
Abstract
Acetate production is one of the most striking differences between Escherichia coli K12 and BL21 strains. Transcription of acetate metabolism genes is regulated. Additionally, acetyl-CoA synthetase, which activates acetate to acetyl-CoA, is regulated by post-translational acetylation. The aim of this study was to understand the contribution of reversible protein lysine acetylation to the regulation of acetate metabolism in E. coli BL21. The phenotypic differences between both strains were especially important in the presence of acetate. The high expression of acetyl-CoA synthetase (acs) in glucose exponential phase in BL21 allows the simultaneous consumption of acetate and glucose. Lack of catabolite repression also affected its post-translational regulator, the protein acetyltransferase (patZ). The effect of the deletion of cobB (encoding a sirtuin-like protein deacetylase) and patZ genes depended on the genetic background. The deletion of cobB in both strains increased acetate production and decreased growth rate in acetate cultures. The deletion of patZ in BL21 suppressed acetate overflow in glucose medium and increased the growth rate in acetate cultures. Differences on acetate overflow between BL21 and K12 strains are caused by many overlapping factors. Two major contributing effects were identified: (1) the expression of acs during exponential growth is not repressed in the BL21 strain due to concomitant cAMP production and (2) the acetyl-CoA synthetase activity is more tightly regulated by protein acetylation in BL21 than in the K12. Altogether these differences contribute to the lower acetate overflow and the improved ability of E. coli BL21 to consume this metabolite in the presence of glucose.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Undoubtedly, Escherichia coli is the best-known microbial cell factory. Although from the physiological point of view the K12 strain is the best characterized, several other strains which possess specific features that make them better candidates for the development of industrial processes are known. For instance, the BL21 strain is used for heterologous production of proteins that do not require post-translational processing (Baneyx 1999).
The BL21 and K12 strains differ in several aspects. DNA mobile elements (IS) are scattered at different locations in the genomes of both strains, occasionally interrupting specific ORFs. Some of the characteristics of the B strain lineage are explained by the lack of certain genes. This is the case of the absence of flagella because of the loss of the flagella gene cluster due to an IS1-associated 41-kbp deletion and the absence of two proteases (lon and ompT) also due to IS elements (Studier et al. 2009). Although genome sequencing allows understanding some specific features of these strains, it has not been enough to explain metabolic differences, especially in what refers to acetate metabolism (Jeong et al. 2009; Studier et al. 2009). The BL21 strain produces low amounts of acetate during growth on high-glucose-concentration media. Acetate accumulation is determinant for industrial applications of microorganisms, especially for the production of recombinant proteins, since it inhibits growth and decreases protein production yield (Kleman and Strohl 1994; De Mey et al. 2007). Moreover, inefficient glucose metabolization limits overall bioprocess yields (Lara et al. 2008; Negrete et al. 2013).
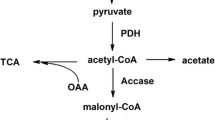
During many years, numerous studies have tried to unravel differences in acetate metabolism in the E. coli K and B strains, which cannot be directly related to the observed genomic differences (El-Mansi et al. 2006; Han et al. 2012; Jeong et al. 2009; Lara et al. 2008; Luli and Strohl 1990; Meier et al. 2012; Negrete et al. 2013; Phue et al. 2005; Phue and Shiloach 2004; Shiloach et al. 1996; Studier et al. 2009; Waegeman et al. 2012; Van de Walle and Shiloach 1998). Research efforts focussed on the two major metabolic pathways which could be responsible for the observed differences in acetate overflow. The first one is the super-pathway of acetate-producing and -consuming routes. There are two major pathways for acetate production in E. coli: the irreversible oxidative decarboxylation of pyruvate, catalysed by pyruvate oxidase (PoxB), and the high-capacity reversible phosphotransacetylase-acetate kinase pathway (Pta-AckA) (Wolfe 2005). There are two acetate assimilation pathways that differ in their affinity towards the substrate. E. coli uptakes high-concentration acetate using the Pta-AckA pathway. At low concentration, acetyl-CoA synthetase is used, given its high affinity towards its substrate (K m = 200 μM for acetate) (Wolfe 2005). Both assimilation pathways transform extracellular acetate into acetyl-CoA that is mainly metabolized by the glyoxylate shunt and the TCA cycle (Fischer and Sauer 2003; Maharjan et al. 2005; Renilla et al. 2012; Walsh and Koshland 1984). The glyoxylate shunt is an anabolic pathway, essential for growth on acetate as the sole carbon source, while the major function of the TCA cycle is the production of metabolic energy. Several authors have stated that a more active glyoxylate shunt may contribute to the lower acetate accumulation in the BL21 strain (Phue et al. 2005; Phue and Shiloach 2004; Son et al. 2011; Waegeman et al. 2011, 2012; Van de Walle and Shiloach 1998) and have reported that glyoxylate shunt genes are more expressed in the B strain (Phue and Shiloach 2004; Van de Walle and Shiloach 1998). Furthermore, flux thought the glyoxylate shunt in BL21 strain is higher than in the K12 (Waegeman et al. 2011, 2012). However, lower levels of glyoxylate shunt transcripts and proteins in the BL21 strain have been reported in a recent transcriptomic and proteomic comparison of BL21 and K12 strains (Marisch et al. 2013).
Interestingly, it has been recently proposed that differences in global regulatory mechanisms could be responsible for the differences on acetate production/consumption between E. coli K12 and BL21. In recent years, differences in the expression of the transcription factor Cra (Son et al. 2011), the levels of the metabolic signal cAMP (Marisch et al. 2013) and global protein acetylation (Weinert et al. 2013) between the K12 and BL21 strains have been described.
The post-translational acetylation of proteins in bacteria contributes to the regulation of bacterial metabolism, especially acetate metabolism (Bernal et al. 2014; Castaño-Cerezo et al. 2011; Gardner and Escalante-Semerena 2009; Mischerikow et al. 2009; Starai et al. 2002). The activity of acetyl-CoA synthetase (Acs) is regulated by acetylation of a specific lysine residue in the vicinity of the substrate binding pocket (Castaño-Cerezo et al. 2014; Crosby et al. 2012b; Gardner et al. 2006; Starai et al. 2002). Many prokaryotic protein acetyltransferases (which catalyse the transfer of an acetyl moiety from acetyl-CoA to the amino group of a lysine residue) belong to the Gcn5-like acetyltransferase family (Crosby et al. 2012a; Gardner and Escalante-Semerena 2008; Lima et al. 2012; Nambi et al. 2010; Thao and Escalante-semerena 2011). In E. coli, the best-known protein acetyltransferase is PatZ (Castaño-Cerezo et al. 2011; Castaño-Cerezo et al. 2014). Acetylation is reversed by deacetylases (Castaño-Cerezo et al. 2011; Castaño-Cerezo et al. 2014; Hayden et al. 2013; Starai et al. 2002; Zhao et al. 2004).
In E. coli K12, acetate metabolism is regulated by the protein acetyltransferase PatZ and the sirtuin CobB (Castaño-Cerezo et al. 2011; Weinert et al. 2013). It has been described that non-enzymatic acetylation of proteins by acetyl-phosphate is the main mechanism of protein acetylation in E. coli. These authors also described that protein acetylation was more abundant in the K12 strain than in BL21 because of elevated acetyl phosphate levels linked to higher acetate production (Weinert et al. 2013). However, the contribution of protein acetylation to the metabolism of E. coli BL21 was not described.
The main objective of this study is to shed light on the role of lysine acetylation in the metabolic differences observed between both E. coli K and B strains, especially focussing on their contribution to acetate metabolism. The occurrence of protein acetylation in E. coli K and B strains will be analysed under different environmental conditions. Responses to the deletion of the acetyltransferase patZ and the NAD+-dependent deacetylase cobB will be described.
Materials and methods
Strains and culture conditions
The E. coli K12 (BW25113) and BL21(DE3) strains and their knockout derivatives used in this study are listed in Table S1. The K12 strains were supplied by the KO collection (Keio University, Japan) (Baba et al. 2006). E. coli BL21(DE3) was purchased from Sigma Aldrich (St. Louis, MO, USA), and its knockout strains were constructed using the phage lambda Red recombinase method (Datsenko and Wanner 2000). The plasmids used for the construction of mutants are listed in Table S2.
For the characterization experiments, bacteria were grown using standard M9 minimal medium (pH 7.4) containing 10 mM (NH4)2SO4, 8.5 mM NaCl, 40 mM Na2HPO4, 20 mM KH2PO4, 185 μM FeCl3, 175 μM EDTA, 7 μM ZnSO4, 7 μM CuSO4 · 5 H2O, 7 μM MnSO4 , 7 μM CoCl2 , 1 mM MgSO4, 0.1 mM CaCl2 and 1 μM thiamine · HCl. As carbon source, 20 mM glucose or 10–60 mM acetate was used. Aerobic 50-mL batch cultures were grown in 500-mL flasks at 37 °C on a rotary shaker at 250 rpm. Cultures were inoculated to an initial optical density (OD600) of 0.05 units with exponentially growing precultures.
Enzyme activities
Approximately 20 mg of E. coli cells was harvested from each condition (glucose exponential phase, glucose stationary phase or acetate exponential phase) and centrifuged for 10 min at 4 °C (7000×g). The supernatant was removed, and cells were resuspended in 50 mM phosphate buffer pH 7.5. For the lysis, cells were sonicated on ice (3 cycles, 20 s each), with a probe of 3 mm in diameter of Vibra Cell VC 375 ultrasonic processor (Sonics Materials, Danbury, CT, USA). Protein extract was clarified by centrifugation for 15 min at 20,000×g at 4 °C to remove cell debris. The supernatant was collected after centrifugation and kept at −80 °C until use. Protein concentration was determined by bicinchoninic acid method (BCA) using a commercial kit (Pierce BCA Kit Pierce, Thermo Fisher Scientific, Rockford, IL, USA).
The enzyme activity assays were optimized for the conditions and media. All measurements were carried out in a microplate spectrophotometer Synergy HT (BioTek, Winooski, VT, USA). Specific enzyme activity was defined as μmol of substrate consumed per minute and mg of protein (U/mg). All enzyme activities were measured at 37 °C.
Isocitrate dehydrogenase (Icdh)
The method was described by Aoshima et al. (2003). The measurement buffer was 65 mM potassium phosphate (pH 7.5). The reaction components were 5 mM MgCl2, 2 mM NADP+ and 2.5 mM d,l-isocitrate. The enzyme activity was followed by the increase in NADPH absorbance at 340 nm (ε NADPH = 6.220 M−1 cm−1). One unit of enzyme activity was that required for the generation of 1 μmol of NADPH per minute.
Isocitrate lyase (AceA)
The assay was that described by Aoshima et al. (2003) using the same buffer as above. The reaction mixture was composed of 5 mM MgCl2, 20 mM phenylhydrazine and 5 mM d,l-sodium isocitrate. The enzyme activity was followed by the increase in absorbance at 324 nm due to the reaction of the glyoxylate produced with phenylhydrazine (ε glyoxylate-phenylhydrazone = 16.8 M−1 cm−1). One unit of enzyme activity was taken as that needed to generate 1 μmol of adduct per minute.
Acetyl-CoA synthetase (Acs)
The method used was that established by Lin et al. (2006). The measurement buffer was 100 mM Tris–HCl (pH 7.8). The reaction mixture contained 5 mM d,l-malate, 1 mM ATP, 2.5 mM MgCl2, 0.1 mM coenzyme A, 3 mM NAD+, 2.5 U/mL malate dehydrogenase, 1.25 U/mL citrate synthase and 100 mM sodium acetate. The acetyl-CoA synthetase activity was followed as the increase in NADH absorbance at 340 nm (ε NADH = 6.220 M−1 cm−1). Enzyme activity unit was defined as the enzyme generating 1 μmol of NADH per minute.
Phosphotransacetylase (Pta)
The assay was carried out as in Peng et al. (Peng and Shimizu 2003). The measurement buffer was 250 mM Tris–HCl, pH 7.8. The reaction components were 1 mM MgCl2, 10 mM d,l-malic acid, 3 mM NAD+, 0.5 mM coenzyme A, 2.5 U/mL malate dehydrogenase, 1.25 U/mL citrate synthase and 10 mM acetyl-phosphate. The enzyme activity was followed as the increase in NADH absorbance at 340 nm (ε NADH = 6.220 M−1 cm−1), one unit being taken as the enzyme required for the generation of 1 μmol of NADH per minute.
Acetate kinase (AckA)
The assay was carried as described by Bergmeyer and colleagues with minor modifications (Bergmeyer et al. 1974). The measurement buffer was 250 mM Tris–HCl, pH 7.8. The reaction components were 10 mM MgCl2, 2 mM ADP, 4 mM of NADP+, 10 mM glucose, 2 U/mL hexokinase, 1 U/mL glucose 6-phosphate dehydrogenase and 10 mM acetyl-phosphate. The enzyme activity was followed as the increase in NADPH absorbance at 340 nm (ε NADH = 6.220 M−1 cm−1), one unit being taken as the enzyme required for the generation of 1 μmol of NADPH per minute.
Relative gene expression
Total RNA was isolated from 3×108 cells by Vantage Total RNA purification kit (ORIGENE, MD, USA) according to the manufacturer’s recommendations. Additionally, DNase I digestion of the isolated RNA was performed using the RNase-Free DNase Set (QIAGEN Ibérica, Madrid, Spain) to avoid DNA interferences during PCR steps. Isolated RNA purity and concentration were assessed in a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). RNA quality was evaluated by microfluidic capillary electrophoresis on an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA) using Agilent RNA 6000 Pico kit. Chips were prepared and loaded according to the manufacturer’s instructions. Isolated RNA was stored at−80 °C for no longer than 3 days.
One microgram of high-quality RNA [rRNA ratio (23S/ 16S) at 1.6, RNA integrity number (RIN) > 9.0 and A 260/A 280 ratio > 2.0] was reverse-transcribed with TaqMan Reverse Transcription Reagents (Applied Biosystems, Foster City, CA, USA) according to the manufacturer’s protocol and stored at−20 °C before use. Briefly, a 25-mL reaction mixture was incubated in a Peltier Thermal Cycler 200 (MJ Research Inc., Boston, MA, USA) for 10 min at 25 °C, 30 min at 48 °C and 5 min at 95 °C.
The primers used in this work (Table S3) were designed using the Primer Express Software v3.0 (Applied Biosystems, Foster City, CA, USA) and ordered from Sigma Aldrich (St. Louis, MO, USA). The polA, dnaA and rrsA genes (encoding DNA polymerase I, transcriptional dual regulator and 16S ribosomal RNA, respectively) were used as internal control for relative quantification.
Quantitative PCR was performed in a 7300 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) using Power SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA) according to the manufacturer’s instructions. Briefly, 25-μl reaction mixtures, with 5 ng template cDNA and 7.5 pmol of each primer, were incubated for 2 min at 50 °C, 10 min at 95 °C and 40 PCR cycles (15 s at 95 °C and 1 min at 60 °C). An additional dissociation step (15 s at 95 °C, 30 s at 60 °C and 15 s at 95 °C) was added to assess non-specific amplification. PCRs were run in triplicate. Raw data were transformed into threshold cycle (C t) values. Relative gene expression for each condition compared with the control was calculated by the comparative C t method (∆∆C t).
Analytical methods
Growth analysis
To estimate cell concentration, cells were resuspended in 0.9 % NaCl, and absorbance was measured at 600 nm (Pharmacia Biotech NovaspecPlus Spectrophotometer, Amersham Biosciences, GE Healthcare Europe GmbH, Barcelona, Spain). A 600 values and dry cell weight were correlated.
HPLC analysis of metabolites
Extracellular metabolites (mainly acetate and glucose) were analysed by HPLC (Shimadzu Scientific Instruments, Columbia, MD, USA), equipped with differential refractive (Shimadzu Scientific Instruments, Columbia, MD, USA) and UV (Waters, Milford, MA, USA) detectors, using a cation-exchange column (HPX- 87H, Bio-Rad Labs, Hercules, CA, USA). The mobile phase was 15 mM H2SO4 at 0.5 mL min−1 flow rate and 45 °C.
Determination of growth kinetic parameters was carried out as described by Nielsen (2006).
Western blotting
A total of 20 μg of cell crude protein extract obtained as described above was resolved by SDS-PAGE in 10 % acrylamide gels. Proteins were blotted onto PVDF membranes, which were proofed against a rabbit anti-acetyl-lysine antibody (InmuneChem, Burnaby, Canada) according to the manufacturer’s instructions. A goat anti-rabbit antibody conjugated with HRP (Santa Cruz Biotechnology, Heidelberg, Germany) was used.
Results
Differences in the acetate metabolism in E. coli K and B strains
Phenotypic differences between E. coli K and B strains
Several differences were observed in batch cultures of E. coli K and B strains in glucose and acetate minimal media. The K12 strain grew faster in glucose cultures, while the specific growth rate of the BL21 strain was higher on acetate. The biomass yield of the K12 strain was higher in both culture conditions. Acetate production in glucose cultures was faster in the K12 strain, also reaching a higher concentration (Tables 1 and 2).
Lysine acetylation profiles and the expression of acs, patZ and cobB genes
The global pattern of protein acetylation of both strains was compared under different growth conditions by western blotting using an anti-acetyl-lysine antibody (Fig. 1). This post-translational modification was more abundant in the K12 strain, and the levels of acetylation were especially high in those conditions where the cells were consuming acetate (glucose stationary phase and acetate cultures).

Western blots showing protein lysine acetylation in protein crude cell extracts in E. coli K12 and BL21 strains. For this analysis, bacteria were grown and harvested at different growth phases and carbon source: glucose batch exponential phase (a), glucose batch stationary phase (b) and acetate batch exponential phase (c)
In order to assess if these differences in protein acetylation were due to differences in the expression of the genes involved in this post-translational modification, the expression of acetyl-CoA synthetase (acs), the protein acetyltransferase (patZ) and the sirtuin-like protein deacetylase (cobB) was measured in both strains during growth on glucose and acetate.
In glucose cultures, the expression of the acs gene was higher in BL21 in the exponential phase of growth, with the differences being less pronounced at the onset of the stationary phase. In acetate cultures, the expression of acs was similar in both strains during the exponential growth phase. A similar behaviour was observed in patZ gene expression in glucose cultures (Fig. 2 and Tables S4 and S5), whereas in acetate cultures patZ expression increased after the first 10 h of culture in the BL21 strain (Tables S4 and S5).

Differential gene expression of acs (a), cobB (b) and patZ (c) during glucose cultures in the K12 (1) and BL21 (2) strains. Differential gene expression is expressed with grey bars, bacterial growth with grey circles, acetate concentration in black squares and glucose concentration in white triangles
The expression pattern of cobB was similar between both strains, although its expression increased slightly in the stationary phase in the BL21 strain growing in glucose (Fig. 2). Interestingly, the analysis of the genetic environment of these genes in the BL21 strain revealed that nagK gene, which in E. coli K12 forms a transcriptional unit with cobB (Castaño-Cerezo et al. 2011), is interrupted by two IS elements (Karp et al. 2010; Keseler et al. 2013).
Catabolite repression of acetate consumption
The different expression profile of acs and patZ genes in the BL21 strain suggests that both genes are less tightly regulated by catabolite repression compared with the K12 strain. Also, a recent study showed that BL21 can co-consume glucose and acetate simultaneously, but not other combinations of carbon sources, such as glucose and lactose (Waegeman et al. 2012). For these reasons, growth of both strains on a mixture of glucose and acetate was characterized (Fig. 3). The presence of acetate at high concentrations inhibited the growth of the K12 strain (Table 3). Acetate was not consumed by the K12 strain until glucose was depleted, while 36 % of acetate was consumed by BL21 before glucose exhaustion (Fig. 3). These results indicate that acetate and glucose are simultaneously consumed by the BL21 strain, leading to a lower acetate production. Similar results have been previously reported (Waegeman et al. 2012).

Growth and metabolite production/consumption of the strains K12 (a), BL21 (d) and the knockout mutants cobB (b, e) and patZ (c, f) of each strain, respectively. All strains and mutants were grown aerobically in minimal media supplemented with two carbon sources, glucose (20 mM) and acetate (25 mM). In black circles is represented the biomass (g/L), in green triangles acetate and in pink squares glucose concentration in the media
Analysis of acetate metabolism enzyme activities
Enzymes related with acetate metabolism in E. coli were measured in glucose and acetate cultures (Fig. 4). Differences in the isocitrate dehydrogenase activity were almost negligible in glucose cultures, although its activity was higher in the K12 strain in acetate cultures (Fig. 4a). More differences were shown in isocitrate lyase (Fig. 4b). In all the conditions assayed, its specific activity was significantly higher in the K12 strain than in the BL21, especially in acetate cultures, which is in contrast with previous reports of fluxes in the glyoxylate shunt (Waegeman et al. 2012) .

Enzyme activities related to acetate metabolism measured in cell crude extracts of the E. coli strains BL21 (1) and K12 (2) and their knockout mutants ΔcobB and ΔpatZ. Cells were cultured under different conditions: glucose batch culture exponential phase (black), stationary phase (light grey) and acetate cultures exponential phase (dark grey). Experiments were performed at 20 mM glucose and 60 mM acetate, respectively. The enzymes measured were isocitrate dehydrogenase (a), isocitrate lyase (b), phosphotransacetylase (c), acetate kinase (d) and acetyl-CoA synthetase (e). Represented values are the average of the enzyme activities measured (n = 8). Error bars indicate the standard deviation of the measurements (n = 8). Statistical test (two-way ANOVA) was performed in order to calculate differential significance between mutants and their corresponding parent strains in each condition (p-value < 0.0001 (***) or <0.001 (**) or <0.01(*))
The activity of the main acetate-producing pathway in glucose cultures, the phosphotransacetylase acetate kinase (Pta-AckA) pathway, was clearly higher in E. coli BL21 compared with the K12 strain (Fig. 4c, d). Differences observed in acetate cultures were not significant. The activity of the acetate scavenging acetyl-CoA synthetase activity (Acs) was higher in the BL21 strain in acetate cultures, which is in agreement with its higher growth rate under these conditions (Fig. 4e).
Is lysine acetylation responsible for these metabolic differences?
Differences in the metabolism and the expression of the genes related to lysine acetylation (cobB and patZ) between the B and K strains have been described above. In order to determine if protein lysine acetylation contributes to the differences in the metabolism of these two strains, the physiological effect of the deletion of the genes related with this post-translational modification was studied.
Phenotypic differences between cobB and patZ deletion mutants in K12 and BL21 genetic backgrounds
The phenotypes of the ΔcobB and ΔpatZ mutants of the K12 strain have been previously described (Castaño-Cerezo et al. 2011). The phenotypic changes caused by the deletion of cobB and patZ in the BL21 strain were more dramatic than in the K12. In glucose cultures, acetate production rate increased more than sixfold in BL21 ΔcobB, while the increase was only twofold in K12 ΔcobB. Acetate production was not detected in the BL21 ΔpatZ mutant (Table 1).
In order to further understand the effects of the mutations on acetate metabolism, the mutants were tested to grow on acetate or acetate/glucose mixtures. Experiments with acetate minimal medium were performed at two different concentrations (10 and 60 mM) to distinguish between the two major acetate consumption pathways. The only active pathway for acetate assimilation at low concentrations is the high-affinity acetyl-CoA synthetase (Starai and Escalante-Semerena 2004), while at high acetate concentrations, acetate is also assimilated through the phosphotransacetylase-acetate kinase pathway (Castaño-Cerezo et al. 2011). The growth rate of the BL21 ΔpatZ mutant was higher compared to the parent strain at both acetate concentrations. This effect was especially evident at 10 mM acetate (Table 2). This differs from what was observed in the K12 strain, where the growth rate was slightly slower in the ΔpatZ mutant at both acetate concentrations (Castaño-Cerezo et al. 2011).
In acetate-glucose cultures, the phenotype of the knockout mutants showed the concurrent involvement of lysine acetylation and catabolite repression in the different acetate metabolism of the B and K strains (Fig. 3). We have previously shown the capability of the B strain of consuming glucose and acetate simultaneously. Despite this, the deletion of cobB gene in both K12 and BL21 strains led to acetate production even in these conditions (Table 3). In the case of the deletion of the patZ gene in the K12 and BL21 strains, their phenotypes were completely different. The K12 patZ mutant produced more acetate than the wild-type strain, while the BL21-derived patZ mutant consumed acetate faster than its parent strain (Table 3).
Lysine acetylation differences
Deletion of cobB and patZ affected protein acetylation in both E. coli strains. In glucose cultures, differences in the acetylation levels between all strains were almost negligible in the exponential growth phase (Fig. 5a). More differences were observed in the stationary phase of glucose cultures, with higher protein acetylation in both K and B ΔcobB mutants (Fig. 5b). In acetate cultures, the acetylation levels were different between mutants and strains. Although it has been previously described that protein acetylation level is higher in the cobB mutant under almost all conditions, the protein acetylation levels also increased in the patZ mutant. For each pair of mutants compared, acetylation was always higher in the K12 background. Interestingly, these differences in protein acetylation levels reflected the different growth behaviour and acetate metabolism of the strains, especially the higher growth rate in the BL21 patZ mutant.

Western blot of acetylated proteins under different genomic backgrounds and culture conditions. Bacteria were grown and harvested in glucose batch cultures at exponential phase (a), stationary phase (b) and acetate batch cultures exponential phase (c)
Effect of the deletion of cobB and patZ on enzyme activities related to acetate metabolism
The enzyme activities analysed previously were also measured in the ΔcobB and ΔpatZ mutants. The changes in the mutants were highly dependent on the genetic background, which further evidenced the differences between both strains.
The isocitrate dehydrogenase activity was higher in glucose cultures in the BL21 ΔpatZ mutant, while this mutation had no effect in the K12 genetic background in this condition. In acetate cultures, the differences were more noticeable, with higher activity in the K12 strain and its mutants (Fig. 4, a1–2).
The activity of isocitrate lyase was always higher in the K12 strain, except in the case of the BL21 ΔcobB mutant that showed higher activity in the stationary phase of glucose cultures. In acetate cultures, the presence of this enzyme is essential for cell growth; consequently, the activity was much higher than in glucose cultures in both strains. The most striking difference observed was the opposite effect of the deletion of cobB in both genetic backgrounds. In the BL21 ΔcobB mutant, isocitrate lyase activity was higher than in the parent strain (Fig. 4, b1–2), which can contribute to its better growth compared to the corresponding K12 mutant. We have recently found that acetylation of isocitrate lyase leads to partial inhibition of its activity in E. coli K12 (Castaño-Cerezo et al. 2014).
Pta-AckA is the major acetate-producing pathway in exponentially growing glucose cultures. Under this condition, differences in the Pta and AckA enzyme activities between the knockouts and their wild type were not significant in the exponential phase, and in the stationary phase they are not relevant due to the inability to consume the low acetate concentrations present in the media (Fig. 4c, d). In high-concentration acetate cultures, AckA activity was similar between both strains and knockouts, while the Pta enzyme activity was higher in the K12 strain and their knockouts and also higher compared with the AckA activity (Fig. 4d).
The acetyl-CoA synthetase activity was higher in acetate compared with glucose cultures, but it was also higher in all three BL21 strains compared with K12 strains in this condition. The differences between the BL21 knockouts and their parent strain were almost not noticeable in glucose cultures, whereas in acetate cultures the BL21 ΔcobB mutant showed a higher activity compared to its parent strain. On the other hand, higher differences were observed in K12 mutants in acetate cultures. The ∆cobB mutant showed half of the parent strain activity, while the ∆patZ mutant almost doubled the wild-type activity (Fig. 4, e1–2).
Discussion
It has been recently proposed that global mechanisms of metabolism and physiology regulation could be responsible for the differences on acetate production and consumption between E. coli strains belonging to K and B lineages (Marisch et al. 2013; Son et al. 2011; Weinert et al. 2013). In this work, we have shown that the regulation of acetate metabolism by protein acetylation and the inefficient catabolite repression of acetyl-CoA synthetase gene expression have a major contribution to the phenotype of E. coli BL21.
The expression of acs and patZ genes was higher in the B strain at the early exponential phase of glucose cultures. Both genes are regulated by CRP, which activates their expression in response to the elevation of cAMP levels upon glucose starvation. In order to assess whether differences in the promoter region upstream acs and patZ affecting the affinity for cAMP-CRP binding could be related to the differences observed in expression, the sequences of the CRP binding sites were analysed as described previously (Castaño-Cerezo et al. 2011). Although both regions were slightly different between strains, the scores of the CRP binding sites were the same for both strains (results not shown). The levels of cAMP during exponential growth are different between both strains (Marisch et al. 2013). The higher cAMP levels in E. coli BL21 evidences that acetate metabolism is not efficiently repressed in this strain during exponential growth and could be the major reason for the lower catabolite repression of acs and patZ in the BL21 strain (Fig. 6).

Scheme of the acetate metabolism regulation in the different E. coli strains, K12 (a) and BL21 (b). The acetate production/consumption futile cycle and its regulation in glucose batch cultures at the exponential phase is represented. Purple, red and yellow balls represent glucose, acetate and cAMP, respectively. a During exponential growth phase, E. coli K12 consumes glucose and produces acetate mainly by the Pta-AckA pathway. In this phase, CRP is inactive due to the absence of cAMP, and acetyl-CoA synthetase gene is not expressed. b During exponential growth phase, E. coli BL21 consumes simultaneously acetate and glucose. This specific feature of the B strain is attributed to the activation of the transcription factor CRP due to the higher cAMP concentration. This singularity leads to the expression of acs. This provokes acetate scavenging during glucose consumption, showing a lower acetate concentration in the media
Glucose-insensitive expression of acs in the BL21 strain allows scavenging the acetate overflown during exponential growth on glucose and simultaneous consumption of glucose and acetate on mixed substrate cultures (Fig. 6). The differences in the expression of acetyl-CoA synthetase during exponential growth on glucose explained the different phenotypes displayed when culture medium was supplemented with acetate. Cultures with glucose/acetate mixtures showed the detrimental effects of acetate on growth of the K12 strain (Table 3), while no detrimental effect was observed in the BL21 strain (Shiloach et al. 1996). Acetate uptake was glucose-repressed in the K12 strain, while the BL21 strain simultaneously consumed both carbon sources; in fact, the BL21 strain grew faster on acetate, which can be explained by the higher acetyl-CoA synthetase activity (Fig. 4e). Moreover, the higher differences in growth rates were observed in low-acetate-concentration cultures, where acetyl-CoA synthetase is the major responsible compound for acetate consumption (Castaño-Cerezo et al. 2011; Starai and Escalante-Semerena 2004), which supports this hypothesis.
On top of this mechanism of transcriptional regulation of acetyl-CoA synthetase gene expression, protein activity is regulated by post-translational acetylation. Acetyl-CoA synthetase is reversibly regulated by the acetylation of the side chain of K609. The modifications are catalysed by the protein acetyltransferase PatZ and the NAD+-dependent deacetylase CobB (Bernal et al. 2014; Castaño-Cerezo et al. 2011; Castaño-Cerezo et al. 2014; Starai et al. 2002). Deletion of patZ and cobB genes demonstrated the importance of protein acetylation on acetate metabolism in E. coli BL21. Acetylated (and thus inactive) acetyl-CoA synthetase accumulates as a result of the deletion of cobB, which leads to acetate accumulation. This demonstrated that wild-type E. coli BL21 produces more acetate than the K12 strain but efficiently scavenges it through the acetyl-CoA synthetase activity (Fig. 4).The importance of the acetylation of acetyl-CoA synthetase for the phenotype of E. coli BL21 on acetate was further demonstrated by increased growth of the strain upon deletion of patZ since acetyl-CoA synthetase remained active. Altogether this demonstrates that the low-acetate-producing metabolism of E. coli BL21 is highly dependent on the activity of acetyl-CoA synthetase and its transcriptional (cAMP-CRP) and post-translational (CobB and PatZ) regulators.
The simultaneous consumption of glucose and acetate by the BL21 strain contradicts a previous hypothesis. Differences in acetate production between the K and B strains have been proposed to cause differential protein lysine acetylation, being more abundant in the K12 strain than in the BL21 in the glucose exponential phase (Weinert et al. 2013). They stated that the abundance of protein acetylation in the K12 strain was due to the higher acetate overflow and attributed it to the higher production of acetyl-phosphate. Although acetate overflow is higher in the K12 strain, the BL21 cobB mutant evidences that the B strain produces similar or even higher amounts of acetate than the K strain but, simultaneously, it scavenges acetate efficiently. This simultaneous production and consumption of acetate has been previously reported in glucose-limited chemostat cultures of the K12 strain (Renilla et al. 2012; Valgepea et al. 2010). Altogether this indicates that the more abundant protein acetylation is unlikely caused by the higher acetate production.
Besides this, the role of the protein acetyltransferase PatZ (formerly YfiQ) is still to be deciphered. Deletion mutants in patZ show big phenotypic differences in several conditions. The higher patZ gene expression (this study) and PatZ protein levels (Weinert et al. 2013) in BL21 at the exponential phase of glucose cultures could suggest higher protein acetylation in this strain. However, the deletion of the protein-acetyltransferase in both strains did not decrease the acetylation in any condition (Fig. 5) (Weinert et al. 2013). Therefore, which is the reason of the different phenotype observed between the BL21 and K12 ΔpatZ mutants compared with their parent strains? Molecular studies in proteins homologous to PatZ from other bacteria demonstrate that it is only active on a small number of proteins, mostly acyl-CoA synthetases with conserved acetylation motifs (Crosby and Escalante-Semerena 2014). Therefore, PatZ is responsible for the acetylation of a reduced number of proteins. Nevertheless, the global effect on protein acetylation observed upon deletion of patZ indicates that the role of PatZ could be more important than previously described (Weinert et al. 2013) and may be involved in the control of chemical acetylation occurrence (Castaño-Cerezo et al. 2014). In fact, the higher “unspecific” acetylation of proteins in K12 might contribute to slowing down its growth in acetate cultures.
It is currently accepted that the phenotypic differences between the K and B strains are due to the sum of several factors, including genomic and regulatory differences. Although it is clear that the lax catabolite repression of acetate metabolism in the BL21 strain may be the cause for its lower acetate overflow, the deletion of cobB and patZ in the BL21 strain provoked sharp phenotypic effects, especially on acetate consumption. This indicates that protein lysine acetylation, modulated by these two proteins, has a major contribution to the regulation of acetate metabolism in the K12 and BL21 strains. Deletion of cobB converted BL21 in an acetate-producing strain, while deletion of patZ in BL21 further reduced acetate overflow and improved growth, confirming that acetate overflow and consumption are tightly regulated by protein acetylation. Differences observed in global protein lysine acetylation in K12 and BL21 strains could be caused by further differences in acetyl-CoA metabolism. Altogether this study has shown further insight into the different acetate metabolism in E. coli B and K strains, demonstrating the contribution of differential lysine acetylation and catabolite repression.
References
Aoshima M, Ishii M, Yamagishi A, Oshima T, Igarashi Y (2003) Metabolic characteristics of an isocitrate dehydrogenase defective derivative of Escherichia coli BL21(DE3). Biotechnol Bioeng 84:732–737. doi:10.1002/bit.10832
Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H (2006) Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006.0008
Baneyx F (1999) Recombinant protein expression in Escherichia coli. Curr Opin Biotechnol 10:411–21
Bergmeyer HU, Forster G, Bernt E (1974) Creatine kinase. Methods Enzym. Anal. pp 784–793
Bernal V, Castaño-Cerezo S, Gallego-Jara J, Ecija-Conesa A, de Diego T, Iborra JL, Cánovas M (2014) Regulation of bacterial physiology by lysine acetylation of proteins. N Biotechnol 31:586–95. doi:10.1016/j.nbt.2014.03.002
Castaño-Cerezo S, Bernal V, Blanco-Catalá J, Iborra JL, Cánovas M (2011) cAMP-CRP co-ordinates the expression of the protein acetylation pathway with central metabolism in Escherichia coli. Mol Microbiol 82:1110–1128
Castaño-Cerezo S, Bernal V, Post H, Fuhrer T, Cappadona S, Sánchez-Díaz NC, Sauer U, Heck AJR, Altelaar AFM, Cánovas M (2014) Protein acetylation affects acetate metabolism, motility and acid stress response in Escherichia coli. Mol Syst Biol 10(11):762
Crosby HA, Escalante-Semerena JC (2014) The acetylation motif in AMP-forming acyl-CoA synthetases contains residues critical for acetylation and recognition by the protein acetyltransferase Pat of Rhodopseudomonas palustris. J Bacteriol. doi:10.1128/JB.00004-14
Crosby HA, Rank KC, Rayment I, Escalante-Semerena JC (2012a) Structural insights into the substrate specificity of the Rhodopseudomonas palustris protein acetyltransferase RpPat: identification of a loop critical for recognition by RpPat. J Biol Chem 287:41392–404. doi:10.1074/jbc.M112.417360
Crosby HA, Pelletier DA, Hurst GB, Escalante-Semerena JC (2012b) System-wide studies of N-lysine acetylation in Rhodopseudomonas palustris reveals substrate specificity of protein acetyltransferases. J Biol Chem 287:15590–15601. doi:10.1074/jbc.M112.352104
Datsenko KA, Wanner BL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi:10.1073/pnas.120163297
De Mey M, De Maeseneire S, Soetaert W, Vandamme E (2007) Minimizing acetate formation in E. coli fermentations. J Ind Microbiol Biotechnol 34:689–700. doi:10.1007/s10295-007-0244-2
El-Mansi M, Cozzone AJ, Shiloach J, Eikmanns BJ (2006) Control of carbon flux through enzymes of central and intermediary metabolism during growth of Escherichia coli on acetate. Curr Opin Microbiol 9:173–179. doi:10.1016/j.mib.2006.02.002
Fischer E, Sauer U (2003) A novel metabolic cycle catalyzes glucose oxidation and anaplerosis in hungry Escherichia coli. J Biol Chem 278:46446–46451
Gardner JG, Escalante-Semerena JC (2008) Biochemical and mutational analyses of AcuA, the acetyltransferase enzyme that controls the activity of the acetyl coenzyme A synthetase (AcsA) in Bacillus subtilis. J Bacteriol 190:5132–5136
Gardner JG, Escalante-Semerena JC (2009) In Bacillus subtilis, the sirtuin protein deacetylase, encoded by the srtN gene (formerly yhdZ), and functions encoded by the acuABC genes control the activity of acetyl coenzyme A synthetase. J Bacteriol 191:1749–1755
Gardner JG, Grundy FJ, Henkin TM, Escalante-Semerena JC (2006) Control of acetyl-coenzyme a synthetase (AcsA) activity by acetylation/deacetylation without NAD+ involvement in Bacillus subtilis. J Bacteriol 188:5460–5468
Han M-J, Lee SY, Hong SH (2012) Comparative analysis of envelope proteomes in Escherichia coli B and K-12 strains. J Microbiol Biotechnol 22:470–8
Hayden JD, Brown LR, Gunawardena HP, Perkowski EF, Chen X, Braunstein M (2013) Reversible acetylation regulates acetate and propionate metabolism in Mycobacterium smegmatis. Microbiology 159:1986–99. doi:10.1099/mic. 0.068585-0
Jeong H, Barbe V, Lee CH, Vallenet D, Yu DS, Choi S-H, Couloux A, Lee S-W, Yoon SH, Cattolico L, Hur C-G, Park H-S, Ségurens B, Kim SC, Oh TK, Lenski RE, Studier FW, Daegelen P, Kim JF (2009) Genome sequences of Escherichia coli B strains REL606 and BL21(DE3). J Mol Biol 394:644–52. doi:10.1016/j.jmb.2009.09.052
Karp PD, Paley SM, Krummenacker M, Latendresse M, Dale JM, Lee TJ, Kaipa P, Gilham F, Spaulding A, Popescu L, Altman T, Paulsen I, Keseler IM, Caspi R (2010) Pathway Tools version 13.0: integrated software for pathway/genome informatics and systems biology. Brief Bioinform 11:40–79. doi:10.1093/bib/bbp043
Keseler IM, Mackie A, Peralta-Gil M, Santos-Zavaleta A, Gama-Castro S, Bonavides-Martínez C, Fulcher C, Huerta AM, Kothari A, Krummenacker M, Latendresse M, Muñiz-Rascado L, Ong Q, Paley S, Schröder I, Shearer AG, Subhraveti P, Travers M, Weerasinghe D, Weiss V, Collado-Vides J, Gunsalus RP, Paulsen I, Karp PD (2013) EcoCyc: fusing model organism databases with systems biology. Nucleic Acids Res 41:D605–12. doi:10.1093/nar/gks1027
Kleman GL, Strohl WR (1994) Acetate metabolism by Escherichia coli in high-cell-density fermentation. Appl Environ Microbiol 60:3952–3958
Lara AR, Caspeta L, Gosset G, Bolívar F, Ramírez OT (2008) Utility of an Escherichia coli strain engineered in the substrate uptake system for improved culture performance at high glucose and cell concentrations: an alternative to fed-batch cultures. Biotechnol Bioeng 99:893–901. doi:10.1002/bit.21664
Lima BP, Thanh Huyen TT, Bäsell K, Becher D, Antelmann H, Wolfe AJ (2012) Inhibition of acetyl phosphate-dependent transcription by an acetylatable lysine on RNA polymerase. J Biol Chem 287:32147–60. doi:10.1074/jbc.M112.365502
Lin H, Castro N, Bennett G, San K-Y (2006) Acetyl-CoA synthetase overexpression in Escherichia coli demonstrates more efficient acetate assimilation and lower acetate accumulation: a potential tool in metabolic engineering. Appl Microbiol Biotechnol 71:870–874. doi:10.1007/s00253-005-0230-4
Luli GW, Strohl WR (1990) Comparison of growth, acetate production, and acetate inhibition of Escherichia coli strains in batch and fed-batch fermentations. Appl Environ Microbiol 56:1004–1011
Maharjan RP, Yu P-LL, Seeto S, Ferenci T, Prasad Maharjan R (2005) The role of isocitrate lyase and the glyoxylate cycle in Escherichia coli growing under glucose limitation. Res Microbiol 156:178–183. doi:10.1016/j.resmic.2004.09.004
Marisch K, Bayer K, Scharl T, Mairhofer J, Krempl PM, Hummel K, Razzazi-Fazeli E, Striedner G (2013) A comparative analysis of industrial Escherichia coli K-12 and B strains in high-glucose batch cultivations on process-, transcriptome- and proteome level. PLoS ONE 8:e70516. doi:10.1371/journal.pone.0070516
Meier S, Jensen PR, Duus JØ (2012) Direct observation of metabolic differences in living Escherichia coli strains K-12 and BL21. Chembiochem 13:308–10. doi:10.1002/cbic.201100654
Mischerikow N, Spedale G, Maarten Altelaar AF, Marc Timmers HT, Pim Pijnappel WWM, Heck AJR (2009) In-depth profiling of post-translational modifications on the related transcription factor complexes TFIID and SAGA. J Proteome Res 8:5020–5030
Nambi S, Basu N, Visweswariah S (2010) cAMP-regulated protein lysine acetylases in Mycobacteria. J Biol Chem 285:24313–24323. doi:10.1074/jbc.M110.118398
Negrete A, Majdalani N, Phue J-N, Shiloach J (2013) Reducing acetate excretion from E. coli K-12 by over-expressing the small RNA SgrS. N Biotechnol 30:269–73. doi:10.1016/j.nbt.2011.11.007
Nielsen J (2006) Microbial process kinetics. In Basic biotechnology. Ratledge, C.(ed.). Cambridge: Cambridge University Press, pp. 127-149
Peng L, Shimizu K (2003) Global metabolic regulation analysis for Escherichia coli K12 based on protein expression by 2-dimensional electrophoresis and enzyme activity measurement. Appl Microbiol Biotechnol 61:163–178
Phue J-N, Shiloach J (2004) Transcription levels of key metabolic genes are the cause for different glucose utilization pathways in E. coli B (BL21) and E. coli K (JM109). J Biotechnol 109:21–30. doi:10.1016/j.jbiotec.2003.10.038
Phue JN, Noronha SB, Hattacharyya R, Wolfe AJ, Shiloach J (2005) Glucose metabolism at high density growth of E. coli B and E. coli K: differences in metabolic pathways are responsible for efficient glucose utilization in E. coli B as determined by microarrays and northern blot analyses. Biotechnol Bioeng 90:805–820
Renilla S, Bernal V, Fuhrer T, Castaño-Cerezo S, Pastor JM, Iborra JL, Sauer U, Cánovas M (2012) Acetate scavenging activity in Escherichia coli: interplay of acetyl-CoA synthetase and the PEP-glyoxylate cycle in chemostat cultures. Appl Microbiol Biotechnol 95:2109–2124. doi:10.1007/s00253-011-3536-4
Shiloach J, Kaufman J, Guillard AS, Fass R (1996) Effect of glucose supply strategy on acetate accumulation, growth, and recombinant protein production by Escherichia coli BL21 (lambda DE3) and Escherichia coli JM109. Biotechnol Bioeng 49:421–428
Son Y-J, Phue J-N, Trinh LB, Lee SJ, Shiloach J (2011) The role of Cra in regulating acetate excretion and osmotic tolerance in E. coli K-12 and E. coli B at high density growth. Microb Cell Fact 10:52. doi:10.1186/1475-2859-10-52
Starai VJ, Escalante-Semerena JC (2004) Acetyl-coenzyme A synthetase (AMP forming). Cell Mol Life Sci 61:2020–2030
Starai VJ, Celic I, Cole RN, Boeke JD, Escalante-Semerena JC (2002) Sir2-dependent activation of acetyl-CoA synthetase by deacetylation of active lysine. Science 298(80):2390–2392. doi:10.1126/science.1077650
Studier FW, Daegelen P, Lenski RE, Maslov S, Kim JF (2009) Understanding the differences between genome sequences of Escherichia coli B strains REL606 and BL21(DE3) and comparison of the E. coli B and K-12 genomes. J Mol Biol 394:653–80. doi:10.1016/j.jmb.2009.09.021
Thao S, Escalante-semerena JC (2011) Biochemical and thermodynamic analyses of Salmonella enterica Pat, a multidomain, multimeric N(ε)-lysine acetyltransferase involved in carbon and energy metabolism. MBio 2:1–8. doi:10.1128/mBio.00216-11.Editor
Valgepea K, Adamberg K, Nahku R, Lahtvee P-J, Arike L, Vilu R (2010) Systems biology approach reveals that overflow metabolism of acetate in Escherichia coli is triggered by carbon catabolite repression of acetyl-CoA synthetase. BMC Syst Biol 4:166
Van de Walle M, Shiloach J (1998) Proposed mechanism of acetate accumulation in two recombinant Escherichia coli strains during high density fermentation. Biotechnol Bioeng 57:71–78
Waegeman H, Beauprez J, Moens H, Maertens J, De Mey M, Foulquie-Moreno M, Heijnen J, Charlier D, Soetaert W (2011) Effect of iclR and arcA knockouts on biomass formation and metabolic fluxes in Escherichia coli K-12 and its implications on understanding the metabolism of Escherichia coli BL21 (DE3). BMC Microbiol 11:70
Waegeman H, Maertens J, Beauprez J, De Mey M, Soetaert W (2012) Effect of iclR and arcA deletions on physiology and metabolic fluxes in Escherichia coli BL21 (DE3). Biotechnol Lett 34:329–37. doi:10.1007/s10529-011-0774-6
Walsh K, Koshland DE (1984) Determination of flux through the branch point of two metabolic cycles. The tricarboxylic acid cycle and the glyoxylate shunt. J Biol Chem 259:9646–9654
Weinert BTTT, Iesmantavicius V, Wagner SAAA, Schölz C, Gummesson B, Beli P, Nyström T, Choudhary C, Scho C, Nystro T (2013) Acetyl-phosphate is a critical determinant of lysine acetylation in E. coli. Mol Cell 51:1–8. doi:10.1016/j.molcel.2013.06.003
Wolfe AJ (2005) The acetate switch. Microbiol Mol Biol Rev 69:12–50
Zhao K, Chai X, Marmorstein R (2004) Structure and substrate binding properties of CobB, a Sir2 homolog protein deacetylase from Escherichia coli. J Mol Biol 337:731–741
Acknowledgments
We wish to thank José María Pastor (Dept. of Biochemistry and Molecular Biology B and Immunology) for helpful discussions, Marta Abrisqueta, Elena Martín-Orozco and David Cerezo (Dept. of Biochemistry and Molecular Biology B and Immunology, University of Murcia) for their help with western blotting and Professor Kerry Smith (Clemson University, South Carolina) for his assistance with the acetate kinase assay. S. Castaño-Cerezo is a recipient of a Ph.D. fellowship from Fundación Séneca (CARM, Murcia). V. Bernal acknowledges a post-doctoral contract from Universidad de Murcia (Programa Propio). This work has been partly funded by MICINN BIO2011-29233-C02-01 and Fundación Séneca-CARM 08660/PI/08 projects.
Conflict of interest
The authors declare that there are no conflicts of interest.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 60 kb)
Rights and permissions
About this article

Cite this article
Castaño-Cerezo, S., Bernal, V., Röhrig, T. et al. Regulation of acetate metabolism in Escherichia coli BL21 by protein Nε-lysine acetylation. Appl Microbiol Biotechnol 99, 3533–3545 (2015). https://doi.org/10.1007/s00253-014-6280-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-014-6280-8