWB170 complex and to the free CWB170 module, respectively. The apparent protein masses derived from the experimentally determined partition coefficients (K av , see methods) are indicated for each protein. The column void volume (V 0) and the masses of standard proteins are also indicated. b SDS-PAGE analysis of the AFC and SEC steps of EC300 and mEC300. Lanes: T total protein extract, FT flow through, AFC affinity chromatography peak fraction, SE1 and SE2 EC300 SEC peak fractions, SE3 mEC300 SEC peak fraction. The full length EC300 (34 kDa) and the CWB170 (12 kDa) polypeptides are indicated by white and black arrows, respectively
Elimination of the internal start site in EC300 coding sequence, with the consequent abolishment of synthesis of the extra CWB170-containing ~12 kDa polypeptide, resulted in a dramatic decrease of the lytic activity of the mutated protein (mEC300). Yet, coincubation of mEC300 with increasing amounts of independently purified CWB170 (Proença et al. 2014) progressively restored at least part of the lytic activity lost by the mutated protein (Fig. 3), further supporting the heterooligomeric nature of the fully active EC300. The amount of mEC300/CWB170 active complexes formed in these conditions was sufficient for their detection in the highly sensitive spot test assay of Fig. 3 but insufficient to induce detectable lysis of dense cell suspensions, probably reflecting inefficient formation of active complexes. This phenomenon and the rescue of lytic activity upon incubation with purified CWB170 were also observed in identical assays with the corresponding mutant mLys170 of the parental endolysin (Proença et al. 2014).

Impact of CWB170 polypeptide in EC300 activity. A fixed amount of purified mEC300 (10 μg, 0.31 nmol) was coincubated with CWB170 at the indicated mEC300: CWB170 molar ratios for 1 h at room temperature. After this period, each protein mixture was spotted on a dense lawn of live E. faecalis cells prepared in agarized protein buffer. The image shows the lysis halos developed after overnight incubation at 37 °C. Lysis halos from individually spotted mEC300 (0.31 nmol), EC300 (0.31 nmol), and CWB170 (1.86 nmol) are shown in the bottom row
During EC300 heterologous production, the CWB170 fragment seemed to accumulate in large excess when compared to the amount detected during Lys170 synthesis in the same conditions (Proença et al. 2014); this is probably the cause of the second peak observed during EC300 SEC, which is composed of free CWB170 (SE2 in Fig. 2). The elution profile of this nonassociated form of CWB170, with an apparent mass of 36 kDa (Fig. 2a), is very similar to that of an independently expressed, recombinant form of CWB170, which we have shown to form homotetramers (Proença et al. 2014).
All the experiments described below were carried out with EC300 from fractions of SEC peak SE1 (Fig. 2), which in fact correspond to a complex of full length EC300 associated with CWB170 subunits. Data from SEC (Fig. 2a) and cross-linking experiments (not shown) indicate that the stoichiometry of the EC300 complex should be identical to that proposed for Lys170 (Proença et al. 2014), that is, one full length EC300 for three CWB170 subunits. In addition, mEC300 seems monomeric in solution but, as described for mLys170 (Proença et al. 2014), the protein appears to exhibit an extended conformation as it elutes during SEC with an apparent mass (46 kDa) higher than the expected for the monomer (34 kDa) (Fig. 2a).
EC300 has superior lytic activity when compared to Lys170
The lytic activity of EC300 and of the parental endolysin Lys170 was tested with E. faecalis cells collected from exponentially growing cultures. Both enzymes could efficiently lyse cells resuspended in a buffered solution, although EC300 provoked faster and more extensive lysis than the endolysin (Fig. 4a). However, Lys170 could neither induce lysis nor even arrest growth of cell suspensions prepared in fresh TSB culture medium, whereas EC300 was still able to elicit lysis in these conditions (Fig. 4b). To rule out the possibility of selective inhibition of Lys170 by TSB components, cell suspensions were simultaneously treated with the endolysin and nisin, a well-known lantibiotic that causes cell death by inducing the formation of pores in the bacterial cytoplasmic membrane (Hasper et al. 2004; Wenzel et al. 2012). Cells killed by the nisin action were revealed to be fully susceptible to the lytic action of Lys170, indicating that TSB components do not significantly interfere with endolysin activity (Fig. 4b). The results also indicated that in nutritional media, E. faecalis cells are intrinsically resistant to Lys170 attack from the outside but still susceptible to the chimeric enzyme.

Comparison of EC300 and Lys170 lytic activities in liquid medium. Cells from exponentially growing E. faecalis strain 1518/05 were suspended either in a buffered solution (a) or in TSB (b) and OD600 variation followed after addition of the lytic enzymes. EC300 and Lys170 were added at 10 μg/ml in a and 50 μg/ml in b. Nisin concentration, either alone or in combination with Lys170, was 2 μg/ml. The “cells” and “buffer” curves correspond to controls with no additions or with added protein buffer, respectively
EC300 spectrum of activity against enterococcal clinical strains
The spectrum of EC300 antibacterial activity was evaluated on a panel of typed, multidrug-resistant E. faecalis, and E. faecium clinical strains (Table 1). These strains displayed a high-level resistance to gentamicin and included vancomycin-resistant enterococci (VRE) of clonal complexes E. faecalis-CC2 and E. faecium-CC17, which have been described as highly prevalent in nosocomial settings and disseminated worldwide (Kuch et al. 2012; Mato et al. 2009; Top et al. 2008). Four quantities of EC300 (10, 3.3, 1.1, and 0.37 μg) were spotted on soft-agar TSA lawns that had been inoculated with cells from exponentially growing cultures of each strain of the panel (see methods). As positive control for EC300 activity, we used a lawn of E. faecalis clinical isolate 1518/05, which we had observed previously to be susceptible to the chimera.
Growth of E. faecium strains appeared unaffected by any of the spotted EC300 amounts. In contrast, growth inhibition could be detected in 97 % of E. faecalis strains for the highest tested quantity of the chimeric lysin and 40 % for the lowest (Fig. 5). Note that in these assay conditions, 10 μg of the endolysin Lys170 produced only a moderate/slight growth inhibition in three of the E. faecalis strains (Table S2). The two lytic enzymes produce indistinguishable lysis halos on dense lawns of the control E. faecalis strain 1518/05 prepared in agarized assay buffer (Fig. 5a), as it would be expected after the results of Fig. 4a. Of the four vancomycin-resistant E. faecalis strains tested, two (EHCP 267 and EHCP 389) seemed to be much more susceptible to EC300 than the others (V583 and EHCP 3) (Fig. 6).

Susceptibility of a panel of typed E. faecalis strains to the EC300 growth inhibition activity. a Lytic activity of Lys170 and EC300 against live cells of strain 1518/05 incorporated as a dense lawn in an agarized buffer. b Representative growth inhibition halos, classified (−) to (+++), obtained in a soft-agar TSA lawn of strain EHCP 78 after spotting the indicated EC300 quantities. Spots of 10 μg of Lys170 and of 10 μl of protein buffer were also tested. c Percentage of strains susceptible to the different amounts of EC300 (growth inhibition evaluated on soft-agar TSA lawns)

Evaluation of EC300 capacity to inhibit growth of four vancomycin-resistant E. faecalis strains (EHCP 3, EHCP 389, EHCP 267, and V583). The amounts of tested EC300 and the assay conditions were as in Fig. 5b. Spots of 10 μg of Lys170 and of 10 μl of protein buffer were also tested as controls
Discussion
The work here presented was prompted by a couple of observations with a few endolysins we have studied recently: (1) the endolysins were able to lyse target bacteria suspended in media that keep cell viability without supporting growth (e.g. buffered solutions) and (2) the same endolysins could induce efficient lysis of target cells suspended in growth-promoting media (e.g. culture media) only if bacteria were first or concomitantly killed by another agent, like for example the lantibiotic nisin. Nisin was previously shown to dramatically enhance the lytic activity of endolysins (Catalão et al. 2010; García et al. 2010; Nascimento et al. 2008) and to trigger the activity of bacterial autolysins (Frias et al. 2009; Lamsa et al. 2012; Severina et al. 1998). These facts, associated to the observation that most studies on the lytic action of endolysins are performed in conditions where target bacteria are in a state of reduced metabolic activity, led us to consider the possibility that bacteria under suitable growth conditions may offer resistance to endolysin lytic activity. We postulate that the killing action of holins, which precedes always endolysin attack in the natural context of phage infection, might have a general function of abolishing such resistance. Although we have not addressed this hypothesis in our study, if confirmed, it may simply indicate that at least some endolysins may not be able to efficiently attack well fit bacteria. Phage VALs and bacteriolysins may stand as better suited alternatives as they are naturally adapted to act from the outside on dividing bacteria. In fact, VALs are responsible for the phenomenon of “lysis from without,” which is characterized by the ability of some phages to induce premature lysis when added to host cells at high multiplicities (Abedon 2011).
The idea of taking advantage of the particular features of bacteriolysins and VALs has been explored recently, particularly when targeting Staphylococcus aureus. The mature form of lysostaphin or just its CWB domain (SH3b) has been fused to endolysin CDs to generate chimeras with improved antibacterial activity or extended spectrum (Donovan et al. 2006; Idelevich et al. 2011; Schmelcher et al. 2012b). The fusion of the lysostaphin SH3b domain to CDs isolated from VALs of staphylococcal phages resulted in lytic enzymes with improved killing/lytic performance when compared to the parental VALs; one possible explanation for this is that the SH3b domain contributes to a more efficient recognition and/or binding to target cells (Paul et al. 2011; Rodríguez-Rubio et al. 2012b). With very few known exceptions (Takác and Bläsi 2005), VALs seem to lack the CWB domains typically found in endolysins. This probably results from the fact that in the virus particle, VAL cell wall targeting is accomplished by other components of the virion structure, such as the receptor binding proteins (Rodríguez-Rubio et al. 2013).
In line with the ideas explained above, we have for the first time engineered a bacteriolysin-like enzyme (EC300) aimed at killing E. faecalis in growth supporting conditions. The chimera EC300, which combined a M23 peptidase CD from a VAL and an endolysin CWB domain (CWB170), showed a general increase of the lytic and killing performance when compared to the parental endolysin (Lys170). The superior antibacterial activity of EC300 was particularly evident in bacterial culture media where it could inhibit growth of almost all tested E. faecalis clinical strains, whereas Lys170 produced only some detectable inhibitory activity in three strains (Table S2). In a previous study, where the endolysin was tested against dense lawns of bacteria prepared in a soft-agar physiologic buffer, more than 90 % of the same strains were susceptible to the endolysin (Proença et al. 2012). Thus, the particular domain configuration M23Orf73-CWB170 seems to render EC300 much less sensitive to the mechanism that retrains Lys170 activity during E. faecalis active growth. However, the chimerical enzyme seems to follow the trend of the endolysin regarding the spectrum of activity (Proença et al. 2012), as it seems to act only against E. faecalis. EC300 was tested against four vancomycin-resistant E. faecalis strains in the growth-promoting conditions, with two of them appearing much more susceptible to the chimera (Fig. 6). Given the reduced number of tested VRE strains and considering that a wide spectrum of relative activity was also observed for vancomycin-susceptible strains (Table S2), we could not establish any obvious correlation between glycopeptide resistance and susceptibility to EC300.
M23-like peptidase domains are present in a wide variety of proteins such as PG hydrolases of bacterial origin and eukaryotic cell proteins but are rarely found in endolysins. One exception seems to be the endolysin of the staphylococcal phage 2638A, which has been described as having an N-terminal M23 peptidase domain, a central amidase CD and a C-terminal SH3b CWB domain. Interestingly, in the assayed conditions, the amidase CD of this endolysin was shown to contribute to most of the enzyme’s lytic activity (Abaev et al. 2013).
Another interesting feature of EC300 results from the fact that, similarly to Lys170, the fully active chimerical enzyme is a complex made of EC300 full-length polypeptide associated with independently produced CWB170 subunits. Although the exact stoichiometry of the EC300 multimer was not determined, the available evidences strongly suggest that it should have the same configuration as the Lys170 multimer (Proença et al. 2014), that is, being made of one molecule of the full-length EC300 complexed with three of CWB170. This will mean that EC300 assembles one M23 endopeptidase CD with four copies of the CWB170, which provides to the lytic enzyme high affinity to the cell wall (Proença et al. 2014). In addition, due to its multimeric nature, EC300 is a protein of almost 70 kDa; this will certainly be an advantage for the study of its effectiveness in animal infection models since, as observed for the dimeric form of the pneumococcal endolysin Cpl-1 (Resch et al. 2011), it should reduce renal clearance (proteins smaller than 60–65 kDa tend to be rapidly eliminated by glomerular filtration in humans; Maack et al. 1979).
The rather promiscuous modular structure of endolysins themselves has also been intensively explored to engineer chimeras with increased solubility and with changed and/or extended lytic spectra when compared to parental endolysins (Croux et al. 1993; Daniel et al. 2010; Fernandes et al. 2012; Mao et al. 2013; Pastagia et al. 2011; Schmelcher et al. 2011; Yang et al. 2014). The results obtained with EC300 suggest that fusing CDs from VALs to CWB domains of cognate endolysins may constitute an additional strategy to generate enzymes with improved features. The next step will be to evaluate if the enhanced lytic properties of EC300 observed in vitro will translate into a better therapeutic efficacy in a murine model of enterococcal bacteraemia.
References
Abaev I, Foster-Frey J, Korobova O, Shishkova N, Kiseleva N, Kopylov P, Pryamchuk S, Schmelcher M, Becker SC, Donovan DM (2013) Staphylococcal phage 2638A endolysin is lytic for Staphylococcus aureus and harbors an inter-lytic-domain secondary translational start site. Appl Microbiol Biotechnol 97:3449–3456. doi:10.1007/s00253-012-4252-4
Abedon ST (2011) Lysis from without. Bacteriophage 1:46–49. doi:10.4161/bact.1.1.13980
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. doi:10.1093/nar/25.17.3389
Anantharaman V, Aravind L (2003) Evolutionary history, structural features and biochemical diversity of the NLPC/P60 superfamily of enzymes. Genome Biol 4:R11. doi:10.1186/gb-2003-4-2-r11
Arias CA, Murray BE (2008) Emergence and management of drug-resistant enterococcal infections. Expert Rev Anti Infect Ther 6:637–655. doi:10.1586/14787210.6.5.637
Besemer J, Borodovsky M (2005) GeneMark: web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res 33(Web Server issue):W451–W454. doi:10.1093/nar/gki487
Boulanger P, Jacquot P, Plançon L, Chami M, Engel A, Parquet C, Herbeuval C, Letellier L (2008) Phage T5 straight tail fiber is a multifunctional protein acting as a tape measure and carrying fusogenic and muralytic activities. J Biol Chem 283:13556–13564. doi:10.1074/jbc.M800052200
Cabré F, Canela EI, Canela MA (1989) Accuracy and precision in the determination of Stokes radii and molecular masses of proteins by gel filtration chromatography. J Chromatogr 472:347–356. doi:10.1016/S0021-9673(00)94133-5
Catalão MJ, Gil F, Moniz-Pereira J, Pimentel M (2010) The mycobacteriophage Ms6 encodes a chaperone-like protein involved in the endolysin delivery to the peptidoglycan. Mol Microbiol 77:672–686. doi:10.1111/j.1365-2958.2010.07239.x
Catalão MJ, Gil F, Moniz-Pereira J, São-José C, Pimentel M (2013) Diversity in bacterial lysis systems: bacteriophages show the way. FEMS Microbiol Rev 37:554–571. doi:10.1111/1574-6976.12006
Chung CT, Niemela SL, Miller RH (1989) One-step preparation of competent Escherichia coli: transformation and storage of bacterial cells in the same solution. Proc Natl Acad Sci U S A 86:2172–2175. doi:10.1073/pnas.86.7.2172
Cotter PD, Hill C, Ross RP (2005) Bacteriocins: developing innate immunity for food. Nat Rev Microbiol 3:777–788. doi:10.1038/nrmicro1273
Croux C, Ronda C, Lopez R, Garcia JL (1993) Interchange of functional domains switches enzyme specificity: construction of a chimeric pneumococcal-clostridial cell wall lytic enzyme. Mol Microbiol 9:1019–1025. doi:10.1111/j.1365-2958.1993.tb01231.x
Daniel A, Euler C, Collin M, Chahales P, Gorelick KJ, Fischetti VA (2010) Synergism between a novel chimeric lysin and oxacillin protects against infection by methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 54:1603–1612. doi:10.1128/AAC. 01625-09
Donovan DM, Dong S, Garrett W, Rousseau GM, Moineau S, Pritchard DG (2006) Peptidoglycan hydrolase fusions maintain their parental specificities. Appl Environ Microbiol 72:2988–2996. doi:10.1128/aem. 72.4.2988-2996.2006
Ebina T, Toh H, Kuroda Y (2009) Loop-length dependent SVM prediction of domain linkers for high-throughput structural proteomics. Biopolymers 92:1–8. doi:10.1002/bip.21105
Emori TG, Gaynes RP (1993) An overview of nosocomial infections, including the role of the microbiology laboratory. Clin Microbiol Rev 6:428–442. doi:10.1128/CMR.6.4.428
Fenton M, Ross P, McAuliffe O, O’Mahony J, Coffey A (2010) Recombinant bacteriophages lysins as antibacterials. Bioengineered Bugs 1:9–16. doi:10.4161/bbug.1.1.9818
Fernandes S, Proença D, Cantante C, Silva FA, Leandro C, Lourenço S, Milheiriço C, de Lencastre H, Cavaco-Silva P, Pimentel M, São-José C (2012) Novel chimerical endolysins with broad antimicrobial activity against Methicillin-Resistant Staphylococcus aureus. Microb Drug Resist 8:333–343. doi:10.1089/mdr.2012.0025
Fisher K, Phillips C (2009) The ecology, epidemiology and virulence of Enterococcus. Microbiology 155:1749–1757. doi:10.1099/mic. 0.026385-0
French GL (2010) The continuing crisis in antibiotic resistance. Int J Antimicrob Agents 3:S3–S7. doi:10.1016/S0924-8579(10)70003-0
Frias MJ, Melo-Cristino J, Ramirez M (2009) The autolysin LytA contributes to efficient bacteriophage progeny release in Streptococcus pneumoniae. J Bacteriol 191:5428–5440. doi:10.1128/JB.00477-09
García P, Martínez B, Rodríguez L, Rodríguez A (2010) Synergy between the phage endolysin LysH5 and nisin to kill Staphylococcus aureus in pasteurized milk. Int J Food Microbiol 141:151–155. doi:10.1016/j.ijfoodmicro.2010.04.029
Gilmore MS, Lebreton F, van Schaik W (2013) Genomic transition of enterococci from gut commensals to leading causes of multidrug-resistant hospital infection in the antibiotic era. Curr Opin Microbiol 16:10–16. doi:10.1016/j.mib.2013.01.006
Gu J, Xu W, Lei L, Huang J, Feng X, Sun C, Du C, Zuo J, Li Y, Du T, Li L, Han W (2011) LysGH15, a novel bacteriophage lysin, protects a murine bacteremia model efficiently against lethal methicillin-resistant Staphylococcus aureus infection. J Clin Microbiol 49:111–117. doi:10.1128/JCM. 01144-10
Hammerum AM (2012) Enterococci of animal origin and their significance for public health. Clin Microbiol Infect 18:619–625. doi:10.1111/j.1469-0691.2012.03829.x
Hasper HE, de Kruijff B, Breukink E (2004) Assembly and stability of nisin-lipid II pores. Biochemistry 43:11567–1175. doi:10.1021/bi049476b
Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR (1989) Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77:51–59. doi:10.1016/0378-1119(89)90358-2
Huycke MM, Spiegel CA, Gilmore MS (1991) Bacteremia caused by hemolytic, high-level gentamicin-resistant Enterococcus faecalis. Antimicrob Agents Chemother 35:1626–1634. doi:10.1128/AAC.35.8.1626
Idelevich EA, von Eiff C, Friedrich AW, Iannelli D, Xia G, Peters G, Peschel A, Wanninger I, Becker K (2011) In vitro activity against Staphylococcus aureus of a novel antimicrobial agent, PRF-119, a recombinant chimeric bacteriophage endolysin. Antimicrob Agents Chemother 55:4416–4419. doi:10.1128/AAC. 00217-11
Khan H, Flint SH, Yu PL (2013) Determination of the mode of action of enterolysin A, produced by Enterococcus faecalis B9510. J Appl Microbiol 115:484–494. doi:10.1111/jam.12240
Klein G (2003) Taxonomy, ecology and antibiotic resistance of enterococci from food and the gastro-intestinal tract. Int J Food Microbiol 88:123–131. doi:10.1016/S0168-1605(03)00175-2
Kropinski AM, Mazzocco A, Waddell TE, Lingohr E, Johnson RP (2009) Enumeration of bacteriophages by double agar overlay plaque assay. Methods Mol Biol 501:69–76. doi:10.1007/978-1-60327-164-6_7
Kuch A, Willems RJ, Werner G, Coque TM, Hammerum AM, Sundsfjord A, Klare I, Ruiz-Garbajosa P, Simonsen GS, van Luit-Asbroek M, Hryniewicz W, Sadowy E (2012) Insight into antimicrobial susceptibility and population structure of contemporary human Enterococcus faecalis isolates from Europe. J Antimicrob Chemother 67:551–555. doi:10.1093/jac/dkr544
Kumar JK (2008) Lysostaphin: an antistaphylococcal agent. Appl Microbiol Biotechnol 80:555–561. doi:10.1007/s00253-008-1579-y
Lamsa A, Liu WT, Dorrestein PC, Pogliano K (2012) The Bacillus subtilis cannibalism toxin SDP collapses the proton motive force and induces autolysis. Mol Microbiol 84:486–500. doi:10.1111/j.1365-2958.2012.08038.x
Loeffler JM, Djurkovic S, Fischetti VA (2003) Phage lytic enzyme Cpl-1 as a novel antimicrobial for pneumococcal bacteremia. Infect Immun 71:6199–61204. doi:10.1128/IAI. 71.11.6199-6204.2003
Maack T, Johnson V, Kau ST, Figueiredo J, Sigulem D (1979) Renal filtration, transport, and metabolism of low-molecular-weight proteins: a review. Kidney Int 16:251–270. doi:10.1038/ki.1979.128
Mao J, Schmelcher M, Harty WJ, Foster-Frey J, Donovan DM (2013) Chimeric Ply187 endolysin kills Staphylococcus aureus more effectively than the parental enzyme. FEMS Microbiol Lett 342:30–36. doi:10.1111/1574-6968.12104
Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Lu F, Marchler GH, Mullokandov M, Omelchenko MV, Robertson CL, Song JS, Thanki N, Yamashita RA, Zhang D, Zhang N, Zheng C, Bryant SH (2011) CDD: a conserved domain database for the functional annotation of proteins. Nucleic Acids Res 39:D225–D229. doi:10.1093/nar/gkq1189
Mato R, Almeida F, Pires R, Rodrigues P, Ferreira T, Santos-Sanches I (2009) Assessment of high-level gentamicin and glycopeptide-resistant Enterococcus faecalis and E. faecium clonal structure in a Portuguese hospital over a 3-year period. Eur J Clin Microbiol Infect Dis 28:855–859. doi:10.1007/s10096-009-0704-x
Murray BE (1990) The life and times of the Enterococcus. Clin Microbiol Rev 3:46–65. doi:10.1128/CMR.3.1.46
Nascimento JG, Guerreiro-Pereira MC, Costa SF, São-José C, Santos MA (2008) Nisin-triggered activity of Lys44, the secreted endolysin from Oenococcus oeni phage fOg44. J Bacteriol 190:457–461. doi:10.1128/JB.01195-07
Nelson DC, Schmelcher M, Rodriguez-Rubio L, Klumpp J, Pritchard DG, Dong S, Donovan DM (2012) Endolysins as antimicrobials. Adv Virus Res 83:299–365. doi:10.1016/B978-0-12-394438-2.00007-4
Noguchi H, Taniguchi T, Itoh T (2008) MetaGeneAnnotator: detecting species-specific patterns of ribosomal binding site for precise gene prediction in anonymous prokaryotic and phage genomes. DNA Res 15:387–396. doi:10.1093/dnares/dsn027
Oechslin F, Daraspe J, Giddey M, Moreillon P, Resch G (2013) In vitro characterization of PlySK1249, a novel phage lysin, and assessment of its antibacterial activity in a mouse model of Streptococcus agalactiae bacteremia. Antimicrob Agents Chemother 57:6276–6283. doi:10.1128/AAC. 01701-13
Pastagia M, Euler C, Chahales P, Fuentes-Duculan J, Krueger JG, Fischetti VA (2011) A novel chimeric lysin shows superiority to mupirocin for skin decolonization of methicillin-resistant and -sensitive Staphylococcus aureus strains. Antimicrob Agents Chemother 55:738–744. doi:10.1128/AAC. 00890-10
Paul VD, Rajagopalan SS, Sundarrajan S, George SE, Asrani JY, Pillai R, Chikkamadaiah R, Durgaiah M, Sriram B, Padmanabhan S (2011) A novel bacteriophage tail-associated muralytic enzyme (TAME) from Phage K and its development into a potent antistaphylococcal protein. BMC Microbiol 11:226. doi:10.1186/1471-2180-11-226
Paulsen IT, Banerjei L, Myers GS, Nelson KE, Seshadri R, Read TD, Fouts DE, Eisen JA, Gill SR, Heidelberg JF, Tettelin H, Dodson RJ, Umayam L, Brinkac L, Beanan M, Daugherty S, DeBoy RT, Durkin S, Kolonay J, Madupu R, Nelson W, Vamathevan J, Tran B, Upton J, Hansen T, Shetty J, Khouri H, Utterback T, Radune D, Ketchum KA, Dougherty BA, Fraser CM (2003) Role of mobile DNA in the evolution of vancomycin-resistant Enterococcus faecalis. Science 299:2071–2074. doi:10.1126/science.1080613
Piuri M, Hatfull GF (2006) A peptidoglycan hydrolase motif within the mycobacteriophage TM4 tape measure protein promotes efficient infection of stationary phase cells. Mol Microbiol 62:1569–1585. doi:10.1111/j.1365-2958.2006.05473.x
Poh CH, Oh HM, Tan AL (2006) Epidemiology and clinical outcome of enterococcal bacterium in an acute care hospital. J Infect 52:383–386. doi:10.1016/j.jinf.2005.07.011
Proença D, Fernandes S, Leandro C, Silva FA, Santos S, Lopes F, Mato R, Cavaco-Silva P, Pimentel M, São-José C (2012) Phage endolysins with broad antimicrobial activity against Enterococcus faecalis clinical strains. Microb Drug Resist 18:322–332. doi:10.1089/mdr.2012.0024
Proença D, Velours C, Leandro C, Garcia M, Pimentel M, São-José C (2014) A two-component, multimeric endolysin encoded by a single gene. Mol Microbiol 95:739–753.doi:10.1111/mmi.12857
Resch G, Moreillon P, Fischetti VA (2011) A stable phage lysin (Cpl-1) dimer with increased antipneumococcal activity and decreased plasma clearance. Int J Antimicrob Agents 38:516–521. doi:10.1016/j.ijantimicag.2011.08.009
Rodríguez-Rubio L, Gutiérrez D, Martínez B, Rodríguez A, Götz F, García P (2012a) The tape measure protein of the Staphylococcus aureus bacteriophage vB_SauS-phiIPLA35 has an active muramidase domain. Appl Environ Microbiol 78:6369–6371. doi:10.1128/AEM. 01236-12
Rodríguez-Rubio L, Martínez B, Rodríguez A, Donovan DM, García P (2012b) Enhanced staphylolytic activity of the Staphylococcus aureus bacteriophage vB_SauS-phiIPLA88 HydH5 virion-associated peptidoglycan hydrolase: fusions, deletions, and synergy with LysH5. Appl Environ Microbiol 78:2241–2248. doi:10.1128/AEM. 07621-11
Rodríguez-Rubio L, Martínez B, Donovan DM, Rodríguez A, García P (2013) Bacteriophage virion-associated peptidoglycan hydrolases: potential new enzybiotics. Crit Rev Microbiol 39:427–434. doi:10.3109/1040841X.2012.723675
Sahm DF, Kissinger J, Gilmore MS, Murray PR, Mulder R, Solliday J, Clarke B (1989) In vitro susceptibility studies of vancomycin-resistant Enterococcus faecalis. Antimicrob Agents Chemother 33:1588–1591. doi:10.1128/AAC.33.9.1588
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
São-José C, Parreira R, Vieira G, Santos MA (2000) The N-terminal region of the Oenococcus oeni bacteriophage fOg44 lysin behaves as a bona fide signal peptide in Escherichia coli and as a cis-inhibitory element, preventing lytic activity on oenococcal cells. J Bacteriol 182:5823–5831. doi:10.1128/JB.182.20.5823-5831.2000
Sava IG, Heikens E, Huebner J (2010) Pathogenesis and immunity in enterococcal infections. Clin Microbiol Infect 16:533–540. doi:10.1111/j.1469-0691.2010.03213.x
Schaberg DR, Culver DH, Gaynes RP (1991) Major trends in the microbial etiology of nosocomial infection. Am J Med 91:72s–75s. doi:10.1016/0002-9343(91)90346-Y
Schmelcher M, Tchang VS, Loessner MJ (2011) Domain shuffling and module engineering of Listeria phage endolysins for enhanced lytic activity and binding affinity. Microb Biotechnol 4:651–662. doi:10.1111/j.1751-7915.2011.00263.x
Schmelcher M, Donovan DM, Loessner MJ (2012a) Bacteriophage endolysins as novel antimicrobials. Future Microbiol 7:1147–1171. doi:10.2217/fmb.12.97
Schmelcher M, Powell AM, Becker SC, Camp MJ, Donovan DM (2012b) Chimeric phage lysins act synergistically with lysostaphin to kill mastitis-causing Staphylococcus aureus in murine mammary glands. Appl Environ Microbiol 78:2297–2305. doi:10.1128/AEM. 07050-11
Severina E, Severin A, Tomasz A (1998) Antibacterial efficacy of nisin against multidrug-resistant Gram-positive pathogens. J Antimicrob Chemother 41:341–347. doi:10.1093/jac/41.3.341
Shankar N, Baghdayan AS, Gilmore MS (2002) Modulation of virulence within a pathogenicity island in vancomycin-resistant Enterococcus faecalis. Nature 417:746–750. doi:10.1038/nature00802
Stockdale SR, Mahony J, Courtin P, Chapot-Chartier MP, van Pijkeren JP, Britton RA, Neve H, Heller KJ, Aideh B, Vogensen FK, van Sinderen D (2013) The lactococcal phages Tuc 2009 and TP901–1 incorporate two alternate forms of their tail fiber into their virions for infection specialization. J Biol Chem 288:5581–5590. doi:10.1074/jbc.M112.444901
Sudiarta IP, Fukushima T, Sekiguchi J (2010) Bacillus subtilis CwlP of the SP-{beta} prophage has two novel peptidoglycan hydrolase domains, muramidase and cross-linkage digesting DD-endopeptidase. J Biol Chem 285:41232–41243. doi:10.1074/jbc.M110.156273
Takác M, Bläsi U (2005) Phage P68 virion-associated protein 17 displays activity against clinical isolates of Staphylococcus aureus. Antimicrob Agents Chemother 49:2934–2940. doi:10.1128/AAC. 49.7.2934-2940.2005
Theuretzbacher U (2012) Accelerating resistance, inadequate antibacterial drug pipelines and international responses. Int J Antimicrob Agents 39:295–299. doi:10.1016/j.ijantimicag.2011.12.006
Thumm G, Götz F (1997) Studies on prolysostaphin processing and characterization of the lysostaphin immunity factor (Lif) of Staphylococcus simulans biovar staphylolyticus. Mol Microbiol 23:1251–1265. doi:10.1046/j.1365-2958.1997.2911657.x
Top J, Willems R, Bonten M (2008) Emergence of CC17 Enterococcus faecium: from commensal to hospital-adapted pathogen. FEMS Immunol Med Microbiol 52:297–308. doi:10.1111/j.1574-695X.2008.00383.x
Uchiyama J, Rashel M, Takemura I, Wakiguchi H, Matsuzaki S (2008) In silico and in vivo evaluation of bacteriophage ϕEF24C, a candidate for treatment of Enterococcus faecalis infections. Appl Environ Microbiol 74:4149–4163. doi:10.1128/AEM. 02371-07
Vinga I, Baptista C, Auzat I, Petipas I, Lurz R, Tavares P, Santos MA, São-José C (2012) Role of bacteriophage SPP1 tail spike protein gp21 on host cell receptor binding and trigger of phage DNA ejection. Mol Microbiol 83:289–303. doi:10.1111/j.1365-2958.2011.07931.x
Wenzel M, Kohl B, Münch D, Raatschen N, Albada HB, Hamoen L, Metzler-Nolte N, Sahl HG, Bandow JE (2012) Proteomic response of Bacillus subtilis to lantibiotics reflects differences in interaction with the cytoplasmic membrane. Antimicrob Agents Chemother 56:5749–5757. doi:10.1128/AAC. 01380-12
Werner G, Coque TM, Franz CM, Grohmann E, Hegstad K, Jensen L, van Schaik W, Weaver K (2013) Antibiotic resistant enterococci-tales of a drug resistance gene trafficker. Int J Med Microbiol 303:360–379. doi:10.1016/j.ijmm.2013.03.001
Yang H, Zhang Y, Yu J, Huang Y, Zhang XE, Wei H (2014) Novel chimeric lysin with high-level antimicrobial activity against methicillin-resistant Staphylococcus aureus in vitro and in vivo. Antimicrob Agents Chemother 58:536–542. doi:10.1128/AAC. 01793-13
Acknowledgments
D. Proença’s work has been supported through the Ph.D fellowship SFRH/BDE/51076/2010 from Fundação para a Ciência e a Tecnologia (FCT, MCTES, Portugal). Typed enterococcal strains were kindly provided by Rosário Mato, except E. faecalis stains V583 and MMH594.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 385 kb)
Rights and permissions
About this article

Cite this article
Proença, D., Leandro, C., Garcia, M. et al. EC300: a phage-based, bacteriolysin-like protein with enhanced antibacterial activity against Enterococcus faecalis . Appl Microbiol Biotechnol 99, 5137–5149 (2015). https://doi.org/10.1007/s00253-015-6483-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-015-6483-7