
Abstract
Xyloglucan-specific endo-β-1,4-glucanases (Xegs, EC 3.2.1.151) exhibit high catalytic specificity for β-1,4 linkages of xyloglucan, a branched hemicellulosic polysaccharide abundant in dicot primary cell walls and present in many monocot species. In nature, GH12 Xegs are not associated with carbohydrate-binding modules (CBMs), and here, we have investigated the effect of the fusion of the xyloglucan-specific CBM44 on the structure and function of a GH12 Xeg from Aspergillus niveus (XegA). This fusion presented enhanced catalytic properties and conferred superior thermal stability on the XegA. An increased k cat (chimera, 177.03 s−1; XegA, 144.31 s−1) and reduced KM (chimera, 1.30 mg mL−1; XegA, 1.50 mg mL−1) resulted in a 1.3-fold increase in catalytic efficiency of the chimera over the parental XegA. Although both parental and chimeric enzymes presented catalytic optima at pH 5.5 and 60 °C, the thermostabilitiy of the chimera at 60 °C was greater than the parental XegA. Moreover, the crystallographic structure of XegA together with small-angle X-ray scattering (SAXS) and molecular dynamics simulations revealed that the spatial arrangement of the domains in the chimeric enzyme resulted in the formation of an extended binding cleft that may explain the improved kinetic properties of the CBM44-XegA chimera.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Glycoside hydrolases (GHs) constitute a large group of carbohydrate-active enzymes found in all domains of life, and to date, more than 130 GH families have been described (CAZy database, http://www.cazy.org/). These enzymes have been extensively studied not only because of their fundamental role in carbon cycling but also due to their industrial and biotechnology applications (Kirk et al. 2002). Xyloglucan-specific endo-β-1,4-glucanases (Xegs; EC 3.2.1.151) demonstrate hydrolytic specificity for xyloglucan (XyG), the most abundant hemicellulose component in the primary cell walls of dicots and also present in almost half of all monocot species. XyGs are comprised of a β-1-4-D-glucan backbone in which up to 75 % of the glucose residues are substituted at the O6 position with mono-, di- or triglycosyl side chains (Benko et al. 2008). Xyloglucans and Xegs are important not only in plant cell wall physiology and metabolism but also for biotechnological applications such as fruit juice clarification, textile processing, cellulose fibre modification, pharmaceutical delivery, food additives and biofuel production, and there has recently been a growing interest both in the xyloglucan polymer and the enzymes that modify it (Gloster et al. 2007).
Xegs have been identified in glycosyl hydrolase families GH5, 12, 16, 44 and GH74 (Powlowski et al. 2009), and GH12 Xegs are characterized by a canonical β-jelly roll fold in which two arched β-sheets form the main body of the protein, where residues of the inner β-sheet define the active-site cleft. This cleft is comprised of six binding sites for branched or unbranched sugars (denominated as −4 to +2, (Davies et al. 1997)), where the −1 (glycone) subsite specifically accommodates an unbranched β-D-glucopyranose and the +1 (aglycone) subsite binds the branched [(1→6)-α-D-xylopyranose]-β-D-glucopyranose. Saccharide binding to the other subsites in the cleft is less stringent, and in addition to the α-D-xylopyranose modification, branches can be extended to include a second saccharide which can be either a β-D-galactopyranose or an α-L-arabinofuranose, and the galactose residues may be further modified by α-L-fucopyranose (Carpita and McCann 2000). The hydrolysis products therefore vary among XyGs derived from different sources and depend on the degree and type of branching modifications. Hydrolysis occurs by a double-displacement retaining mechanism in which conserved glutamic acid residues act as the nucleophile (Glu116 in Hypocrea jecorina Cel12A) and the acid/base (Glu200 in H. jecorina Cel12A) (Okada et al. 2000).
In addition to the catalytic domain, many glycoside hydrolases include an additional carbohydrate-binding domain (CBM), which might assist polysaccharide recognition and binding, increasing the enzyme concentration on the substrate surface and thereby improving the rate of hydrolysis (Guillen et al. 2010). Although some GH74 Xegs contain a CBM, these modules showed no significant affinity for xyloglucan, indicating that they contribute to xyloglucan hydrolysis by localizing the enzyme within the cell wall polymer matrix (Ichinose et al. 2012; Ishida et al. 2007). To date, three CBMs with high xyloglucan affinity have been identified. The CBM65A and CBM65B are associated with the Eubacterium cellulosolvens endoglucanase cel5A gene (Luis et al. 2013) and a polycystic kidney disease (PKD)-CBM44 with the celJ gene of Clostridium thermocellum (Najmudin et al. 2006); however, to our knowledge, there are no reports of a GH12 Xeg associated with a CBM. The CBM44 domain from the celJ gene of C. thermocellum shows a high affinity for xyloglucan and somewhat lower affinities for β-glucan, lichenan and glucomannan. The crystal structure revealed a β-jelly roll fold consisting of two five-stranded anti-parallel β-sheets, whose concave face defines a deep binding cleft (Najmudin et al. 2006). Site-directed mutagenesis has confirmed the role of three tryptophan residues located in this cleft in determining xyloglucan affinity, and in particular, mutation of Trp194 abolished xyloglucan binding to the CBM44 (Najmudin et al. 2006).
Here, we describe the effect of the fusion of a high xyloglucan affinity CBM (PKD-CBM44) on the structure and function of the GH12 from Aspergillus niveus (XegA). Kinetic characterization of the resulting chimera revealed an increase in catalytic efficiency, indicating that the association of XyG-specific CBMs to the GH12 XegA yields a specialized and productive enzyme. A combination of small-angle X-ray scattering (SAXS) and molecular dynamics simulations (MDS) reveals that the relative orientation of the CBM and Xeg domains in the chimera results in the formation of an extended xyloglucan binding cleft, which indicated the superior catalytic efficiency of the chimeric enzyme. Moreover, the crystal structure of XegA revealed a stable dimeric arrangement, which is consistent with the results of a previous study (Damasio et al. 2012) which suggested that enzyme dimerization impaired catalytic activity.
Materials and methods
Design and construction of the PKD-CBM44-XegA chimera
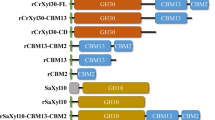
The continuous DNA sequence encoding the PKD and family 44 CBM domains of the CelJ from C. thermocellum (GenBank Gene ID: 4808226, KEGG entry: Cthe_0624) was amplified by polymerase chain reaction (PCR) using genomic DNA from C. thermocellum strain ATCC 27405 as the template with primers Fw and Rv (see Table 1). The xegA gene of A. niveus (GenBank Acc No JN222918.1) was amplified using the pExPYR-xegA plasmid as template (Damasio et al. 2012). The amplified fragment contained a 50 bp intron that was removed by overlap PCR (Wurch et al. 1998) for expression of the enzyme in Escherichia coli. In the first step, two PCR reactions were performed using the primer pairs X1-X2 and X3-X4. Primers X2 and X3 contain complementary regions (shown in bold), while X1 and X4 contain the restriction sites for NheI and BamHI (underlined) respectively. Products from the second step were mixed in an equimolar ratio, and finally, a third reaction using primers X1 and X4 amplified the intron-deleted xegA coding sequence. The PKD-CBM44 and XegA coding sequences were cloning into the pET28a(+) expression vector (EMD Millipore, Billerica, MA, USA) using the respective restriction sites and used as templates to construct the PKD-CBM44-XegA chimera coding sequence (see Fig. 1) using the overlap PCR methodology described above and the primer pairs P1-P2 and P3-P4. In this case, the complementary regions of P2 and P3 (shown in bold) encode a peptide linker (GGSGG) inserted between the individual domains of the chimera. The final coding sequence for the PKD-CBM44-XegA chimera was cloned into pET28a(+) using NheI and HindIII cleavage sites (underlined) and confirmed by nucleotide sequencing.

Schematic flow diagram showing the construction of the PKD-CBM44-XegA chimera by overlap PCR. The top right part shows the elimination of the single intron (grey box marked with the letter “I”) from the XegA gene. The remaining lower part shows the amplification of the PKD-CM44 coding sequence and the fusion with the XegA coding sequence to yield the final PKD-CBM44-XegA coding sequence including the linker sequence (shown as the black box). See the “Materials and methods” section for further details
Expression and purification of individual and fused proteins
All recombinant proteins were expressed in E. coli Rosetta (DE3) pLysS (EMD Millipore, Billerica, MA, USA) transformed with pET28a(+) their respective coding sequences and grown in HDM medium (containing per litre, 25 g yeast extract, 15 g tryptone and 1.2 g of MgSO4 supplemented with 40 μg mL−1 kanamycin and 34 μg mL−1 chloramphenicol). The cells were grown at 37 °C to an A600 of 0.6, and expression was induced with 0.5 mM IPTG for 5 h at 30 °C. Cells were harvested by centrifugation (8000g/4 °C/30 min), and the cell pellets were resuspended in 15 mL of lysis buffer (100 mM HEPES pH 7.5, 1 mM phenylmethylsulfonyl fluoride, 300 mM NaCl, 40 mM imidazole and 1 % (v/v) Triton X-100). Cell disruption was performed by sonication (pulses of 30 s, 40 % duty cycle, total of 6 min) on ice, and cell debris were removed by centrifugation. The supernatant was applied to a nickel affinity column (Promega, Madison, WI, USA), and protein purification was performed following manufacturer’s recommendations using 60 and 250 mM of imidazole in wash and elution buffers, respectively. The eluted proteins were analyzed by 12 % SDS–PAGE and visualized with Coomassie blue (Sigma-Aldrich, São Paulo, Brazil) staining. Protein concentrations were determined by A280 measurements using extinction coefficients calculated by ProtParam software (Gasteiger et al. 2005).
Circular dichroism spectroscopy
Far-UV CD spectra of individual and chimeric proteins were measured between 190 and 250 nm in 10 mM phosphate buffer pH 7.0 at 25 °C with a Jasco J-810 spectropolarimeter (Jasco Inc. Tokyo, Japan) using 1 mm path-length cuvettes and a protein concentration of 0.2 mg mL−1. A total of six spectra were collected and averaged for each protein, and the averaged spectrum was corrected by subtraction of a buffer blank and converted to mean residue molar ellipticity (θ; deg cm2 dmol−1).
Enzyme assays and biochemical characterization
Xyloglucanase activity was assayed using the xyloglucan extracted from Hymenaea courbaril seeds as substrate (Tine et al. 2006) and measuring total reducing sugar release by the 3,5-dinitrosalicylic acid (DNS) method (Miller 1959). The effect of pH and temperature on xyloglucan hydrolysis by the purified XegA and PKD-CBM44-XegA was measured in 50 mM phosphate–citrate buffer over the pH and temperature ranges of 2.0–7.0 and 40–75 °C respectively. The thermal stability of the purified enzymes were determined by incubating samples at 50 and 60 °C, and aliquots were withdrawn at determined time intervals over the course of the experiment. After immediate cooling on ice, residual activities were determined at 60 °C using the DNS assay. The kinetic parameters were determined over a substrate range of 0.25–10 mg mL−1 at pH 5.5 and 60 °C. The reaction mixture consisted of 90 μL substrate in 50 mM buffer and 10 μL enzyme solution and were incubated for 10 min. The reaction was stopped by adding 100 μL of the DNS solution and immediately boiled for 5 min. The reducing sugars released as a result of enzyme activity were measured at 540 nm (Versamax microplate reader, Molecular Devices, Sunnyvale, CA, USA), and one activity unit (U) was defined as the amount of enzyme releasing 1 μmol of reducing sugar from xyloglucan per minute (using glucose as the reference). All assays were performed in triplicate.
Mass spectrometric analysis of XegA and PKD-CBM44-XegA hydrolysis profile
The hydrolysis profiles were evaluated using 450 μL of a 5 mg mL−1 H. courbaril xyloglucan suspension in 50 mM ammonium acetate buffer, pH 5.5 and purified XegA or PKD-CBM44-XegA to a final enzyme concentration of 1.0 μM for 24 h at 30 °C. Samples were subsequently boiled for 10 min to denature the enzymes and centrifuged at 10,000g for 5 min. The supernatant was diluted 10-fold in MeOH/H2O 8:2 (v/v) solution and injected into an ESI source by a ACQUITY H-Class UPLC system (Waters Corporation, Milford, MA, USA) at 100 μL min−1. The electrospray ionization mass spectrometry experiments (ESI-MS) were accomplished on a Xevo TQ-S MS spectrometer (Waters Corporation, Milford, MA, USA) equipped with a Z-spray source operating in positive and negative modes. Data were collected and analyzed on the software MassLynx 4.0 (Waters Corporation, Milford, MA, USA). Argon was used as the collision gas. For ESI-MS, the data were acquired in the full scan mode. The analytical parameters were as follows: capillary voltage, 3.2 kV; cone voltage, 40 V; source temperature, 150 °C; and desolvation temperature (N2), 300 °C.
Crystallization and data collection
The XegA sample obtained from the nickel-affinity column was further purified by size-exclusion chromatography on a Superdex 200 (16/60) column (GE Healthcare, São Paulo, Brazil) to achieve high purity and homogeneity. Samples were pooled and concentrated in 20 mM Hepes pH 7.5 using Amicon Ultra-15 filter units to a concentration of 12 mg mL−1. Crystallization screening was performed by the vapour diffusion method using commercially available kits (SaltRX, Crystal Screen and Crystal Screen 2, Hampton Research, Aliso Viejo, CA, USA; Precipitant Synergy, Wizard I and II—Rigaku Corporation, Tokyo, Japan; PACT and JCSG—Qiagen/Nextal, Germantown, DM, USA). Sitting drops, containing 0.5 μL of XegA and 0.5 μL of mother liquor, were prepared using a HoneyBee 963 robot (Genomic Solutions Ltd, Huntingdon, UK) and equilibrated against 80 μL of reservoir at 18 °C. Automated imaging of crystallization plates was carried out using the Rock Imager Robot (Formulatrix, Bedford, Ma, USA). Clusters of needles were obtained after 2 days in 25 % (w/v) PEG4000, 200 mM ammonium sulfate and 100 mM sodium acetate buffer, pH 4.6. Elongated plate-shaped crystals were obtained after 4 days by a combination of pH reduction to 3.6, precipitant concentration to 20 % and preparation of 1 μL hanging drops. A single crystal was flash cooled in a nitrogen gas stream at 100 K, and a complete X-ray diffraction data set was collected at the W01B-MX2 beamline (Brazilian Synchrotron Light Laboratory, Campinas, Brazil), using a Mar Mosaic 225 mm charged-coupled device (CCD) detector. Processing and scaling were carried out using HKL2000 (Otwinowski and Minor 1997). Information concerning the crystal lattice and the statistics of data processing are shown in Table 2.
Structure determination
The three-dimensional structure of XegA was solved by molecular replacement using the MOLREP program (Vagin and Teplyakov 2010) and the atomic coordinates of the endo-β-1,4-glucanase from Aspergillus aculeatus (Protein Data Bank code 3VL8 showing 63 % amino acid identity with XegA) as the search model. The Matthews coefficient calculation yielded 2.11 Å3 Da−1 for the two monomers of XegA in the asymmetric unit (ASU). Structure refinement consisted of restrained and overall B-factor refinement with the REFMAC5 program (Murshudov et al. 1997) using automatic weighting between X-ray and geometry terms. After each cycle of refinement, the model was inspected and manually adjusted within the (2F o − F c) and (F o − F c) electron density maps using the COOT program (Emsley et al. 2010). Solvent molecules were manually added at positive peaks above 3.0 σ in the Fourier difference maps. The local and global quality of the structure was evaluated using MolProbity (Chen et al. 2010). The final model comprises residues from A18 to A238. N-terminal residues in the histidine tag were not observed in the electron density map and were not modelled. The structure refinement statistics are summarized in Table 2.
Small-angle X-ray scattering (SAXS)
SAXS data for PKD-CBM44-XegA were collected using a 165 mm MarCCD detector at the D02A/SAXS2 beamline (Brazilian Synchrotron Light Laboratory, Campinas, Brazil). The radiation wavelength was set to 1.48 Å, and the sample-to-detector distance was set to 1482.4 mm to give a scattering vector range from 0.11 to 2.23 nm−1 (q = 4.π.sinθ/λ, where 2θ is the scattering angle). Protein samples at 2, 4 or 8 mg mL−1, in 50 mM NaCl and 20 mM Tris–HCl, pH 7.5, were centrifuged for 10 min at 20,000g (4 °C) and filtered to remove any aggregates. Measurements were carried out at room temperature (~23 °C). Buffer scattering was recorded and subtracted from the corresponding sample scattering. The integration of SAXS patterns was performed using the Fit2D software (Hammersley et al. 1997), and the curves were scaled with respect to protein concentration. Fitting of the experimental data together with evaluation of the pair-distance distribution function p(r) were performed using the GNOM program (Svergun 1992). The ab initio dummy atom model was calculated using simulated annealing procedure in the DAMMIN program (Svergun 1999). An averaged model was generated from 10 independent runs using the DAMAVER program suite (Volkov and Svergun 2003).
Molecular dynamics simulation (MDS)
The crystal structures of PKD-CBM44 domains (PDB ID 2C26) and the XegA (PDB ID 4NPR) were manually fitted into the SAXS-derived envelop using the PyMOL Molecular Graphics System (Version 1.5.0.4, Schrödinger, LLC), taking into account the short linkers between the proteins (SATG and GGSGG). The superposition was refined using the Collage program of the Situs package (Wriggers 2010); the polypeptides were linked, and the loop between the C-terminus of the PKD-CBM44 to the N-terminus of the XegA was modelled using the SwissPDB program (Guex and Peitsch 1997). The final coordinates served as the starting structure for molecular dynamics simulation.
MDS of the chimera in aqueous solution were performed over a period of 50 ns using the GROMACS 4.6.5 package (Hess et al. 2008). The system was defined by a cubic box of 14.1 nm each side containing the chimera whose atomic coordinates were obtained as described in the previous paragraph. The inter- and intramolecular interactions were modelled using the GROMOS 43A2 force field (Schuler and van Gunsteren 2000) and the SPC water model (Berendsen et al. 1981). The SETTLE algorithm (Miyamoto and Kollman 1992) was used to constrain 89,037 water molecules and maintain their geometry.
The protein bond lengths involving hydrogen atoms were constrained by means of the LINCS algorithm (Hess et al. 1997). The system included 26 Na+ ions to maintain neutrality, and a constant temperature of 298 K was maintained using the V-rescale thermostat (Bussi et al. 2007) with a coupling time of 0.2 ps in the NVT ensemble. The residues were ionized considering a pH of 7.0. Lennard-Jones and electrostatic interactions were calculated within a cut-off of 1.4 nm. The particle-mesh Ewald (PME) technique was used for the treatment of electrostatic interactions (Essmann et al. 1995). Periodic boundary conditions together with the minimum image convention were applied with neighbour list actualization in 10 steps (Allen and Tildesley 1987). The equations of motion were integrated with a time step of 2.0 fs using the “leapfrog” algorithm (van Gunsteren and Berendsen 1988).
The limited memory Broyden-Fletcher-Goldfarb-Shanno quasi-Newtonian minimizer (l-bfgs) available in the GROMACS-4.6.5 package was used to optimize the protein structure in a step prior to solvent thermalization. The thermalization step itself was performed for 100 ps with the atomic coordinates frozen, and successive time step increases (0.0002 to 0002 ps). The root mean square deviation (RMSD) and radius of gyration were calculated using the g_rms and g_gyrate programs supplied with the GROMACS package and data supplied by coordinates collected during the MDS. The CRYSOL program (Svergun et al. 1995) was used to calculate the scattering curves of the high and the low resolution models of the simulated PKD-CBM44-XegA chimera.
Results
Rational design and spectroscopic characterization of the PKD-CBM44-XegA chimera
The coding sequences for the PKD-CBM44 and XegA were fused by overlap PCR to yield the PKD-CBM44-XegA coding sequence with a GGSGG peptide linker that allows a high degree relative inter-domain movement (Fig. 1). The linker sequence was chosen on the basis of previous studies showing that small rich glycine linkers allow mobility without disturbing the native protein conformation and consequently the activity of individual enzymes (Cota et al. 2013). Moreover, a short linker decreases the probability of proteolytic attack in the inter-domain region during heterologous protein expression and purification.
The chimera and the individual proteins were successfully produced as soluble proteins in E. coli (DE3) Rosetta and purified to homogeneity (Fig. 2a). The recombinant proteins have a molecular weight of 26, 30 and 56 kDa for XegA, PKD-CBM44 and the chimeric PKD-CBM44-XegA respectively, including the additional His-tag sequence added to the N-terminus. The far-UV CD spectra of all enzymes (Fig. 2b) presented two main bands at 200 nm (positive) and at 215–218 nm (negative), indicating that these proteins are rich in β-sheet structures and this is in agreement with the high β-sheet content observed in the crystal structures. A positive band around 230 nm is also observed in the PKD-CBM44 spectrum and can be associated with exciton coupling between aromatic residues, especially tryptophans, which are in proximity in the tertiary structure (Grishina and Woody 1994). An analysis of the three-dimensional structure of the protein (PDBID: 2C26) and the final PKD-CBM44-XEGA structure reveals that W194 and W198 are separated by a distance of ~10 Å and that W168 and W209 by a distance of less than 5 Å. This characteristic band has been previously observed in a GH16 hydrolase, the β-1,3-1,4-glucanase from Bacillus subtilis (Furtado et al. 2013) and CtCBM22 (Furtado and Ward, unpublished data). These results indicate that the conformation of the domains is not altered in the chimera and that the protein adopts a native-like structure, which is further corroborated by functional studies.

a SDS–PAGE analysis of the purified proteins. After Ni2+-affinity purification, proteins were analyzed on a 12 % SDS–polyacrylamide gel and subsequently stained with Coomassie blue. M, molecular weight standards (as indicated on the figure); lane 1, PKD-CBM44; lane 2, XegA; lane 3, PKD-CBM44-XegA chimera. b Far-UV circular dichroism spectra of the purified PKD-CBM44-XegA chimera (solid line), PKD-CBM44 (dashed line) and XegA (dotted line). See “Materials and methods” for further experimental details
The fusion of PKD-CBM44 with XegA increases the catalytic efficiency and thermal stability
The influences of pH and temperature on xyloglucanase activity of the recombinant XegA and chimeric PKD-CBM44-XegA were investigated (Fig. 3a, b), and both enzymes displayed a maximum activity at pH 5.5 and 60 °C. These values are similar to those observed for the native XegA expressed in A. nidulans (Damasio et al. 2012). Although presenting the same optimum catalytic temperature, the PKD-CBM44-XegA showed higher activity than XegA at temperatures above 60 °C, presenting 90 and 70 % of activity at 65 and 70 °C respectively, as compared to 50 e 35 % relative activity in XegA at these temperatures. This difference is also apparent in the thermostability results, in which XegA retained 35 % xyloglucanase activity after 5 min incubation at 60 °C, in comparison with the chimera that retained 70 % activity under the same conditions (Fig. 3c). On incubation at 50 °C, both enzymes retained 80–85 % activity after 150 min incubation. Incubation of pre-treated sugar cane bagasse (Bragatto et al. 2013) with the PKD-CBM44-XegA chimera increased reducing sugar release by 15 % after 12 h incubation at 50 °C as compared with the XegA alone or an equimolar mixture of XegA + CBM44 (data not shown). This increase is similar to that reported for the soluble xyloglucan (Fig. 3b), and since the effects of enzyme treatment are only apparent after long incubation times, we surmise that the xyloglucan is extracted from the biomass and subsequently hydrolyzed in the soluble form.

Characterization of the xyloglucanase activity for XegA and PKD-CBM44-XegA chimera. Effects of a pH and b temperature on the catalytic activity of the PKD-CBM44-XegA chimera (open circles) and XegA (closed circles). c Thermal inactivation of the PKD-CBM44-XegA chimera (open symbols) and XegA (closed symbols) at 50 °C (circles) and 60 °C (squares). All assays were performed in triplicate (error bars shown) using xyloglucan extracted from Hymenaea courbaril seeds as substrate. See “Materials and methods” for further experimental details
Kinetic parameters for the xyloglucanase activity were determined for both XegA and PKD-CBM44-XegA at 60 °C and pH 5.5. The specific activities were measured in U nmol−1 of protein, and Table 3 presents the values of the measured kinetic parameters. The chimeric PKD-CBM44-XegA displayed V max and k cat values 1.25-fold higher than the parental XegA and a slight decrease in the K M value from 1.5 and 1.3 mg mL−1 respectively, and as a result, the catalytic efficiency (k cat/K M) of xyloglucan hydrolysis was 30 % greater for the chimera.
To evaluate the effect of the fusion of the PKD-CBM44 domains on the XegA cleavage pattern of H. courbaril xyloglucan, the hydrolysis products of parental enzyme and chimera were investigated by mass spectrometry (Fig. 4a, b). The profile of the hydrolysis products were identical for both enzymes and is consistent with the function proposed for the CBM that is to maintain the proximity of catalytic domain to the carbohydrate, without altering its mode of catalysis. The oligomers generated by hydrolysis with XegA and PKD-CBM44-XegA were the same as those previously reported (Tine et al. 2006) using the Cel12A (EGIII) endoglucanase from Trichoderma viride and include the XXXXG oligosaccharide, a typical H. courbaril sequence not present in Tamarindus indica xyloglucan (Buckeridge et al. 1997).

Electrospray ionization-mass spectrometry of oligosaccharides derived from Hymenaea courbaril seed xyloglucan after hydrolysis with either a the PKD-CBM44-XegA chimera or b XegA. Molecular masses of the hydrolysis products together with the corresponding oligosaccharide are shown, where the letter G represents an unbranched β-D-glucopyranose and the letter X represents a branched [(1→6)-α-D-xylopyranose]-β-D-glucopyranose
The inactive dimeric arrangement of XegA
Previous studies have shown that the XegA expressed in A. niveus is catalytically inactive at low temperature (Damasio et al. 2012), and with the goal of further understanding this effect, the crystal structure of XegA was solved at 2.5 Å resolution. The crystal contained two XegA monomers per asymmetric unit and 42 % solvent content. The XegA displays the canonical jelly roll fold with the substrate binding cleft and active site located in the concave face (Fig. 5a). In common with other GH12 members, XegA performs hydrolysis by a two-step mechanism, in which Glu133 acts as the nucleophile and Glu219 acts as the acid/base, with retention of the anomeric carbon configuration of the glycone group (Fig. 5b). Other key residues conserved in the active site of structurally characterized GH12 enzymes include Asp115 and Asp119 (Fig. 5b, supplementary Fig. S1), which play a crucial role in maintaining the pKa and orientation of the catalytic residues (Khademi et al. 2002). In addition, Trp41 and Trp26 stack against sugar moieties at −2 and −4 subsites (Fig. 5b) of the glucose backbone and function to immobilize the substrate for nucleophilic attack (Gloster et al. 2007; Yoshizawa et al. 2012). The Tyr37 residue stacks against xylose side chain at −3 subsite (Fig. 5b, supplementary Fig. S1), and replacement of this residue by alanine increases the hydrolysis of other polysaccharides, such as β-glucan, reducing the enzyme specificity (Yoshizawa et al. 2012), and this tyrosine has therefore been considered a key residue for xyloglucan recognition by Xegs.

Crystal structure of XegA. a Canonical GH12 β-jellyroll fold showing the β-sheets in green and the α-helices in blue. b Active site residues involved in substrate recognition and catalysis. The oligosaccharide was docked using the atomic coordinates of a GH12 Xeg from Aspergillus aculeatus complexed with a xyloglucan fragment (PDBID: 3VL9). c The XegA dimer present in the crystallographic asymmetric unit, in which one molecule is represented as a surface coloured according to electrostatic potential and the other as a ribbon cartoon. Dimer formation occludes substrate access to the enzyme active sites. d Details of the XegA dimer interface, which is stabilized by hydrophobic (left) and polar (right) interactions
Analyses of the protein-protein interface of the two molecules present in the asymmetric unit of XegA crystal using PISA (http://www.ebi.ac.uk/msd-srv/prot_int/pistart.html) suggest the formation of a stable dimeric structure with a buried area of 1480 Å2 and ΔGdiss of 5.2 kcal mol−1. One XegA molecule is rotated 180° with respect to the other, implying that the two protein molecules are oriented face to face (Fig. 5c). The dimer is stabilized by hydrophobic (between Trp26A and Tyr132B, Trp26A and Leu209B, Ala44A and Leu168B and Trp41 and Pro170, see Fig. 5d, left) and polar interactions including direct and water-mediated hydrogen bonds (Fig. 5d, right). At the present time, the biological implications of XegA dimerization and inactivation are incompletely understood.
The PKD-CBM44-XegA chimera is a compact monomer in solution
SAXS experiments were performed in order to experimentally estimate the structural arrangement of the three domains that compose the PKD-CBM44-XegA chimera. Although XegA is a dimer, as shown previously (Damasio et al. 2012) and in the present crystal structure, the chimera is a monomer in solution with a maximum molecular dimension of ~9.5 nm and a radius of gyration of ~3 nm (Fig. 6a, inset). The averaged low-resolution envelope presented three well-defined lobes into which the structures of PKD, CBM44 and XegA could be readily fitted (Fig. 6b). The relative positions of the three domains so obtained were used as the starting model for molecular dynamics simulations of the chimera. The values for the RMSD increased rapidly over the initial 16 ns of the simulation (Supplementary Fig. S2A) and thereafter stabilized at a value of 0.9–1.0 nm. The 50-ns simulated model preserved all characteristics observed in the arrangement obtained from SAXS data, and its examination indicates that multiple interdomain interactions are formed between the three domains (Fig. 6b, c). These inter-domain contacts suggest that the relative movement between the three domains is restricted and that the orientation between the CBM44 and XEG domains is maintained at least over the time course of the simulation. The contacts between the PKD and CBM44 domains may suggest that in the complete C. thermocellum CelJ protein, the relative mobility between these two domains is restricted and that the PKD domain serves as a linker to regulate the relative orientation and distance between the CBM44 and catalytic domains.

PKD-CBM44-XegA analysis by small-angle X-ray scattering (SAXS) and molecular dynamics simulations (MDS). a Experimental SAXS curve obtained for PKD-CBM44-XegA (open circles) and calculated scattering curves for high (blue line) and low (red line) resolution structural models using the atomic coordinates of the chimera from MDS. Inset: Normalized distance distribution p(r) function. b Different views of the SAXS-derived envelope (white surface) superposed on the atomic coordinates of the PKD-CBM44-XegA chimera from MDS, showing the PKD (purple ribbon), CBM44 (blue ribbon) and XegA (green ribbon) domains. c Structural model of PKD-CBM44-XegA (white cartoon) highlighting key residues in the XegA substrate binding and active sites (solid spheres, carbons in green), which are in alignment with the xyloglucan binding cleft of CBM44. Key residues involved in carbohydrate binding in CBM44 are shown as solid spheres with carbon atoms in blue
In this model, the carbohydrate-binding surfaces of the CBM44 and XegA domains are aligned to form an extended binding cleft (Fig. 6c). The carbohydrate-binding surfaces of XegA includes the active site, and among other residues includes the catalytic acids Glu133 and Glu219, tryptophan residues Trp26 and Trp41 that interact with the main chain glucopyranoside residues, and Tyr37 that interacts with the xylose side chain (Fig. 6c). The carbohydrate-binding cleft of PKD-CBM44 is formed by several polar amino acid residues, in which the hydrophobic platform provided by three tryptophans, Trp189, Trp194 and Trp198, is essential for the carbohydrate recognition and binding (Najmudin et al. 2006).
Discussion
In the present study, we have reported that the chimeric enzyme comprised of the PKD-CBM44 domains of the CelJ protein from C. thermocellum with the GH12 XegA from A. niveus presents an increased catalytic efficiency as compared to the XegA catalytic domain alone, although the cleavage pattern against xyloglucan extracted from H. courbaril seeds, optimum catalytic temperature and pH optima of the chimeric and parental XegA enzymes remained essentially unaltered.
The chimera showed highest xyloglucanase activity at temperatures above 60 °C, which is higher than for the parental enzyme, and is probably due to structural stabilization resulting from the contacts with the PKD and CBM domains. The PKD domain derives its name from the extracellular segment of cell surface glycoprotein polycystin-1 that is encoded by the pkd1 gene (Bycroft et al. 1999). The PKD domain has also been identified in a number of bacterial glycoside hydrolases; however, the function in prokaryotes remains poorly understood. It has been reported that the PKD domain from the chitinase A of Alteromonas sp. strain O-7 mediates chitin binding through tryptophan residues (Orikoshi et al. 2005). The PKD domain in the celJ gene of C. thermocellum displays an immunoglobulin-like β-sandwich fold comprising 3- and 4-stranded β-sheets packed face to face (Najmudin et al. 2006), and our MDS results demonstrate protein-protein contacts between the PKD and CBM44 domains which in the intact native CelJ may play a role in the overall cellulosome function. Although removal of the PKD domain had no effect on the affinity of the CBM44 for xyloglucan (Najmudin et al. 2006), our MDS results indicate that the PKD and CBM44 form an interdomain interface with the XegA which may contribute to the stability of the chimera. Thermal stabilization of the catalytic domain by fusion with a CBM has previously been reported for the Cryptococcus sp. S-2 lipase (Thongekkaew et al. 2012). This enzyme was fused to a CBD (cellulose binding domain) from a H. reesei cellobiohydrolase retaining 100 % activity after 120 min incubation at 50 °C, whereas the native enzyme is not stable at this temperature (Thongekkaew et al. 2012). In another study, a β-mannanase from Aspergillus usamii fused to a CBM1 (Tang et al. 2013) also presented a significant increase in optimum catalytic temperature and thermostability as compared to the parental enzyme, showing that this is a robust approach to increase of thermotolerance of catalytic domains.
Kinetics studies showed a 30 % increase in the catalytic efficiency for xyloglucan hydrolysis of the chimeric enzyme. The altered catalytic proprieties of PKD-CBM44-XegA are unusual since most studies have shown that the fusion of a CBM to catalytic domains enhanced their activities only against insoluble substrates (Charnock et al. 2000). A significant increase in catalytic activity of a xylanase from Thermotoga maritima against soluble xylan after fusion with a CBM2 has also been observed (Kittur et al. 2003); however, such reports are uncommon in the literature. Indeed, the GH11 xylanase from A. niger fused with the CBM from thermophilic T. maritima (TmCBM9-1_2) showed a 4.2-fold increase in specific activity against the insoluble oat-spelt xylan as compared to on soluble birchwood xylan (Liu et al. 2011).
The crystal structure of the XegA domain was solved to a resolution of 2.5 Å, and showed a homodimeric structure in which the interface between the two protein molecules occludes substrate access to the enzyme active site supporting previous studies in which dimerization impairs catalytic activity. In contrast, the PKD-CBM44-XegA chimera is a monomer in solution, where the compact structure shown by SAXS is stabilized by a network of interdomain contacts as predicted by molecular dynamics simulations.
As previously noted (Najmudin et al. 2006), the CBM44 domain shares high structural similarity with the xylan-binding CBM15 family, and the crystal structure of the CBM15 from xylanase 10C from Pseudomonas cellulosa has previously been solved as a complex with xylopentose (Szabo et al. 2001). Superposition of the CBM15-xylopentose structure with the CBM44 domain in the simulated chimera reveals that many binding cleft residues are structurally conserved including the Trp189 and Trp194 in the CBM44 with Trp176 and Trp181 residues in the CBM15, respectively (data not shown). By analogy, this implies that the orientation of polysaccharide binding is also conserved between the two CBMs and since the reducing end of the xylopentose in the superposed CBM15 structure points towards the −4 subsite in the substrate binding cleft of XegA in the chimera structure. Then, we presume that the direction of the xyloglucan chain is maintained between the CBM44 domain and the XegA domain substrate binding clefts.
The favourable alignment of the polysaccharide-binding sites of CBM44 and XegA domains indicates that in the PKD-CBM44-XegA chimera, the CBM-xyloglucan interaction facilitates the positioning of the substrate in the active site of the hydrolase, and the relative orientations of domains in the PKD-CBM44-XegA results in the favourable alignment of the CBM44 xyloglucan binding cleft with the substrate binding cleft of the XegA, and we suggest that this structural feature underlies the improved catalytic performance of the chimera.
References
Allen MP, Tildesley DJ (1987) Computer simulation of liquids. Claredon Press, Oxford
Benko Z, Siika-aho M, Viikari L, Reczey K (2008) Evaluation of the role of xyloglucanase in the enzymatic hydrolysis of lignocellulosic substrates. Enzym Microb Technol 43(2):109–114
Berendsen HJC, Postma JPM, van Gunsteren WF, Hermans J (1981) Interaction models for water in relation to protein hydration. Intermol Forces:331–342
Bragatto J, Segato F, Squina FM (2013) Production of xylooligosaccharides (XOS) from delignified sugarcane bagasse by peroxide-HAc process using recombinant xylanase from Bacillus subtilis. Ind Crop Prod 51:123–129
Buckeridge MS, Crombie HJ, Mendes CJM, Reid JSG, Gidley MJ, Vieira CCJ (1997) A new family of oligosaccharides from the xyloglucan of Hymenaea coubaril L. (Leguminosae) cotyledons. Carbohydr Res 303(2):233–237
Bussi G, Donadio D, Parrinello M (2007) Canonical sampling through velocity rescaling. J Chem Phys 126(1):014101
Bycroft M, Bateman A, Clarke J, Hamill SJ, Sandford R, Thomas RL, Chothia C (1999) The structure of a PKD domain from polycystin-1: implications for polycystic kidney disease. EMBO J 18(2):297–305
Carpita NC, McCann MC (2000) The cell wall. In: Buchanan BB, Gruissem W, Jones R (eds) Biochemistry and molecular biology of plants. Amercian Society of Plant Physiologists, Rockville, pp 52–109
Charnock SJ, Bolam DN, Turkenburg JP, Gilbert HJ, Ferreira LMA, Davies GJ, Fontes CMGA (2000) The X6 “thermostabilizing” domains of xylanases are carbohydrate-binding modules: structure and biochemistry of the Clostridium thermocellum X6b domain. Biochem 39(17):5013–5021
Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D 66:12–21
Cota J, Oliveira LC, Damasio ARL, Citadini AP, Hoffmam ZB, Alvarez TM, Codima CA, Leite VBP, Pastore G, de Oliveira-Neto M, Murakami MT, Ruller R, Squina FM (2013) Assembling a xylanase-lichenase chimera through all-atom molecular dynamics simulations. Biochim Biophys Acta Protein Proteomics 1834(8):1492–1500
Damasio ARL, Ribeiro LFC, Ribeiro LF, Furtado GP, Segato F, Almeida FBR, Crivellari AC, Buckeridge MS, Souza TACB, Murakami MT, Ward RJ, Prade RA, Polizeli MLTM (2012) Functional characterization and oligomerization of a recombinant xyloglucan-specific endo-β-1,4-glucanase (GH12) from Aspergillus niveus. Biochim Biophys Acta Protein Proteomics 1824(3):461–467
Davies GJ, Wilson KS, Henrissat B (1997) Nomenclature for sugar-binding subsites in glycosyl hydrolases. Biochem J 321(2):557–559
Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of Coot. Acta Crystallogr D 66:486–501
Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG (1995) A smooth particle mesh Ewald method. J Chem Phys 103(19):8577–8593
Furtado GP, Ribeiro LF, Lourenzoni MR, Ward RJ (2013) A designed bifunctional laccase/β-1,3-1,4-glucanase enzyme shows synergistic sugar release from milled sugarcane bagasse. Protein Eng Des Sel 26(1):15–23
Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A (2005) Protein identification and analysis tools on the ExPASy server. In: Walker JM (ed) The proteomics protocols handbook. Humana Press, New York, pp 571–607
Gloster TM, Ibatullin FM, Macauley K, Eklöf JM, Roberts S, Turkenburg JP, Bjørnvad ME, Jørgensen PL, Danielsen S, Johansen KS, Borchert TV, Wilson KS, Brumer H, Davies GJ (2007) Characterization and three-dimensional structures of two distinct bacterial xyloglucanases from families GH5 and GH12. J Biol Chem 282(26):19177–19189
Grishina IB, Woody RW (1994) Contributions of tryptophan side-chains to the circular-dichroism of globular proteins—exciton couplets and coupled oscillators. Faraday Discuss 99:245–262
Guex N, Peitsch MC (1997) SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18(15):2714–2723
Guillen D, Sanchez S, Rodriguez-Sanoja R (2010) Carbohydrate-binding domains: multiplicity of biological roles. Appl Microbiol Biotechnol 85(5):1241–1249
Hammersley AP, Brown K, Burmeister W, Claustre L, Gonzalez A, McSweeney S, Mitchell E, Moy JP, Svensson SO, Thompson AW (1997) Calibration and application of an X-ray image intensifier/charge-coupled device detector for monochromatic macromolecular crystallography. J Synchrotron Radiat 4:67–77
Hess B, Bekker H, Berendsen HJC, Fraaije JGEM (1997) LINCS: a linear constraint solver for molecular simulations. J Comput Chem 18(12):1463–1472
Hess B, Kutzner C, van der Spoel D, Lindahl E (2008) GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput 4(3):435–447
Ichinose H, Araki Y, Michikawa M, Harazono K, Yaoi K, Karita S, Kaneko S (2012) Characterization of an endo-processive type xyloglucanase having a β-1,4-glucan-binding module and an endo-type xyloglucanase from Streptomyces avermitilis. Appl Environ Microbiol 78(22):7939–7945
Ishida T, Yaoi K, Hiyoshi A, Igarashi K, Samejima M (2007) Substrate recognition by glycoside hydrolase family 74 xyloglucanase from the basidiomycete Phanerochaete chrysosporium. FEBS J 274(21):5727–5736
Khademi S, Zhang DC, Swanson SM, Wartenberg A, Witte K, Meyer EF (2002) Determination of the structure of an endoglucanase from Aspergillus niger and its mode of inhibition by palladium chloride. Acta Crystallogr D 58:660–667
Kirk O, Borchert TV, Fuglsang CC (2002) Industrial enzyme applications. Curr Opin Biotechnol 13(4):345–351
Kittur FS, Mangala SL, Rus’d AA, Kitaoka M, Tsujibo H, Hayashi K (2003) Fusion of family 2b carbohydrate-binding module increases the catalytic activity of a xylanase from Thermotoga maritima to soluble xylan. FEBS Lett 549(1–3):147–151
Liu LW, Cheng J, Chen HG, Li XQ, Wang SY, Song AD, Wang MD, Wang B, Shen JW (2011) Directed evolution of a mesophilic fungal xylanase by fusion of a thermophilic bacterial carbohydrate-binding module. Process Biochem 46(1):395–398
Luis AS, Venditto I, Temple MJ, Rogowski A, Basle A, Xue J, Knox JP, Prates JAM, Ferreira LMA, Fontes CMGA, Najmudin S, Gilbert HJ (2013) Understanding how noncatalytic carbohydrate binding modules can display specificity for xyloglucan. J Biol Chem 288(7):4799–4809
Miller GL (1959) Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal Chem 31(3):426–428
Miyamoto S, Kollman PA (1992) Settle—an analytical version of the shake and rattle algorithm for rigid water models. J Comput Chem 13(8):952–962
Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D 53:240–255
Najmudin S, Guerreiro CIPD, Carvalho AL, Prates JAM, Correia MAS, Alves VD, Ferreira LMA, Romao MJ, Gilbert HJ, Bolam DN, Fontes CMGA (2006) Xyloglucan is recognized by carbohydrate-binding modules that interact with β-glucan chains. J Biol Chem 281(13):8815–8828
Okada H, Mori K, Tada K, Nogawa M, Morikawa Y (2000) Identification of active site carboxylic residues in Trichoderma reesei endoglucanase Cel12A by site-directed mutagenesis. J Mol Catal B Enzym 10(1–3):249–255
Orikoshi H, Nakayama S, Hanato C, Miyamoto K, Tsujibo H (2005) Role of the N-terminal polycystic kidney disease domain in chitin degradation by chitinase A from a marine bacterium, Alteromonas sp. strain O-7. J Appl Microbiol 99(3):551–557
Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. Macromol Crystallogr A 276:307–326
Powlowski J, Mahajan S, Schapira M, Master ER (2009) Substrate recognition and hydrolysis by a fungal xyloglucan-specific family 12 hydrolase. Carbohydr Res 344(10):1175–1179
Schuler LD, van Gunsteren WF (2000) On the choice of dihedral angle potential energy functions for n-alkanes. Mol Simul 25(5):301–319
Svergun DI (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J Appl Crystallogr 25:495–503
Svergun DI (1999) Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys J 76(6):2879–2886
Svergun D, Barberato C, Koch MHJ (1995) CRYSOL—a program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J Appl Crystallogr 28:768–773
Szabo L, Jamal S, Xie H, Charnock SJ, Bolam DN, Gilbert HJ, Davies GJ (2001) Structure of a family 15 carbohydrate-binding module in complex with xylopentaose. Evidence that xylan binds in an approximate 3-fold helical conformation. J Biol Chem 276(52):49061–49065
Tang CD, Li JF, Wei XH, Min R, Gao SJ, Wang JQ, Yin X, Wu MC (2013) Fusing a carbohydrate-binding module into the Aspergillus usamii β-mannanase to improve its thermostability and cellulose-binding capacity by in silico design. PLoS One 8(5):e64766
Thongekkaew J, Ikeda H, Iefuji H (2012) Increases thermal stability and cellulose-binding capacity of Cryptococcus sp. S-2 lipase by fusion of cellulose binding domain derived from Trichoderma reesei. Biochem Biophys Res Com 420(1):183–187
Tine MAS, Silva CO, de Lima DU, Carpita NC, Buckeridge MS (2006) Fine structure of a mixed-oligomer storage xyloglucan from seeds of Hymenaea courbaril. Carbohydr Polym 66(4):444–454
Vagin A, Teplyakov A (2010) Molecular replacement with MOLREP. Acta Crystallogr D 66:22–25
van Gunsteren WF, Berendsen HJC (1988) A leap-frog algorithm for stochastic dynamics. Mol Simul 1(3):173–185
Volkov VV, Svergun DI (2003) Uniqueness of ab initio shape determination in small-angle scattering. J Appl Crystallogr 36:860–864
Wriggers W (2010) Using Situs for the integration of multi-resolution structures. Biophys Rev 2(1):21–27
Wurch T, Lestienne F, Pauwels PJ (1998) A modified overlap extension PCR method to create chimeric genes in the absence of restriction enzymes. Biotechnol Tech 12(9):653–657
Yoshizawa T, Shimizu T, Hirano H, Sato M, Hashimoto H (2012) Structural basis for inhibition of xyloglucan-specific endo-β-1,4-glucanase (XEG) by XEG-protein inhibitor. J Biol Chem 287(22):18710–18716
Acknowledgments
This work was supported by research grants FAPESP 2010/18850-2, CNPq 307795/2009-8 (RJW), FAPESP 2013/13309-0 and 2014/07135-1 (MTM), FAPESP 2012/20549-4 and 2013/18910-3 (ARLD), FAPESP stipends, FAPESP 2010/07133-8 (LR), and CAPES stipend (GPF).
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Gilvan P. Furtado and Camila R. Santos have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 779 kb)
Rights and permissions
About this article

Cite this article
Furtado, G.P., Santos, C.R., Cordeiro, R.L. et al. Enhanced xyloglucan-specific endo-β-1,4-glucanase efficiency in an engineered CBM44-XegA chimera. Appl Microbiol Biotechnol 99, 5095–5107 (2015). https://doi.org/10.1007/s00253-014-6324-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-014-6324-0