
Abstract
Corynebacterium glutamicum ORF NCgl0328, designated noxA, encodes an NADH oxidase enzyme. The noxA gene, which was preferentially expressed in the log growth phase, was found to be under the control of the whcA, whcB, and whcE genes, which play regulatory roles in cells under oxidative stress. While noxA transcription was minimal in whcE-deleted mutant cells (ΔwhcE) during growth, its transcription was maximal even in the stationary phase in ΔwhcA cells. The transcription levels of noxA in ΔwhcB and whcB-overexpressing cells were comparable to the levels only in the log growth phase in ΔwhcA and whcA-overexpressing cells, respectively. Direct binding of purified WhcA to the promoter region of noxA was observed in vitro. The DNA-protein interaction was only possible in the presence of the reducing agent dithiothreitol. A noxA-deleted mutant strain and a strain overexpressing the noxA gene (P180-noxA) were established, and these strains were found to exhibit defective cell growth. The ΔnoxA and P180-noxA strains were sensitive to the redox-cycling oxidant menadione, suggesting a role of noxA in redox balancing. Accordingly, the purified NoxA enzyme exhibited NADH-oxidizing activity. Taken together, these data show that noxA plays a role in oxidative stress responses and also that the gene is under direct control of the WhcA protein, which was shown to be a regulatory DNA-binding protein. Furthermore, the involvement and roles of the whcA, whcB, and whcE genes in regulating the expression of noxA were demonstrated.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Corynebacterium glutamicum is a gram-positive organism belonging to the class Actinobacteria, which includes the genera Mycobacterium and Streptomyces (Gao and Gupta 2012). C. glutamicum is widely used for the industrial production of amino acids by fermentation processes (Leuchtenberger et al. 2005). During the course of fermentation, microorganisms encounter a variety of cellular stresses including oxidative stress, which is harmful to the fermenting cells. In the past, the oxidative stress response pathways of C. glutamicum were analyzed by our group, and it was found that several whiB-like genes play important regulatory roles in the oxidative stress responses of C. glutamicum (Choi et al. 2009; Kim et al. 2005; Lee et al. 2012, 2013).
The whiB gene was originally identified and characterized in Streptomyces coelicolor as a developmental regulatory gene required for the sporulation of aerial hyphae (Davis and Chater 1992). Actinobacteria species are known to possess whiB-like genes that function in diverse cellular processes, including cell division, differentiation, pathogenesis, starvation survival, and stress response (Zheng et al. 2012). Typically, WhiB-like proteins possess a redox-sensitive Fe-S cluster coordinated to four conserved cysteine residues (Alam et al. 2007; Crack et al. 2009; Jakimowicz et al. 2005; Rybniker et al. 2010; Singh et al. 2007; Smith et al. 2010). This cluster plays an important role in controlling protein activity. Upon exposure of the protein to oxygen, for example, the cluster can be lost and the coordinating cysteine thiols are oxidized, resulting in a loss of protein activity (Crack et al. 2009; Singh et al. 2009). Some WhiB-like proteins possess a helix-turn-helix DNA-binding motif, indicating a role of these proteins as transcription factors (Smith et al. 2010).
C. glutamicum possesses four whiB-like genes: whcE (whiB1), whcD (whiB2), whcB (whiB3), and whcA (whiB4). Of these homologues, three (whcE, whcA, and whcB) have been studied and characterized (Choi et al. 2009; Kim et al. 2005; Lee et al. 2012). The whcE gene plays a positive role in the survival of cells exposed to oxidative and heat stresses (Kim et al. 2005), whereas the whcA gene plays a negative role in the expression of genes associated with the oxidative stress response (Choi et al. 2009). The WhcA protein interacts with SpiA, an oxidoreductase-like protein (Park et al. 2011, 2012), and the interaction is labile to oxidants. The whcB gene plays a regulatory role in the stationary phase of bacterial growth, particularly in electron transfer reactions (Lee et al. 2012). Furthermore, an analysis of the whc gene expression network by Lee et al. (2013) revealed that, although the whc genes are paralogues, they play distinct regulatory roles during cellular responses to oxidative stress. After all, the oxidative stress response of C. glutamicum is regulated in a complex manner involving a hierarchy of several whc genes (Lee et al. 2013). Since whcB lies at the top of the hierarchy, it is considered the primary regulator of whc gene transcription.
Despite recent progress, many questions regarding the mechanisms of oxidative stress responses in C. glutamicum remain unanswered. In our previous study, the open reading frame (ORF) NCgl0328 (noxA) was detected in analyses involving proteomes of whc mutants, but the function and regulation of the noxA gene in the oxidative stress response pathway have not been clarified yet (Choi et al. 2009; Lee et al. 2012). In this study, the involvement and roles of the whcA, whcB, and whcE genes in regulating the expression of noxA were demonstrated. Direct binding of WhcA to the promoter region of the noxA gene was demonstrated for the first time. Furthermore, we report that the purified NoxA protein contains NADH-oxidizing activity. Finally, we propose a role for the noxA gene in the oxidative stress response and cell physiology of C. glutamicum.
Materials and methods
Bacterial strains and growth conditions
C. glutamicum AS019E12 (Follettie and Sinskey 1986; Yoshihama et al. 1985), a rifampicin-resistant and restriction-deficient derivative of the ATCC13059 strain, was utilized in generating the HL1393 and HL1458 strains. C. glutamicum HL1393 carries a ΔNCgl0328 (ΔnoxA) mutation, whereas C. glutamicum HL1458 carries the noxA-overexpressing plasmid pSL531 (i.e., P180-noxA). C. glutamicum HL1171 (Choi et al. 2009), HL1312 (Lee et al. 2012), and HL810 (Kim et al. 2005) carry a ΔwhcA mutation, a ΔwhcB mutation, and a ΔwhcE mutation, respectively, and C. glutamicum HL1176, HL1313, and HL1108 carry the pSL432 (Choi et al. 2009), pSL469 (Lee et al. 2012), and pSL395 (Kim et al. 2005) plasmids, respectively. Plasmids pSL432 (i.e., P180-whcA), pSL469 (i.e., P180-whcB), and pSL395 (i.e., P180-whcE) allow for the overexpression of whcA, whcB, and whcE genes, respectively. Cultivation of Escherichia coli was performed at 37 °C in Luria-Bertani complex medium (Sambrook et al. 2001). C. glutamicum cells were cultured at 30 °C in MB (Follettie et al. 1993) or MCGC (von der Osten et al. 1989). Glucose was added as a carbon source in the minimal medium at a concentration of 1 % (w/v). Antibiotics were added at the following concentrations (μg ml−1): 50 ampicillin, 20 chloramphenicol, and 25 kanamycin.
Construction of plasmids and strains
Standard molecular cloning, transformation, and electrophoresis procedures were employed. Plasmids were introduced into C. glutamicum cells by electroporation (Follettie et al. 1993). All the restriction enzymes and DNA-modifying enzymes were purchased and used according to the manufacturer’s instructions (Takara Bio). PCR was carried out as previously described (Kim et al. 2005). The nucleotide sequence of noxA (NCgl0328, GenBank GeneID 1021176) was obtained from the published C. glutamicum genome sequences (GenBank accession no. NC_003450.3; Ikeda and Nakagawa 2003; Kalinowski et al. 2003). The C. glutamicum ΔnoxA mutant strain was generated according to the method by Schäfer et al. (1994), as follows: the primary PCR products amplified with the F1 5′-CACAGCACCCCACTTCACATAACC-3′, R1 5′-CCGCCGCGAGGTTTTCCACGCGGGCAACC-3′, and F2 5′-CGTGGAAAACCTCGCGGCGGCTTTTCTA-3′, R2 5′-TTCGATGAGGGTTACTTTGCTGTC-3′ primer pairs were used as templates for secondary PCR. The amplified fragment was cloned into the pGEM-T-easy vector (Promega), after which, the fragment resulting from the digestion with EcoRI was isolated and inserted into the EcoRI-digested pK19mobsacB plasmid (Schäfer et al. 1994). The resulting plasmid, pSL511, was transformed into E. coli ET12567 (MacNeil et al. 1992), and the plasmids from this strain were electro-transformed into C. glutamicum. Subsequent steps were conducted as described (Hwang et al. 2002). The chromosomal deletion of the noxA gene (a 114-bp internal deletion to begin 254 bp downstream of the translation start site and end 214 bp upstream of stop codon) in C. glutamicum was validated using PCR, and the mutant bacterial strain was designated HL1393. Plasmid pSL531 was constructed by amplifying the noxA gene using primers 5′-AAAACTGCAGCCAACACATAAAAAGG-3′ and 5′-AAAACTGCAGCCGGTCACAAGCAAAG-3′, followed by ligation of the amplified DNA at the PstI site in the pSL360 plasmid (Park et al. 2004), which allows for the overexpression of the cloned genes. The pSL521 plasmid encoding the maltose-binding protein (MBP)-WhcA-His6 fusion protein was constructed by amplifying the whcA gene using the primers 5′-ATGACGTCTGTGATTCCAGAGC-3′ and 5′-CGCGGATCCTTAGTGGTGATGGTGATGATGAACCCCGGCGATC-3′, followed by the ligation of the amplified DNA at the BamHI site of the pMAL-c2 vector (New England Biolabs). The pSL544 plasmid expressing the His6-NoxA protein was constructed by introducing the BamHI digested fragment, which was amplified from the noxA gene using primers 5′-AATGGGTCGCGGATCCATGTCACTTTCAGTCGT-3′ and 5′-GCTCGAATTCGGATCCGAGTTAGTAGCTGTTGTC-3′, into the pET28a vector (Novagen).
RNA analysis
5′ Rapid Amplification of cDNA Ends (RACE) was carried out using a 5′/3′ RACE Kit, 2nd Generation (Roche Diagnostics). Total RNA was isolated from the cells at the exponential phase (OD600 of 5–15) or stationary phase (OD600 of 20) using a NucleoSpin RNA II Kit (Macherey-Nagel). First-strand complementary DNA (cDNA) synthesis was conducted as described previously (Park et al. 2008). cDNA synthesis was carried out using a DyNAmo cDNA Synthesis Kit (Finnzymes), and real-time quantitative PCR (RT-qPCR) was performed as described (Hong et al. 2014), using THUNDERBIRD SYBR qPCR Mix (TOYOBO) and a CFX96™ Real-Time PCR Detection System (Bio-Rad). All the reactions were performed in triplicate. The PCR conditions consisted of an activation step of 15 min at 95 °C, followed by 40 cycles at 95 °C for 20 s, at 60 °C for 20 s, and at 72 °C for 40 s. Data were collected at the 72 °C step of each cycle. Normalized expression and standard error values were calculated using the CFX Manager software ver. 1.5 (Bio-Rad), which employs the ΔΔCt method. Verification of RT-qPCR products was performed by melting curve and peak analyses. Gene expression levels were normalized to the levels of the 16S rRNA gene, which was used as an endogenous control. The primers used for the detection of whcA, whcB, whcE, trxB, and noxA were as follows: whcA, 5′-ATCGCCCTTGTTATTGCTACCGGA-3′ and 5′-AGTAGCTGTTGTCGATGCGCCTAT-3′; whcB, 5′-ATTGCCTCACCAGCTTCCCG-3′ and 5′-TCGCCGTCCGGGTGATAGAA-3′; whcE, 5′-ACGAAGCAATCTGCCGTGAA-3′ and 5′-AGCGGTTGCAGACCATCTTT-3′; trxB, 5′-ACCCAACTTGGTGGTCAGATGGAA-3′ and 5′-TTGAGCAGCGGAACCATAGACCAT-3′; noxA, 5′-ATCGCCCTTGTTATTGCTACCGGA-3′ and 5′-AGTAGCTGTTGTCGATGCGCCTAT-3′.
Enzyme assays and protein purification
Cells grown under aerobic conditions to mid-exponential phase were harvested by centrifugation, after which, crude cellular extracts were prepared as previously described (Kim et al. 2004). Protein concentrations were determined by the Bradford method, using bovine serum albumin solutions as standards (Bradford 1976). NADH oxidase activity in the crude cellular extracts was determined by a photometric assay at 340 nm (molar extinction coefficient of 6.22 \( \times \) 103 M−1 cm−1) using 50 mM potassium phosphate buffer (pH 7.0) containing 10 μM flavin mononucleotide (FMN), 100 μM NADH (Sigma–Aldrich), and a limiting amount of crude extract. β-NADH and FMN were used as the electron donor and the electron acceptor, respectively. The reaction mixture lacking NADH was used as the blank.
The MBP-WhcA-His6 fusion protein was purified as follows. E. coli BL21(DE3) (Merck Biosciences) harboring pSL521 was cultivated in LB. Proteins were induced by treating cells with 0.3 mM IPTG at an OD600 of 0.5. The cells were cultivated for additional 1 h to fully induce the proteins. The cells were harvested by centrifugation, resuspended in a binding buffer (MBPTrap HP, GE Healthcare), and lysed by sonication on ice (15 times for 3 s with a 6-s rest interval at output level 30 %, VCX-400; Sonics & Materials Inc.). The lysate was centrifuged at 10,000×g for 60 min at 4 °C to remove the cell debris. The resulting crude extract was retained for purification. The MBP-WhcA-His6 fusion protein was purified on an amylose column (MBPTrap HP, GE Healthcare) and subsequently on a Ni2+-NTA column (HiTrap His-FF, GE Healthcare) as described in the Recombinant Protein Purification Handbook (GE Healthcare). The His6-NoxA fusion protein was purified on a Ni2+-NTA column (HiTrap His-FF, GE Healthcare) after it was overexpressed as described above. Subsequently, the proteins were analyzed by 17 % sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE).
DNA-protein affinity purification
Potential DNA-protein interactions were analyzed using a Dynabeads-based magnetic separation protocol (Life Technologies). First, the noxA promoter region spanning nucleotides −211 to +70 relative to the transcriptional start site was amplified by PCR using primers 5′-AAGCACCCAACTCGCCAAA-3′ and 5′-GTGGCGCGGCGGTTGGTAAT-3′. The ORF of the noxA gene amplified with primers 5′-GGCCTCCCATCAGAAACAA-3′ and 5′-ATCCTCACGCCCGCCGAGACC-3′ was used as the control DNA. When needed, the primers were appropriately labeled with biotin, according to the manufacturer’s instructions (Life Technologies). Next, approximately 30 μg of each biotin-labeled PCR product was mixed with 5 μl (6.7 × 107 beads) streptavidin-coated beads (Dynabeads M-280 Streptavidin) in mobilization buffer (10 mM Tris, 1 mM ethylenediaminetetraacetic acid (EDTA), 2 M NaCl, pH 7.5), and the mixture was incubated at room temperature for 3 h to allow for coupling between biotin-labeled DNA and streptavidin-coated beads. Uncoupled DNA was removed by washing the mixture with binding buffer (100 mM HEPES, pH 7.6, 5 mM EDTA, 50 mM [NH4]2SO4, 5 mM dithiothreitol (DTT)], 1 % Tween 20 [w/v], 150 mM KCl). Subsequently, the coupled beads were resuspended in 600 μl binding buffer, and the binding reaction was carried out using 200 μg purified WhcA protein at 15 °C for 3 h with gentle shaking. Salmon sperm DNA was used as a competitor DNA for the reaction. Unbound and nonspecifically bound proteins were removed by magnetic separation using a magnet particle concentrator (Life Technologies), followed by several washes with the binding buffer. Finally, bound proteins were eluted with a binding buffer containing NaCl (0.6 or 1 M). Eluted proteins were separated by 17 % SDS-PAGE and stained with Coomassie Brilliant Blue G-250.
Electrophoretic mobility shift assay
The 220-bp DNA fragment that includes the promoter region of the noxA gene (from nucleotides −211 to +9 relative to the transcriptional start site) was amplified using the primers 5′-AAGCACCCAACTCGCCAAA-3′ and 5′-TGTTGGAAACAGAGTCATGGTAGG-3′. The binding reaction was performed in a total volume of 20 μl (100 mM HEPES, pH 7.6, 5 mM EDTA, 50 mM [NH4]2 SO4, 5 mM DTT, 1 % Tween 20 [w/v], 150 mM KCl) in the presence of 1 μg poly(dI-dC). Binding experiments were also carried out in the absence of DTT. The DNA-protein mixture was incubated at 30 °C for 30 min and was analyzed via electrophoresis using 8 % native polyacrylamide gels. DNA bands were visualized using GelRed nucleic acid stain (Biotium).
Sensitivity to oxidants
The sensitivity of C. glutamicum cells to diamide (N,N,N′,N′-tetramethylazodicarboxamide), menadione (2-methyl-1,4-naphthoquinone sodium bisulfite), or H2O2 was assessed on MB plates as follows. C. glutamicum cells (100 μl), which had been cultivated to mid-exponential phase, were mixed with 0.8 % (v/v) top agar and poured onto MB plates. Paper disks (6.0 mm, Whattman) with 20 μl each of 1 M diamide, 1 M menadione, or H2O2 (50 % w/v) were placed on the plates, after which the plates were incubated at 30 °C for 24 h, until the formation of a clear zone around the disks.
Results
Expression of the noxA gene during growth
The NCgl0328 gene (noxA) of C. glutamicum is annotated to encode NADH oxidase, which belongs to the nitroreductase family of proteins. Theoretically, NADH oxidase (EC 1.6.99.3) catalyzes the oxidation of NADH by simultaneously reducing molecular O2 to either H2O2 in a two-electron reduction or directly to H2O in a four-electron reduction. These reactions use FMN as a cofactor. Accordingly, a putative FMN-binding site was identified on the N-terminal and central domains of the noxA-encoded protein. Since the NoxA protein was identified by 2D-PAGE (Choi et al. 2009; Lee et al. 2012) and suggested to be involved in the oxidative stress response networks, the molecular, regulatory, and physiological properties of the gene were further analyzed.
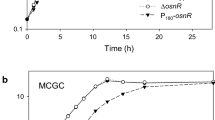
First of all, we determined the transcription start site of the noxA gene using the 5′ RACE PCR technique. As shown in Fig. 1a, the noxA transcription start site was identified as a guanine base located 27 bp upstream of the translation start codon (ATG). This agreed with the results of Pfeifer-Sancar et al. (2013), which used RNA-Seq for determination of transcription start sites. The putative promoter sequences TTGCAC (-35) and TACCAT (-10) were located in the region upstream of the transcription start site. Purine-rich sequences presumed to be ribosome-binding sites were also located between the ATG start codon and the +1 transcription start site. Next, the transcription profile of noxA was assessed and compared with the profiles of the whcA, whcE, and whcB genes, which play regulatory roles in oxidative stress response of C. glutamicum. As shown in Fig. 1b, the transcription of noxA was found to peak in the log phase and to decrease as the cells entered the stationary phase, at which point, the transcription level was only 30 % of the level observed in log phase. Transcription of the whcA, whcE, and whcB genes, on the other hand, was found to increase by up to two-–threefold, as the cells entered stationary phase. As suggested by Choi et al. (2009), these data may indicate that the expression of noxA is repressed by the whcA gene during the stationary phase of C. glutamicum.

Promoter region of the noxA gene and transcription of whcA, whcB, whcE, and noxA during growth. a The transcriptional start site (+1) was experimentally determined by 5′ RACE. The translational initiation codon (ATG) for the noxA gene is shown in bold. The predicted promoter sequences of −10 and −35, shown in bold, were predicted from known consensus sequences. b Cells were grown in MCGC media, and mRNA levels were measured by RT-qPCR. Gene expression levels were normalized to the levels of the 16S rRNA gene, which was used as an endogenous control. Data represent three independent experiments. Filled circles indicate the growth of C. glutamicum. Bars indicate relative mRNA levels of noxA, whcA, whcE, and whcB. OD optical density
The roles of whc genes during noxA expression
To investigate the potential involvement of the whc genes in noxA gene expression, the transcription levels of noxA were assessed in detail under various genetic conditions. The levels of noxA transcription were measured in whc-deleted and whc-overexpressing cells. Overexpression of whc was achieved by employing the P180 promoter, which induces overexpression of the cloned gene regardless of growth phase (Park et al. 2004). Typically, a 20-fold overexpression of whcA, whcE, and whcB was achieved as verified by RT-qPCR. First, noxA transcription was monitored in whcA-deleted (ΔwhcA) and whcA-overexpressing (P180-whcA) cells. As shown in Fig. 2a, noxA transcription was minimal in P180-whcA cells while it was maximal in ΔwhcA cells regardless of the growth phase. These data clearly indicate that noxA is negatively regulated by whcA. Next, the transcription levels of noxA in ΔwhcE and P180-whcE cells were assessed. While the messenger RNA (mRNA) levels of noxA were minimal in ΔwhcE cells (Fig. 2b), the transcription of noxA in P180-whcE cells was nearly maximal, with decreased transcription being observed in the log phase. Overall, the levels of noxA transcription in P180-whcE cells were low compared with the transcription levels in the ΔwhcA cells, indicating an additional regulation of gene expression by a factor other than whcE. Nevertheless, these data clearly show not only that noxA is regulated by the whc genes but also that the whcA and whcE genes play negative and positive roles, respectively, in the expression of noxA.

Transcription of noxA and whcA in C. glutamicum strains. Cells were grown in glucose MCGC media, and mRNA levels were measured by RT-qPCR. Mean values of the mRNA levels and standard deviations are shown based on three independent experiments. Representative growth curves from three independent experiments are shown. Line graphs indicate cell growth and bars indicate relative mRNA levels. a filled circle indicates wild-type, empty circle ΔwhcA, and filled inverted triangle P180-whcA; b filled circle indicates wild-type, empty circle ΔwhcE, filled inverted triangle P180-whcE; c and d filled circle indicates wild-type, empty circle ΔwhcB, filled inverted triangle P180-whcB
Next, the transcription levels of noxA were monitored in ΔwhcB and P180-whcB cells to elucidate the role of whcB, whose regulatory role in oxidative stress responses is not clear. As shown in Fig. 2c, the mRNA expression levels of noxA were repressed in P180-whcB cells, especially in the log phase. Repression of noxA in P180-whcB cells was less evident in the stationary phase. Accordingly, the transcription levels of noxA were monitored in the ΔwhcB cells, and stimulation of noxA transcription was only observed during transition from exponential to stationary growth phase. Since whc transcription is organized into a hierarchy and whcB lies at the top of the hierarchy (Lee et al. 2012), we determined whether the observed repression by whcB is exerted via whcA. As shown in Fig. 2d, the expression of whcA was low in ΔwhcB cells. Furthermore, the expression of whcA in P180-whcB cells was comparable to that in the wild-type strain, except in the stationary phase, during which, the transcription of whcA was decreased. Taken together, these findings suggest that the effect of whcB on noxA expression may be an indirect one exerted via whcA. It was thus investigated whether WhcA binds to the promoter region of the noxA gene in vitro.
Binding of WhcA to the promoter region of noxA
The potential binding of WhcA to the promoter region of noxA was assessed using DNA-protein affinity purification to visualize bound protein and electrophoretic mobility shift assay (EMSA) to visualize the bound DNA. The WhcA protein was overexpressed as an MBP-WhcA-His6 fusion protein and then isolated for in vitro analysis. The addition of histidine residues at the C-terminus of the protein was essential for the stability of the purified protein (unpublished data). Next, WhcA was assessed for potential binding to the promoter region of noxA (from −211 to +70) by DNA-protein affinity purification. WhcA was allowed to interact with DNA, after which, it was eluted from the DNA for analysis by SDS-PAGE. As shown in Fig. 3a, while the 56.5-kDa WhcA protein was found to bind to the 281-bp DNA fragments harboring the noxA promoter region, and could be eluted from the DNA, the protein did not bind to the ORF region of the gene, suggesting the specificity of DNA-binding. The potential binding of WhcB was also assessed; however, specific binding of the WhcB protein to the noxA promoter region was not observed (data not shown). These findings indicate that WhcA functions as a transcriptional repressor for the noxA gene. Moreover, these findings provide further evidence of whcB exerting its effect via whcA.

DNA binding of the purified WhcA protein to the promoter region of the noxA gene. DNA affinity purification (a) and electrophoretic mobility shift assays (EMSA, b) were conducted and analyzed by SDS-PAGE (a) and native polyacrylamide gel electrophoresis (b), respectively. The arrow indicates MBP-WhcA-His6. M molecular weight marker, L loaded protein, W washed fraction, E1 protein eluted with 0.6 M NaCl, E2 protein eluted using 1 M NaCl
The potential binding of MBP and WhcA to the noxA promoter region was also assessed using EMSA to visualize shifted DNA. The target DNA region (bases −211 to +9 relative to the transcriptional start site) was PCR-amplified as described in the “Materials and methods” section. As shown in Fig. 3b, purified WhcA protein was found to bind to the promoter region of the noxA gene, as evidenced by shifted DNA bands. Binding of MBP to the noxA promoter region was not observed, suggesting the specificity of the observed protein-DNA binding. Furthermore, shifted bands were not observed in the absence of DTT. Considering the role of WhcA as a repressor of oxidative stress response genes, the binding of the protein only under reducing conditions (i.e., in the presence of DTT) is anticipated. It is also interesting to note that SpiA, which is known to interact with WhcA, was not needed for the DNA binding (see “Discussion”).
Growth properties of the noxA deletion or overexpression strains
In previous experiments, retarded growth rates were observed in the host cells, when the transcription of noxA was decreased to a minimum level (Fig. 2), suggesting that noxA transcription is required for proper cell function. Furthermore, the involvement of multiple whc genes in the regulation of noxA transcription indicates that tight regulation of the noxA gene is important for cell physiology. To further analyze these phenomena, a noxA-deleted mutant strain (ΔnoxA) of C. glutamicum was generated by gene disruption, and a C. glutamicum strain overexpressing noxA (P180-noxA) was established. Subsequently, the growth rates of both the strains were monitored. Internal deletion of noxA in the ΔnoxA strain was confirmed by PCR (data not shown) and the approximately 20-fold overexpression of noxA in the P180-noxA strain was confirmed by RT-qPCR (data not shown). First, the growth properties of both strains were assessed using minimal or complex medium. In the minimal medium, the ΔnoxA mutant strain showed retarded growth with a doubling time of 2.4 h (Fig. 4a), compared with the 2.3 h doubling time measured for the wild-type strain. Furthermore, the P180-noxA strain showed much slower growth with a doubling time of 2.5 h, suggesting that excess noxA gene expression during the growth phase may interfere with proper cell function. The growth data shown suggests that the noxA-encoded protein may function as an enzyme whose cellular concentration must be strictly controlled for proper cell function. Differences in growth rates among the strains were not observed when the cells were grown in a complex medium (data not shown). As suggested by protein sequence homology, the noxA-encoded protein may function as an NADH oxidase.

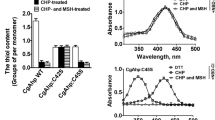
Growth characteristics of C. glutamicum wild-type and noxA mutants and sensitivity of the C. glutamicum strains to oxidants. a Cells were grown on glucose MCGC minimal media. Data represent three independent experiments. filled circle indicates wild-type, empty circle ΔnoxA, filled inverted triangle P180-noxA. b A paper disk containing diamide, menadione, or H2O2 was placed on each MB plate containing lawns of C. glutamicum cells and incubated at 30 °C for 24 h. Numbers denote diameter (mm) of the growth-inhibiting zones. Data represent three independent experiments
Response of noxA mutants to oxidative stress
Because the noxA gene was found to be under the control of the whc genes, it was assumed that noxA plays a role in oxidative stress responses. This hypothesis was tested by challenging the ΔnoxA and P180-noxA C. glutamicum strains with various stress-causing oxidants including diamide, menadione, and H2O2. The sensitivities of the strains to the reagents were monitored using an agar-diffusion test. As shown in Fig. 4b, the P180-noxA strain was found to be highly sensitive to menadione (but not to H2O2 or diamide) compared with the wild-type strain, as shown by a larger zone of growth inhibition for P180-noxA. Sensitivity of the ΔnoxA strain to the oxidants was detected, but to a lesser extent than that of the P180-noxA strain. Sensitivity of the ΔnoxA and P180-noxA strains to the redox-cycling compound menadione, but not to H2O2, may suggest a role for the noxA gene in redox balance rather than in the detoxification of reactive oxygen species (see “Discussion”). Assuming that the noxA-encoded protein may be membrane-associated, the sensitivity of the ΔnoxA and P180-noxA cells to detergents (Triton X-100 and Tween 20) was assessed. No differences were observed among the strains (data not shown).
The increased sensitivity of the ΔnoxA and P180-noxA strains to oxidants is thought to possibly be due to a faulty oxidation repair system. Thioredoxin reductase reduces oxidized thioredoxin using NADPH as a cofactor, and this reduced thioredoxin functions as a general protein disulfide reductant, which can reactivate previously oxidized proteins (Holmgren 1985). The mRNA levels of ORF NCgl0663, which is assumed to be the trxB gene encoding thioredoxin reductase in C. glutamicum, were thus measured, and only marginal differences in ORF NCgl0663 expression levels were observed among the C. glutamicum wild-type, ΔnoxA, and P180-noxA strains in the log phase (data not shown). This result therefore excludes the possibility that the defective cell growth observed was caused by a defective expression of the trxB gene.
NADH oxidase activity
Because noxA has been annotated to encode NADH oxidase, and our findings suggest a role for noxA in redox balancing, the noxA-encoded protein was tested for NADH oxidase activity. Because the presence of multiple NADH oxidases in the annotated genome of C. glutamicum could potentially hinder our analysis, NADH oxidase activity was measured and compared in the cell extracts of wild-type, ΔnoxA, and P180-noxA strains. In the ΔnoxA mutant strain, the activity decreased by approximately 70 % relative to the wild-type strain (Table 1). The activity in the P180-noxA strain, however, increased by 20-fold. These data suggest that the noxA-encoded protein (NoxA) possesses NADH oxidase activity and NoxA accounts for the majority of the NADH-oxidizing activity in C. glutamicum.
To determine if NADH was oxidized specifically by NoxA and not by other enzymes present in the host cell, the NoxA enzyme was purified. First, the noxA gene was cloned in to the T7 promoter-based plasmid pET28a to give pSL544 and was subsequently expressed in E. coli as an NH2-terminal His6-tagged protein (His6-NoxA). Analyses carried out with the extracts of E. coli harboring pSL544 revealed high levels of NADH oxidase activity (Table 2). Crude extracts from E. coli transformed with pSL544 were analyzed by SDS-PAGE. A protein of 24.8 kDa, a size consistent with the predicted molecular mass for the His6-NoxA protein, was identified in crude extracts (Fig. 5). The enzyme was purified 3.6-fold to homogeneity with 89 % yield by Ni2+-NTA affinity chromatography (Table 2). Purified His6-NoxA showed a single band on SDS-PAGE stained with Coomassie Brilliant Blue (Fig. 5). The purified His6-NoxA dominated the NADH oxidase activity detected in E. coli cell-free extracts (Table 2). Purified His6-NoxA showed high activity in oxidizing NADH in the presence of FMN. The purified protein was colorless and did not display activity in the absence of FMN. These results clearly demonstrate that the NADH oxidase activity observed in crude extracts of the C. glutamicum wild-type and P180-noxA cells (Table 1) corresponds to that of the NoxA protein.

SDS-PAGE analysis of expression and purification of His6-NoxA. The purified His6-NoxA (lane 4, 19 μg) and molecular-weight standards (lane 1) are indicated along with their corresponding molecular masses. Lane 1 molecular mass standards, lane 2 crude extract (12 μg), lane 3 column flow-through fractions (8 μg), lane 4 His6-NoxA after Ni2+-NTA chromatography. SDS-PAGE gel (17 %) was stained with Coomassie Brilliant Blue R-250
Discussion
In this study, the role of whcA in the regulation of its target gene, noxA, was assessed. In accordance with the noxA transcription data from the ΔwhcA and P180-whcA strains of C. glutamicum, purified WhcA protein was shown to bind to the promoter region of noxA. The previously reported presence of the helix-turn-helix DNA-binding motif of WhcA is in agreement with our data. It is interesting to note that SpiA is not required for the DNA-binding of WhcA. Park et al. (2011, 2012) postulated that the WhcA protein forms a complex with the SpiA protein and that the SpiA-WhcA protein complex then binds to its target promoters to repress gene expression in the absence of oxidative stress. However, it was clearly shown in this study that the WhcA protein in fact presents in its DNA-binding configuration and binds to its target gene (noxA). Because Park et al. noted that SpiA played both positive and negative roles and that some whcA-regulated genes are not under the control of the spiA gene, we can speculate that the noxA gene is not likely to be part of the spiA-regulon. This hypothesis is further supported by the fact that, while the growth of spiA mutants is affected by both menadione and diamide (Park et al. 2011), the growth of noxA mutants is only affected by menadione. Moreover, unlike in the case of spiA mutants, the expression of the trxB gene encoding thioredoxin reductase was not affected in the noxA mutants—a finding that further supports our hypothesis. Furthermore, the fact that a reducing agent (DTT) is required for the binding of WhcA to DNA is noteworthy, as it suggests that a reducing environment, which is the proposed condition under which WhcA binds DNA as a repressor, is also necessary for the WhcA protein to maintain its DNA-binding activity.
The regulation of noxA gene expression by whcE and whcB appears to be different from the regulation by whcA. Although the transcription of noxA was minimal in the ΔwhcE strain, the stimulatory effect achieved by the overexpression of whcE gene was rather limited, suggesting the presence of an additional regulatory component. It is possible that the limited effect is due to the presence of an intact chromosomal copy of whcA, which exerts a negative regulatory effect. The overexpression of whcE likely stimulates the expression of whcA, and the stimulatory effect of whcE may be suppressed by the WhcA protein. Overall, our findings, including the evidence for the action of whcB being conveyed via whcA, are in agreement with the hypothesis proposed by Lee et al. (2013).
The involvement of multiple whc genes in the regulation of noxA expression suggests that tight regulation of noxA gene expression is essential. The severe retardation of the growth rates observed for the P180-whcA, P180-whcB, and ΔwhcE strains were associated with low expression levels of noxA, suggesting the importance of adequate noxA expression. It is also interesting to note that, although the overexpression of noxA in the P180-noxA cells caused severe growth retardation, overexpression of noxA did not cause severe growth defects in ΔwhcA, P180-whcE, and ΔwhcB cells. This could be due to the level of expression achieved in the P180-noxA cells. Approximately 20-fold expression was observed in P180-noxA cells, whereas only deregulation was observed in the P180-whcA, P180-whcB, and ΔwhcE cells.
The sensitivity of the noxA mutant strains to oxidants indicates that NADH oxidase plays a role in oxidative stress responses. It is interesting to note that noxA mutants were only sensitive to menadione, and not to H2O2 or diamide. The redox-cycling compound menadione exerts its toxic effects by stimulating the intracellular production of superoxide radicals (Afanas’ev et al. 1990). The thiol-specific oxidant diamide, on the other hand, specifically oxidizes sulfhydryl groups, such as those in cysteine residues. The comparable levels of trx transcription measured in the ΔnoxA and P180-noxA strains correlates with the diamide insensitivity of the strains. The increased sensitivity of P180-noxA cells to menadione is suggestive of an overproduction of superoxide radicals in C. glutamicum cells.
The noxA gene has been annotated to encode NADH oxidase, a member of the nitroreductase family of proteins. In this study, the noxA-encoded protein was indeed shown to exhibit NADH oxidase activity. The fact that the basal level of NADH oxidase activity measured was relatively high in the ΔnoxA mutant strain is indicative of the presence of multiple NADH-oxidizing enzymes in C. glutamicum. Possible candidates for such additional nitroreductase enzymes include NCgl2735 and NCgl2913, which are found in the genome of C. glutamicum. The similarity of these two gene products with the noxA-encoded protein, however, is low (20–24 %). The observed NADH-oxidizing activity may alternatively result from the electron transfer reaction from NADH to oxygen through the components of the electron transport chain. The measured NADH oxidation activity of NoxA would strongly compete with NADH oxidation by the NADH dehydrogenase (ndh) of the respiratory chain and thus with oxidative phosphorylation. In addition, although this has not been shown in C. glutamicum, many enzymes exhibiting NADH oxidase activity have been reported in other organisms (Kawasaki et al. 2009; Kundu et al. 2012; Singh et al. 2004; Zarepour et al. 2010). NADH oxidase catalyzes the oxidation of NADH to NAD+ using molecular oxygen as the electron acceptor. One type catalyzes the oxidation of NADH by reducing molecular oxygen to H2O2, while the other catalyzes a four-electron reduction of oxygen to H2O with NADH oxidation. Some enzymes also reduce H2O2 to H2O at the expense of NADH. Although NADH oxidases play an important role in scavenging oxygen in anaerobic bacteria, thus providing defense against oxidative stress, common NADH oxidases of mesophilic organisms are assumed to protect cells from oxidative stress by reducing oxygen to water, without the formation of harmful reactive oxygen species (Chenier et al. 2008; Cortial et al. 2010; Derr et al. 2012; Jia et al. 2010; Kang et al. 2013; Kawasaki et al. 2005; Pesakhov et al. 2007; Yang and Ma 2007). Moreover, NADH oxidases from several bacteria also play important roles in recycling pyridine nucleotides such as NAD+/NADH during catabolism (Kang et al. 2013; Sauvageot et al. 2012; Yamamoto et al. 2006; Zhang et al. 2012). The fact that ΔnoxA and P180-noxA cells did not show significant difference in their sensitivities to H2O2 in this study indicates that NADH oxidase may not be involved in H2O2 generation and/or elimination in C. glutamicum, thus excluding the possibility of a role for the enzyme in the detoxification of reactive oxygen species. Furthermore, the growth differences that were observed only in minimal medium suggest that NADH oxidase may contribute to sugar metabolism, probably through the oxidization of NADH, as observed in Enterococcus faecalis (Sauvageot et al. 2012), suggesting a role in NAD+/NADH balance. The finding that both deletion and overexpression of the noxA gene were harmful to the cell growth also provides evidence for a role of noxA in maintaining NAD+/NADH balance. Although unlikely, NADH oxidase from C. glutamicum may play additional roles by transferring electrons to acceptors other than molecular oxygen.
Taken together, our results show that the noxA gene of C. glutamicum plays important roles in oxidative stress responses as well as in NAD+/NADH redox balance. Considering the role of C. glutamicum cells in metabolite overproduction and the importance of cofactor regeneration such as NAD+/NADH cycling in metabolic engineering (Hou et al. 2014; Ji et al. 2013; Rocha-Martin et al. 2011; Zhang et al. 2014), the noxA gene may be of importance in the field of biotechnology. Toward fully elucidating the function of noxA in C. glutamicum physiology, additional investigations are underway.
References
Afanas’ev IB, Korkina LG, Suslova TB, Soodaeva SK (1990) Are quinones producers or scavengers of superoxide ion in cells? Arch Biochem Biophys 281:245–250
Alam MS, Garg SK, Agrawal P (2007) Molecular function of WhiB4/Rv3681c of Mycobacterium tuberculosis H37Rv: a [4Fe−4S] cluster coordinating protein disulphide reductase. Mol Microbiol 63:1414–1431
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Chenier D, Beriault R, Mailloux R, Baquie M, Abramia G, Lemire J, Appanna V (2008) Involvement of fumarase C and NADH oxidase in metabolic adaptation of Pseudomonas fluorescens cells evoked by aluminum and gallium toxicity. Appl Environ Microbiol 74:3977–3984
Choi WW, Park SD, Lee SM, Kim HB, Kim Y, Lee HS (2009) The whcA gene plays a negative role in oxidative stress response of Corynebacterium glutamicum. FEMS Microbiol Lett 290:32–38
Cortial S, Chaignon P, Iorga BI, Aymerich S, Truan G, Gueguen-Chaignon V, Meyer P, Moréra S, Ouazzani (2010) NADH oxidase activity of Bacillus subtilis nitroreductase NfrA1: insight into its biological role. FEBS Lett 584:3916–3922
Crack JC, den Hengst CD, Jakimowicz P, Subramanian S, Johnson MK, Buttner MJ, Thomson AJ, Le Brun NE (2009) Characterization of [4Fe-4S]-containing and cluster-free forms of Streptomyces WhiD. Biochemistry (NY) 48:12252–12264
Davis NK, Chater KF (1992) The Streptomyces coelicolor whiB gene encodes a small transcription factor-like protein dispensable for growth but essential for sporulation. Mol Gen Genet MGG 232:351–358
Derr AM, Faustoferri RC, Betzenhauser MJ, Gonzalez K, Marquis RE, Quivey RG Jr (2012) Mutation of the NADH oxidase gene (nox) reveals an overlap of the oxygen- and acid-mediated stress responses in Streptococcus mutans. Appl Environ Microbiol 78:1215–1227
Follettie M, Sinskey A (1986) Recombinant DNA technology for Corynebacterium glutamicum. Food Technol 40:88–94
Follettie MT, Peoples O, Agoropoulou C, Sinskey A (1993) Gene structure and expression of the Corynebacterium flavum N13 ask-asd operon. J Bacteriol 175:4096–4103
Gao B, Gupta RS (2012) Phylogenetic framework and molecular signatures for the main clades of the phylum Actinobacteria. Microbiol Mol Biol Rev 76:66–112
Holmgren A (1985) Thioredoxin. Annu Rev Biochem 54:237–271
Hong EJ, Park JS, Kim Y, Lee HS (2014) Role of Corynebacterium glutamicum sprA encoding a serine protease in glxR-mediated global gene regulation. PLoS One 9:e93587
Hou J, Suo F, Wang C, Li X, Shen Y, Bao X (2014) Fine-tuning of NADH oxidase decreases byproduct accumulation in respiration deficient xylose metabolic Saccharomyces cerevisiae. BMC Biotechnol 14:13
Hwang BJ, Yeom HJ, Kim Y, Lee HS (2002) Corynebacterium glutamicum utilizes both transsulfuration and direct sulfhydrylation pathways for methionine biosynthesis. J Bacteriol 184:1277–1286
Ikeda M, Nakagawa S (2003) The Corynebacterium glutamicum genome: features and impacts on biotechnological processes. Appl Microbiol Biotechnol 62:99–109
Jakimowicz P, Cheesman MR, Bishai WR, Chater KF, Thomson AJ, Buttner MJ (2005) Evidence that the Streptomyces developmental protein WhiD, a member of the WhiB family, binds a [4Fe-4S] cluster. J Biol Chem 280:8309–8315
Ji XJ, Xia ZF, Fu NH, Nie ZK, Shen MQ, Tian QQ, Huang H (2013) Cofactor engineering through heterologous expression of an NADH oxidase and its impact on metabolic flux redistribution in Klebsiella pneumoniae. Biotechnol Biofuels 6:7
Jia B, Lee S, Pham BP, Cho YS, Yang J, Byeon H, Kim JC, Cheong G (2010) An archaeal NADH oxidase causes damage to both proteins and nucleic acids under oxidative stress. Mol Cells 29:363–371
Kalinowski J, Bathe B, Bartels D, Bischoff N, Bott M, Burkovski A, Dusch N, Eggeling L, Eikmanns BJ, Gaigalat L, Goesmann A, Hartmann M, Huthmacher K, Krämer R, Linke B, McHardy AC, Meyer F, Möckel B, Pfefferle W, Pühler A, Rey DA, Rückert C, Rupp O, Sahm H, Wendisch VF, Wiegräbe I, Tauch A (2003) The complete Corynebacterium glutamicum ATCC 13032 genome sequence and its impact on the production of L-aspartate-derived amino acids and vitamins. J Biotechnol 104:5–25
Kang TS, Korber DR, Tanaka T (2013) Influence of oxygen on NADH recycling and oxidative stress resistance systems in Lactobacillus panis PM1. AMB Expr 3:1–9
Kawasaki S, Watamura Y, Ono M, Watanabe T, Takeda K, Niimura Y (2005) Adaptive responses to oxygen stress in obligatory anaerobes Clostridium acetobutylicum and Clostridium aminovalericum. Appl Environ Microbiol 71:8442–8450
Kawasaki S, Satoh T, Todoroki M, Niimura Y (2009) b-type dihydroorotate dehydrogenase is purified as a H2O2-forming NADH oxidase from Bifidobacterium bifidum. Appl Environ Microbiol 75:629–636
Kim HJ, Kim TH, Kim Y, Lee HS (2004) Identification and characterization of glxR, a gene involved in regulation of glyoxylate bypass in Corynebacterium glutamicum. J Bacteriol 186:3453–3460
Kim TH, Park JS, Kim HJ, Kim Y, Kim P, Lee HS (2005) The whcE gene of Corynebacterium glutamicum is important for survival following heat and oxidative stress. Biochem Biophys Res Commun 337:757–764
Kundu TK, Velayutham M, Zweier JL (2012) Aldehyde oxidase functions as a superoxide generating NADH oxidase: an important redox regulated pathway of cellular oxygen radical formation. Biochemistry 51:2930–2939
Lee JY, Park JS, Kim HJ, Kim Y, Lee HS (2012) Corynebacterium glutamicum whcB, a stationary phase‐specific regulatory gene. FEMS Microbiol Lett 327:103–109
Lee JY, Kim HJ, Kim ES, Kim P, Kim Y, Lee HS (2013) Regulatory interaction of the Corynebacterium glutamicum whc genes in oxidative stress responses. J Biotechnol 168:149–154
Leuchtenberger W, Huthmacher K, Drauz K (2005) Biotechnological production of amino acids and derivatives: current status and prospects. Appl Microbiol Biotechnol 69:1–8
MacNeil DJ, Gewain KM, Ruby CL, Dezeny G, Gibbons PH, MacNeil T (1992) Analysis of Streptomyces avermitilis genes required for avermectin biosynthesis utilizing a novel integration vector. Gene 111:61–68
Park SD, Lee SN, Park IY, Choi JS, Jeong WK, Kim Y, Lee HS (2004) Isolation and characterization of transcriptional elements from Corynebacterium glutamicum. J Microbiol Biotechnol 14:789–795
Park SD, Youn JW, Kim YJ, Lee SM, Kim Y, Lee HS (2008) Corynebacterium glutamicum σE is involved in responses to cell surface stresses and its activity is controlled by the anti-σ factor CseE. Microbiology 154:915–923
Park JS, Shin S, Kim ES, Kim P, Kim Y, Lee HS (2011) Identification of SpiA that interacts with Corynebacterium glutamicum WhcA using a two‐hybrid system. FEMS Microbiol Lett 322:8–14
Park JS, Lee JY, Kim HJ, Kim ES, Kim P, Kim Y, Lee HS (2012) The role of Corynebacterium glutamicum spiA gene in whcA-mediated oxidative stress gene regulation. FEMS Microbiol Lett 331:63–69
Pesakhov S, Benisty R, Sikron N, Cohen Z, Gomelsky P, Khozin-Goldberg I, Dagan R, Porat N (2007) Effect of hydrogen peroxide production and the Fenton reaction on membrane composition of Streptococcus pneumoniae. Biochim Biophys Acta (BBA) Biomembr 1768:590–597
Pfeifer-Sancar K, Mentz A, Rückert C, Kalinowski J (2013) Comprehensive analysis of the Corynebacterium glutamicum transcriptome using an improved RNAseq technique. BMC Genomics 14:888
Rocha-Martin J, Vega D, Bolivar JM, Godoy CA, Hidalgo A, Berenguer J, Guisan JM, Lopez-Gallego F (2011) New biotechnological perspectives of a NADH oxidase variant from Thermus thermophilus HB27 as NAD+-recycling enzyme. BMC Biotechnol 11:101
Rybniker J, Nowag A, Van Gumpel E, Nissen N, Robinson N, Plum G, Hartmann P (2010) Insights into the function of the WhiB‐like protein of mycobacteriophage TM4—a transcriptional inhibitor of WhiB2. Mol Microbiol 77:642–657
Sambrook J, Russell DW, Russell DW (2001) Molecular cloning: a laboratory manual. Cold spring harbor laboratory press Cold Spring. Harbor, New York
Sauvageot N, Ladjouzi R, Benachour A, Rince A, Deutscher J, Hartke A (2012) Aerobic glycerol dissimilation via the Enterococcus faecalis DhaK pathway depends on NADH oxidase and a phosphotransfer reaction from PEP to DhaK via EIIADha. Microbiology 158:2661–2666
Schäfer A, Tauch A, Jäger W, Kalinowski J, Thierbach G, Pühler A (1994) Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145:69–73
Singh R, Wiseman B, Deemagarn T, Donald LJ, Duckworth HW, Carpena X, Fita I, Loewen PC (2004) Catalase-peroxidases (KatG) exhibit NADH oxidase activity. J Biol Chem 279:43098–43106
Singh A, Guidry L, Narasimhulu KV, Mai D, Trombley J, Redding KE, Giles GI, Lancaster JR Jr, Steyn AJ (2007) Mycobacterium tuberculosis WhiB3 responds to O2 and nitric oxide via its [4Fe-4S] cluster and is essential for nutrient starvation survival. Proc Natl Acad Sci U S A 104:11562–11567
Singh A, Crossman DK, Mai D, Guidry L, Voskuil MI, Renfrow MB, Steyn AJ (2009) Mycobacterium tuberculosis WhiB3 maintains redox homeostasis by regulating virulence lipid anabolism to modulate macrophage response. PLoS Pathog 5:e1000545
Smith LJ, Stapleton MR, Fullstone GJ, Crack JC, Thomson AJ, Le Brun NE, Hunt DM, Harvey E, Adinolfi S, Buxton RS (2010) Mycobacterium tuberculosis WhiB1 is an essential DNA-binding protein with a nitric oxide-sensitive iron-sulfur cluster. Biochem J 432:417–427
von der Osten CH, Gioannetti C, Sinskey AJ (1989) Design of a defined medium for growth of Corynebacterium glutamicum in which citrate facilitates iron uptake. Biotechnol Lett 11:11–16
Yamamoto Y, Pargade V, Lamberet G, Gaudu P, Thomas F, Texereau J, Gruss A, Trieu‐Cuot P, Poyart C (2006) The Group B Streptococcus NADH oxidase Nox‐2 is involved in fatty acid biosynthesis during aerobic growth and contributes to virulence. Mol Microbiol 62:772–785
Yang X, Ma K (2007) Characterization of an exceedingly active NADH oxidase from the anaerobic hyperthermophilic bacterium Thermotoga maritima. J Bacteriol 189:3312–3317
Yoshihama M, Higashiro K, Rao EA, Akedo M, Shanabruch WG, Follettie MT, Walker GC, Sinskey AJ (1985) Cloning vector system for Corynebacterium glutamicum. J Bacteriol 162:591–597
Zarepour M, Kaspari K, Stagge S, Rethmeier R, Mendel RR, Bittner F (2010) Xanthine dehydrogenase AtXDH1 from Arabidopsis thaliana is a potent producer of superoxide anions via its NADH oxidase activity. Plant Mol Biol 72:301–310
Zhang GC, Liu JJ, Ding WT (2012) Decreased xylitol formation during xylose fermentation in Saccharomyces cerevisiae due to overexpression of water-forming NADH oxidase. Appl Environ Microbiol 78:1081–1086
Zhang X, Zhang R, Bao T, Rao Z, Yang T, Xu M, Xu Z, Li H, Yang S (2014) The rebalanced pathway significantly enhances acetoin production by disruption of acetoin reductase gene and moderate-expression of a new water-forming NADH oxidase in Bacillus subtilis. Metab Eng 23:34–41
Zheng F, Long Q, Xie J (2012) The function and regulatory network of WhiB and WhiB-like protein from comparative genomics and systems biology perspectives. Cell Biochem Biophys 63:103–108
Acknowledgments
This work was supported by a Korea University Grant to H.-S. Lee.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Park, J.C., Kim, Y. & Lee, HS. Involvement of the NADH oxidase-encoding noxA gene in oxidative stress responses in Corynebacterium glutamicum . Appl Microbiol Biotechnol 99, 1363–1374 (2015). https://doi.org/10.1007/s00253-014-6327-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-014-6327-x