
Abstract
Alkaline pectate lyases have great application potential in the bioscouring of textiles. They are isolated predominantly from bacteria and a few fungi. Here, we report the biochemical characteristics of a novel alkaline pectate lyase PelA from the basidiomycete Volvariella volvacea. The full-length pelA encodes a 321-amino-acid polypeptide containing a putative 18-residue signal peptide and a pectate lyase family 1 catalytic domain. It contains one conserved and one non-conserved potential N-glycosylation site (N-X-S/T) at the residues N95 and N198, respectively. The enzyme showed optimal activity at 60 °C and pH 10, although it was stable between pH 4 and pH 11. Additional Ca2+ was not required to measure PelA activity in vitro, but it could significantly enhance its activity and thermal stability. The V max values using polygalacturonic acid as substrate were increased from 50.71 to 89.96 IU mg−1 by the addition of 0.1 mM Ca2+, whereas the K m values were decreased from 0.681 to 0.514 mg ml−1. Site-directed mutagenesis revealed PelA has only one N-glycan attached to the residue N95. This N-glycan is crucial to its efficient secretion and activity possibly due to its role in maintaining the secondary structure of PelA. Amino acid substitution at the residue N198 had no effect on PelA secretion, but resulted in a slight (5.16 %) to modest (27.37 %) decrease in specific activity and less thermal stability, indicating the amino acid itself is also important for activity due to it being highly conserved and because of its proximity to the catalytic site.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pectate lyases (Pels) are endo-acting enzymes that catalyze the eliminative cleavage of pectate, to produce 4,5-unsaturated galacturonosyl at the non-reducing end (Solbak et al. 2005). The activity of pectate lyase was first discovered by Starr and Moran (1962) in the cultures of Erwinia carotovora and Bacillus polymyxa. Extracellular Pels secreted by plant pathogens are known to play an important role in the decomposition of plant residues during infection, their action resulting in the maceration of plant tissues (Marín-Rodríguez et al. 2002). Pels are now classified into polysaccharide lyase (PL) families 1, 2, 3, 9, and 10 (http://www.cazy.org/Polysaccharide-Lyases.html).
Pectin is a major component within the primary cell walls of all land plants and comprises a range of galacturonic acid-rich polysaccharides (Willats et al. 2001). They play an important role in the structure and function of both primary and secondary cell walls. Consequently, the enzymatic degradation of pectic substances is crucial in several industrial and biotechnological processes. Recently, microbial alkaline pectate lyases have received more attention due to their extensive applications in many fields, such as paper making, coffee and tea fermentation, textile and plant fiber processing, oil extraction, and the treatment of industrial wastewater (Hoondal et al. 2002; Solbak et al. 2005; Zhang et al. 2013; Zou et al. 2014). Alkaline pectate lyases were isolated predominantly from bacteria, such as Bacillus spp. (Hatada et al. 2001; Berensmeier et al. 2004), Erwinia chrysanthemi (Tardy et al. 1997; Jenkins et al. 2004), Klebsiella spp. (Yuan et al. 2011), Paenibacillus spp. (Boland et al. 2010; Li et al. 2014), Streptomyces sp. S27 (Yuan et al. 2012), and a few fungi, such as Penicillium occitanis (Damak et al. 2011) and Aspergillus niger (Benoit et al. 2012). Basidiomycetes, especially white-rot fungi, efficiently degrade all major components of plant cell walls including cellulose, hemicellulose, pectin, and lignin in nature. Although the genome sequencing revealed the existence of multiple genes for putative pectinolytic enzymes in basidiomycetes (Ohm et al. 2010), no encoding pectate lyases have been biochemically characterized to date.
Volvariella volvacea, the edible straw mushroom, is cultured on an industrial scale in many tropical and subtropical regions of Southeast Asia. V. volvacea grows naturally on rice straw and is an atypical white-rot basidiomycetous fungus, which has complete cellulolytic and hemicellulolytic systems but has very low lignolytic activity (Zheng and Ding 2013). Herein, we report the biochemical characteristics of a novel alkaline pectate lyase (PelA) from V. volvacea, functionally expressed in Pichia pastoris. PelA contains two putative N-glycosylation sites, one of which is conserved between fungal enzymes and the other not. As with most of recombinant proteins overexpressed in P. pastoris, the recombinant PelA was glycosylated. We also report the distinctive roles of the highly conserved glycosylation site in its secretion and activity.
Materials and methods
Strains, culture conditions, vectors, and chemicals
V. volvacea V14 (accession no. CMB002) was obtained from the culture collection of CUHK (WDCM68) located in the Biology Department, Chinese University of Hong Kong (Hong Kong, China). Escherichia coli DH5α (Invitrogen, Carlsbad, CA, USA) was used for cloning and P. pastoris KM71H (Invitrogen) was used for protein expression. The pGEM-T vector (Promega, Madison, WI, USA) was used to subclone DNA fragments for sequencing. The pPICZαB vector (Invitrogen) was used for constructing the yeast expression vectors. For the extraction of RNA, the fungus was cultured in rice straw compost as described previously (Ding et al. 2006). The polygalacturonic acid (PGA) was purchased from Sigma-Aldrich (St. Louis, MO, USA).
Cloning of PelA gene
An expressed sequence tag (EST) was obtained previously through the EST sequencing of a V. volvacea complementary DNA (cDNA) library. The library was constructed from the messenger RNA isolated from the mycelia of V. volvacea which had been cultivated on rice straw (Ding et al. 2006). A BLAST search against the recent GenBank releases showed this EST sequence had homology to pectate lyases. The gene-specific primer 5′-GCGCTTGGCGAGGGCAACAATTTCGT-3′ which was based on the EST sequence was designed to generate the 5′-end DNA fragment (coding for PelA). Rapid amplification of the cDNA ends (RACE)-PCR was carried out using the SMART RACE cDNA Amplification Kit (Clontech, Palo Alto, CA, USA). The 5′-cDNA end fragment was subcloned into the pGEM-T vector and sequenced. The full-length cDNA of pelA was then generated by 3′-RACE using the gene-specific primer 5′-ACATGGGGAACAGTCAACAGACATCTGT-3′ designed from the sequence of the extreme 5′-end of pelA and sequenced as above.
Construction of expression vectors and site-directed mutagenesis
The construction of the expression plasmid pPICZαB-PelA was carried out according to the manufacturer’s protocol (Invitrogen, Carlsbad, CA). The cDNA fragment encoding mature PelA, flanked by EcoRI/XbaI restriction sites (underlined) at the 5′ and 3′ ends, respectively, was amplified by PCR using the following oligonucleotides: 5′-TGCCGGAATTCCGATTCCGATCGAACCCTCTGAGCA-3′ and 5′-GCTCTAGATTAATGATGATGATGATGATGAAATGTCAAGGTCTGGCCG-3′ and Pfu polymerase (Agilent-Stratagene; La Jolla, CA, USA). After double digestion, the fragment was ligated at the EcoRI/XbaI sites of the pPICZαB expression vector to yield the construct pPICZαB-PelA in which pelA was under the transcriptional control of the AOX1 (alcohol oxidase) promoter. This construct was transferred into E. coli DH5α and the gene insert was confirmed by DNA sequencing. Site-directed plasmid mutagenesis of the PelA gene was performed by PCR using oligonucleotide primers containing the desired point mutations (see list of primers in Table 1) and by using plasmid pPICZαB-PelA as a template. PCR was performed under the following conditions: one cycle of 94 °C for 5 min, 55 °C for 30 s, and 72 °C for 6 min; 30 cycles of 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 6 min followed by a final extension at 72 °C for 10 min. After gel purification, the amplified product was digested with DpnI to eliminate the template plasmid. The site-directed mutagenesis resulted in the following amino acid changes in PelA (Fig. 1a): N95A, N95D, N95Q, S97A, N198A, N198D, and N198Q within the putative N-glycosylation motifs (N-X-S/T; numbering based on the full length of protein with signal peptide). These mutated constructs were transferred into E. coli DH5α and the gene insert was confirmed by DNA sequencing.

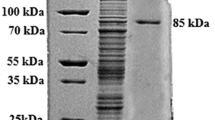
a Schematic structures of PelA and N-glycosylation mutants. b SDS-PAGE of purified PelA and endo H-treated PelA. Lane M, molecular weight markers; lane 1, PelA; lane 2, endo H-treated PelA. c SDS-PAGE of purified PelA and mutants at N95. Lane M, molecular weight markers; lanes 1–5, PelA, N95A, N95D, N95Q, and S97A, respectively. d SDS-PAGE analysis of purified PelA and mutants at N198. Lane M, molecular weight markers; lanes 1–4, PelA, N198A, N198D, and N198Q, respectively
Expression and purification of PelA and N-glycosylation mutants in P. pastoris
All expression plasmids containing a wild-type gene or N-glycosylation site mutants of pelA were then linearized by SacI and transformed into P. pastoris KM71H competent cells by electroporation with a Gene Pulser II apparatus (Bio-Rad, Hercules, CA). Transformants of P. pastoris containing the target DNA were selected on the basis of Zeocin resistance using yeast extract-peptone-dextrose (YPD) agar plates supplemented with 1 M sorbitol and 100 μg ml−1 Zeocin (Invitrogen). The most efficient PelA-producing transformants under the methanol-inducible AOX1 promoter were screened according to the manufacturer’s protocol (Invitrogen). In brief, every transformant was grown in a 250-ml flask containing 50 ml buffered complex glycerol (BMGY) medium at 30 °C and 250 rpm for 16–24 h until the cell density reached an OD600 value of 3–4. Yeast cells from a portion of the culture suspension were harvested by centrifugation and resuspended in 25 ml of buffered methanol complex (BMMY) medium to a final OD600 value of 30. BMGY and BMMY media were prepared according to the Pichia expression system manual from Invitrogen. Pectate lyase activity was determined following an additional 2 days induction at 30 °C and 250 rpm. The transformants exhibiting the highest expression level were selected for the production of wild-type PelA and mutants. The recombinant PelA and mutant enzymes with the 6 × His-tag were purified by affinity chromatography using nickel-nitrilotriacetic acid (Ni-NTA) agarose gel (Qiagen, Valencia, CA, USA) according to the manufacturer’s manual. Endoglycosidase H (endo H)-treated PelA was prepared by the deglycosylation of purified wild-type enzyme using endoglycosidase H (endo-N-acetylglucosaminidase H of Streptomyces plicatus; NEB, Ipswich, MA, USA) according to the manufacturer’s protocol. The enzyme sample (containing 300 μg protein of PelA) in 1 × glycoprotein denaturing buffer was boiled for 10 min. After cooling, PelA was incubated with 2000 NEB units of endo H in 50 mM sodium citrate buffer (pH 5.5) for 12 h at 28 °C. Controls contained an equal amount of PelA, treated under the same conditions with 10 mM ammonium acetate solution (pH 5.5) instead of endo H. Enzyme homogeneity and the molecular weight of purified wild-type PelA, endo H-treated PelA, and N-glycosylation mutants were estimated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE 10 % w/v).
Biochemical characterization of PelA
Pectate lyase activity was determined by spectrophotometrically monitoring an increase in optical density at 235 nm due to the formation of the unsaturated uronide product. Unless otherwise specified, the standard assay mixture contained 0.99 ml of 0.2 % (w/v) polygalacturonic acid (Sigma Chemical Co. type P7276) in 100 mM glycine-NaOH buffer at pH 10.0 and 10 μl of diluted enzyme solution. The reaction mixture was incubated at 60 °C for 10 min, and then the reaction was terminated by adding 0.5 ml of 500 mM HCl. One enzyme unit was defined as the formation of 1 μmol unsaturated polygalacturonic acid per min, with a molar extinction coefficient of 4600 M−1 cm−1 at 235 nm. The protein concentration was determined using a BCA Protein Assay Kit (Thermo Scientific Pierce, Rockford, IL, USA) with bovine serum albumin as standard. Optimal pH and temperature values were determined using the standard assay in the ranges of pH 7.0–12.0 (Tris-HCl pH 7–9 and glycine-NaOH pH 9–12) at a temperature of 30–70 °C, respectively. Thermal stability was determined by measuring the residual activities after preincubating the enzyme at 30–50 °C without substrate for 0–60 min. The pH stability was estimated by measuring the residual activity after incubating the enzyme in universal buffer with pH of 3.0–12.0 at 4 °C for 24 h. The kinetic constants (V max and K m) were determined at 60 °C after a reaction time of 5 min using polygalacturonic acid at concentrations from 0.1 to 2.5 mg ml−1. The V max and K m values were calculated by GraphPad Prism 5.0 software (http://www.graphpad.com/prism/) using non-linear regression.
Effect of metal ions and chemicals on PelA activity and thermal stability
The effect of metal ions and inhibitors on PelA activity was determined under the standard assay condition in the presence of various metal ions, ethylenediaminetetraacetic acid (EDTA), and SDS (each at 0.1 and 0.5 mM, respectively). The Ca2+ requirement for PelA activity was determined under standard conditions in the presence of 0–1 mM CaCl2. To determine the restoring effect of metal ions on PelA activity, EDTA (1.0 mM) was added into the enzyme solution to chelate the divalent ions, the enzyme solution was then dialysed against 0.1 M Tris-HCl (pH 9.0) to remove extra EDTA. Different concentrations of cations (Ca2+, Mg2+, Zn2+, Ni2+, and Mn2+) were then added into enzyme solutions, and enzyme activity was measured under standard conditions. The effect of Ca2+ on the thermal stability of PelA was evaluated using heat treatment for 0–60 min in the presence of 0.1 mM Ca2+. After heat treatment, the enzymes were cooled on ice, and the remaining activity was measured under the standard assay conditions.
Effect of N-glycosylation on PelA secretion, activity, and stability
The effect of N-glycosylation on PelA secretion in P. pastoris was evaluated by comparing the secretion levels of the wild-type PelA with N-glycosylation mutants in BMMY cultures. Aliquots (0.2 ml) of culture media were removed at different time intervals as indicated and centrifuged, and PelA activity was determined in the supernatants. The secretion levels of target proteins (mg ml−1) were defined as the enzyme activity (U ml−1) in the culture supernatant divided by the individual specific activity (U mg−1). Each expression experiment was performed in triplicate. The crude supernatant or those concentrated by precipitation using 10 % trichloroacetic acid were loaded on SDS-PAGE to analyze the integrity of enzyme molecules. The effect of N-glycosylation on PelA activity was evaluated using the wild-type PelA and various N-glycosylation mutants under the standard assay conditions described as above. The effect of N-glycosylation on PelA thermal stability was investigated by determining the residual activity after heating at different temperatures. Samples containing 1 mg ml−1 of the protein variants in phosphate buffer (pH 7.5) were incubated at 20, 30, 35, 40, 45, 50, 55, and 60 °C for 10 min. After heat treatment, the enzymes were cooled on ice, and the remaining activity was measured under standard assay conditions. T m values (defined as the temperature at which 50 % of the initial activity is lost after heat treatment) were determined from the plots of relative inactivation (%) against temperature (°C). All enzymes were purified by Ni-NTA agarose affinity chromatography.
Circular dichroism spectra
Spectra for PelA and its N-glycosylation mutant enzymes (0.2 mg ml−1) were recorded at 25 °C over the range of 190–260 nm with a MOS-500 Circular Dichroism Spectrometer (Bio-Logic, Grenoble, Isere, Rhone-Alpes, France), incorporating a bandwidth of 1 nm and a scanning speed of 120 nm min−1. Each spectrum was measured in triplicate using a quartz cuvette of 2 mm path length (Starna Scientific Ltd, Hainault, Essex, UK) and was corrected by subtracting a buffer blank spectrum. Data were analyzed using the CDNN circular dichroism spectra (CDS) deconvolution software (Applied Photophysics, Leatherhead, Surrey, UK).
Nucleotide sequence accession number
The nucleotide sequence of V. Volvacea pelA has been assigned the GenBank accession no. KJ57378.
Results
Cloning and sequence analysis of pelA
The full-length cDNA of pelA consists of 963 bp encoding 321 amino acids with a putative signal peptide of 18 amino acids. The calculated pI and molecular weight values of PelA are 8.82 and 34,088 Da, respectively. Alignment of the deduced amino acid sequence of pelA with the pectate lyase genes of other organisms showed the highest sequence similarities to pectate lyase B from Coprinopsis cinerea okayama7#130 (accession no. XP_001839398.2), hypothetical protein AGABI1DRAFT_87431 from Agaricus bisporus var. burnettii JB137-S8 (accession no. EKM76092.1), hypothetical protein AGABI2DRAFT_139877 from Agaricus bisporus var. bisporus H97 (accession no. EKV41722.1), and polysaccharide lyase family 1 protein from Auricularia delicata TFB-10046 SS5 (accession no. EJD53916.1), with 73, 69, 68, and 72 % identities, respectively. Two putative N-glycosylation sites (N-X-S/T) at the positions of N95 and N198 were found in the protein sequence using NetNGlyc 1.0 Server (http://www.cbs.dtu.dk/services/NetNGlyc/). Interestingly, multiple sequence alignment revealed that the two N-glycosylation sites are not equally conserved among the different fungal species (Fig. 2). The first one at residue N95 (NTS) is conserved in all listed pectate lyase sequences but is present in a less-conserved region near to the N-terminus. The second site at residue N198 (NAS), present in a conserved region proximity to the catalytic center, is not conserved, although the amino acid itself is highly conserved (Fig. 2).

Alignment of glycosylation sites of PelA with other pectate lyases. Identical residues are shaded in blue and conserved residues are shaded in gray. The two potential N-glycosylation sites are marked with red triangles. The catalytic center was boxed. EKM76092.1, hypothetical protein AGABI1DRAFT_87431 from Agaricus bisporus var. burnettii JB137-S8; EKV41722.1, hypothetical protein AGABI2DRAFT_139877 from Agaricus bisporus var. bisporus H97; EJD53916.1, polysaccharide lyase family 1 protein from Auricularia delicata TFB-10046 SS5; XP-001839398.2, pectate lyase B from Coprinopsis cinerea okayama7#130; EJD06337.1, polysaccharide lyase family 1 protein from Fomitiporia mediterranea MF3/22; ELU39081.1, polysaccharide lyase family 1 protein from Rhizoctonia solani AG-1 IA; XP-003037654.1, polysaccharide lyase family 1 protein from Schizophyllum commune H4-8; EIM82658.1, pectate lyase B from Stereum hirsutum FP-91666 SS1; XP-001402478.1, pectate lyase plyB from Aspergillus niger CBS 513.88; XP-749213.1, pectate lyase A from Aspergillus fumigatus Af293; ENH79763.1, pectate lyase from Colletotrichum orbiculare MAFF 240422; EFX05003.1, pectate lyase A from Grosmannia clavigera kw1407; XP-001265635.1, pectate lyase from Neosartorya fischeri NRRL 18; XP_002566377.1, Pc22g24890 from Penicillium chrysogenum Wisconsin 54-1255; XP-002149699.1, pectate lyase A from Talaromyces marneffei ATCC 18224
Purification and characterization of PelA
The gene encoding PelA from V. volvacea was successfully expressed in P. pastoris. The recombinant protein with a 6 × His-tag at its C-terminus was purified by affinity chromatography in a one-step procedure using Ni-NTA agarose gel. SDS-PAGE analysis revealed that the purified PelA migrated as two bands with a molecular mass of 35 and 37.5 kDa, respectively (Fig. 1b). After treating with endo H, only one protein band with an estimated molecular mass of 35 kDa was observed, suggesting that it was overglycosylated (Fig. 1b). The optimal pH and temperature for PelA were 10.0 and 60 °C, respectively (Fig. 3a, b). The PelA was relatively stable at a pH ranging from 4 to 11, retaining more than 80 % of the initial activity after incubation at 4 °C for 24 h (Fig. 3c). PelA was relatively stable at 40 °C and more than 60 % of the activity was retained after 1.0 h incubation; however, the enzyme activity rapidly decreased at a temperature above 50 °C (Fig. 3d).

Effects of pH (a) and temperature (b) on the activity and effects of pH (c) and temperature (d) on the stability of PelA. Values shown are means of triplicate determinations ± standard error (SE)
Effect of metal ions and chemicals on PelA activity and thermal stability
The effects of metal ions on PelA activity were tested by using polygalacturonic acid as the substrate. Ca2+ when present at a low concentration significantly enhanced its activity, whereas other metal ions had no or only slight inhibition effects (Table 2). The influence of Ca2+ was further investigated by adding CaCl2 at concentrations ranging from 0 to 1 mM at the optimal pH of the enzyme. PelA showed high activity without additional Ca2+, but the relative activity was significantly enhanced in the presence of 0.1–1.0 mM Ca2+ (Fig. 4a). The kinetic constant V max values towards polygalacturonic acid were increased from 50.71 IU mg−1 without Ca2+ to 89.96 IU mg−1 with 0.1 mM Ca2+, whereas K m values were decreased from 0.681 to 0.514 mg ml−1. When EDTA was added, Ca2+ was bound by EDTA, thus the Ca2+ activation occurred at higher concentrations (Fig. 4a). EDTA completely inhibited the PelA at the concentration of 1.0 mM. Unexpectedly, PelA activity was restored to 26.15 IU mg−1 after removing the extra chelated metal ions and EDTA by dialysis (Fig. 4b). The PelA activity was increased to 57.10 IU mg−1 when 0.2 mM Ca2+ was added into the dialyzed enzyme solution. However, other metal ions did not have a similar restoring effect on PelA activity (Fig. 4b), demonstrating the absolute requirement for Ca2+ as a cofactor for its activity. The presence of Ca2+ significantly (P < 0.001) enhanced thermal stability of PelA. The residual activity was 50 % after a 60-min incubation at 50 °C in the presence of Ca2+, whereas less than 10 % of residual activity was retained without the presence of 1.0 mM Ca2+ (Fig. 3d).

The effect of different concentrations of Ca2+ on PelA activity (a) and the restoring effect of metal ions on EDTA-treated PelA activity (b)
Effect of N-glycosylation on PelA secretion, activity, and stability
The two forms of PelA on SDS-PAGE were tentatively attributed to different degrees of glycosylation at the two putative N-glycosylation sites in the protein. Seven PelA mutants were constructed with a single substitution in the N-glycosylation site to determine whether one or both sites were N-glycosylated and to determine the effect on PelA secretion and activity. Considering that the alteration of highly conserved residue N95 may affect the thermostability and catalytic activity of the mutant enzymes, one of them, the S97A mutant, has the substitution of Ser at S97 by Ala. SDS-PAGE analysis of the mutant enzymes of N95A, N95D, N95Q, and S97A showed a single protein band with molecular mass 35 kDa, whereas the mutant enzymes of N198A, N198D, and N198Q appeared in two bands similar to the wild-type PelA. This indicates that only the N95 site was N-glycosylated (Fig. 1b, c). The expression level and biochemical properties of the wild-type PelA and all mutants were compared and the data are listed in Fig. 5 and Table 3.

Comparison of secretion level of PelA with N-glycosylation mutants in P. pastoris. a and c cell density of mutants at N-95 site and N198 site, respectively. b and d secreted PelA by mutants at N-95 site and N198 site, respectively. The single colony was used to inoculate 50 ml of BMGY in a 250-ml Erlenmeyer flask and the culture was incubated with shaking (250 rpm) at 30 °C until the OD600 value reached 3–4. Cells were harvested by centrifugation and resuspended in 25 ml of BMMY in 250-ml Erlenmeyer flasks to a final OD600 value of 30. Cell suspensions were incubated at 30 °C with shaking (250 rpm) and were induced by addition of 100 % methanol every 24 h to a final concentration of 1.0 %. All expression experiments were performed in triplicate. Values shown are means of triplicate determinations ± standard error (SE)
Elimination of the N-glycosylation sites at either N95 or S97 significantly reduced the amount of PelA into the extracellular fraction, and the secreted PelA was reduced from 58.5 % of the wild-type for mutant S97A to 81.1 % for N95Q (Fig. 5). To exclude the possible effect of proteolytic degradation on the protein quantities, the integrity of enzyme molecules in supernatants was analyzed by SDS-PAGE. All expressed products, including PelA and various N-glycolsylation mutants, remained intact in culture supernatants and no proteolytic fragments were generated (Fig. S1 in the Supplementary Material), indicating that the differences in enzyme secretion levels were not related to the proteolytic degradation of the recombinant proteins. Additionally, each of the PelA mutants at N95 or S97 had significantly lower specific activity than the wild type, ranging from 38.64 % of the wild type for mutant N95A to 81.07 % for S97A, respectively (Table 3). These findings indicated that removal of carbohydrate from N95 sites resulted in a significant decrease in secretion level and activity; however, the amount of decrease varies with the residual site selected for mutation and the newly induced residues. As expected from the lack of glycosylation at the position N198, the mutant enzymes of N198A, N198D, and N198Q had a similar expression level in P. pastoris as the wild-type PelA (Fig. 5). In contrast, the mutation at N198 has a slight or modest effect on specific activity, retaining 94.84 % of the wild-type PelA for N198A and 77.22 and 72.43 % for N198D and N198Q, respectively (Fig. 5 and Table 3). Slight changes in optimal pH and temperature values were also observed in mutant enzymes compared with the wild-type PelA (Table 3). Unexpectedly, the mutant enzymes of N198A, N198D, and N198Q had a less thermal stability than that of N95A, N95D, N95Q, and S97A in terms of T m (Fig. S2 in the Supplementary Material and Table 3). These mutants were further studied by CD spectroscopy in order to analyze whether the secondary structure of PelA was affected by the N-glycosylation mutations. The spectrum of the wild-type PelA showed a single minimum at 218 nm, a crossover point at 205–206 nm, and a maximum at 195 nm (Fig. 6), indicating a parallel β-helix structure as other pectate lyases (Kamen et al. 2000). The average CD spectra of N198A, N198D, and N189Q were very similar to that of the wild-type PelA, suggesting that the mutations did not disrupt the secondary structure of PelA and maintained a conformation close to the native protein. A prominent change in the CD spectrum was observed upon suppression of protein glycosylation by the elimination of the N-glycosylation site. The ellipticities at 195 and 218 nm were reduced greatly in the mutants of N95A, N95D, N95Q, and S97A. And furthermore, the crossover point was shifted to 198–199, 199–200, and 205–206 nm for mutants of N95A, N95D, and N95Q, respectively, indicating that suppression of N-glycosylation at this site led to structural alteration and therefore greatly impaired their secretion and the specific activity of the mutants (Fig. 6).

CD spectra of PelA and its mutants of N95A, N95D, N95Q, and S97A (a) and N198A, N198D, and N198Q (b)
Discussion
In nature, saprotrophic basidiomycetes play a central role in the degradation of plant biomass. They have an extensive set of carbohydrate-active enzymes for degrading polysaccharides, including cellulose, hemicellulose, and pectin within the plant cell wall. Genome sequencing revealed that some of them contained abundant genes encoding glycoside hydrolases and polysaccharide lyases for pectin degradation, although the repertoire of carbohydrate-active enzymes (CAZymes, http://www.cazy.org) in basidiomycetous fungal genomes varies according to species and their saprotrophic lifestyle (Martinez et al. 2004; Ohm et al. 2010; Benoit et al. 2012; Floudas et al. 2012). For instance, Schizophyllum commune contains 13 candidate polysaccharide lyases in PL1, PL3, PL4, and PL9 and 7 glycoside hydrolases in GH28, GH88, and GH105 related to pectin degradation (Ohm et al. 2010). Recently, the genome of V. volvacea was sequenced and assembled. It contains 17 candidate polysaccharide lyases in PL1, PL3, and PL4 and 9 glycoside hydrolases in GH28 and GH88 probably for pectin degradation (Bao et al. 2013; Chen et al. 2013). As yet, the biochemical information about the pectin degrading enzymes from basidiomycetesis is relatively scarce compared with bacteria and other fungi. The physiological-biochemical functions of these carbohydrate-active enzymes in pectin degradation in saprotrophic basidiomycetes remain elusive (Antov and Pericin 2001).
Herein, we are first in reporting the biochemical characteristics of a novel alkaline pectate lyase PelA from V. volvacea. It belongs to family 1 polysaccharide lyases and has high identities with hypothetical or uncharacterized proteins from other basidiomycetous fungi. It has an alkaline pH optimum and a wide pH stability. The optimal temperature of PelA (60 °C) is higher than that of other fungal Pels from Aspergillus nidulans (42 °C) (Zhao et al. 2007) and Fusarium oxysporum (50 °C) (Di Pietro and Roncero 1996). A thermoactive fungal pectate lyase Pel 1 from P. occitanis has the same optimal temperature as PelA, but Pel 1 exhibited lower optimal pH and V max values (9.0 and 17.19 IU mg−1, respectively) than PelA (Damak et al. 2011). Pectate lyases with highly alkaline-active, wide pH stability and thermoactivity were only reported from a few bacterial or actinomycetes species, such as alkaliphilic Bacillus sp. strain P-4-N (Hatada et al. 2001), Bacillus subtilis (Soriano et al. 2006), and Streptomyces sp. S27 (Yuan et al. 2012). Such properties related to pH and temperature makes PelA a good candidate for use in many areas, such as paper treatment and textile and plant fiber processing.
All Pels with the exception of PelW from E. chrysanthemi (Shevchik et al. 1999) and Pel from Bacillus pumilus (Klug-Santner et al. 2006) require Ca2+ for in vitro activity. Their requirement of Ca2+ had been reported from the concentrations of 0.1 to 1.0 mM (Payasi et al. 2009). PelA, as other Pels, also needs Ca2+ as a cofactor because EDTA completely inhibited PelA at the concentration of 1.0 mM. But an unusual feature of PelA is the requirement of trace Ca2+ for its activity. Firstly, extra Ca2+ did not need to be added into the reaction mixture for in vitro PelA activity. Furthermore, EDTA inhibition can be eliminated by dialysis against 0.1 M Tris-HCl (pH 9.0) and 85.18 % of initial activity was restored. These data suggested that the trace Ca2+ bound to the substrate is sufficient for PelA activity (Tardy et al. 1997). The pectate lyase Bsp165PelA with trace Ca2+ requirement was previously identified from Bacillus sp. N16-5. However, once inhibited with EDTA, its activity could not be restored by dialysis (Gang et al. 2010; Zheng et al. 2012). The Ca2+ was also found to have a positive effect on the thermostability of PelA. This phenomenon had been previously reported in Pels from other organisms, such as pectate lyase from Bacillus macerans (Morozova et al. 2006).
N-glycosylation is one of the most common posttranslational modifications performed by P. pastoris and plays a crucial role in a number of physiological and biochemical properties of a protein such as folding, stability, intracellular trafficking, or activity (Macauley-Patrick et al. 2005; Trombetta 2003; Skropeta 2009). Although the potential N-glycosylation sites of a protein can be predicted from the consensus sequence N-X-S/T, not all such sites are (fully) occupied (Skropeta 2009). By changing potential N-glycosylation sites by site-directed mutagenesis to prevent glycosylation, we revealed that the wild-type PelA as produced in P. pastoris has N-glycan attached to residue N95 near to the N-terminus. The double band of the recombinant wild-type PelA was thus likely due to heterogeneity of the glycosylation at N95. Whatever mutation was introduced at the N95 or S97 sites, all resulted greatly in a decrease in protein amounts in the supernatants and in specific activities of the non-glycosylated PelA versions compared to the wild-type enzyme, demonstrating that N-glycosylation at N95 is necessary for efficient secretion of PelA in P. pastoris and is important for activity as well. To date, suppression of glycosylation is typically achieved using site-directed conversion of the asparagine (N) residue of the N-X-S/T consensus sequence to the structurally-related glutamine (Q) and aspartic acid (D) residue or to other amino acids such as alanine (A) (Skropeta 2009). This technique has been widely used under the assumption that the asparagine (N) residue itself has no impact on the covalent structure of the target glycoprotein. Our results showed that, besides the occupancy of N-glycosylation at the N95 site, the position of the amino acids selected for site-directed mutagenesis had great impact on the biochemical properties of the mutant enzymes. The substitution at N95 resulted in stronger structural alteration and therefore leads to greater loss of enzyme activity compared to the effect of the substitution S97A. This indicates that the highly conserved residue N95 itself is also very important for protein structure and activity. In this case, S/T is a more suitable candidate mutation site for verifying the role of glycosylation in determining the biochemical properties of a protein. Interestingly, although the substitution at N198 seemed to have no effect on PelA secretion, it still resulted in a slight or even modest decrease in specific activity and less thermal stability, indicating that the highly conserved N198 residue is important to the catalytic activity due to its proximity to catalytic site. The mutants of N95Q and S97A shifted the pH optima towards a basic side by 0.5 units (pH 10.0 to 10.5). The pH at which the enzyme is maximally active is primarily determined by the pK(a)s of the active site residues (Pokhrel et al. 2012). Probably, a substitution of N95 by Q95 or S97 by A97 may cause the disruption of long-range electrostatic interactions and affect the pH profile of enzyme activity. However, further studies are needed to elucidate the reason for the pH shifts of mutants N95Q and S97A.
In conclusion, we report the first biochemical characterization of a novel alkaline pectate lyase PelA from the basidiomycetous fungus V. volvacea. The highly conserved residue N95 acting in glycosylation plays a very important role in enzyme activity and secretion in P. pastoris. PelA possesses favorable characteristics such as highly alkaline and thermoactive, wide pH stability and trace Ca2+ requirement, suggesting it is a good candidate for textile and paper treatment industries.
References
Antov MG, Pericin DM (2001) Production of pectinase by Polyporus squamosus in aqueous two-phase system. Enzyme Microb Technol 28:467–472
Bao D, Gong M, Zheng H, Chen M, Zhang L, Wang H, Jiang J, Wu L, Zhu Y, Zhu G, Zhou Y, Li C, Wang S, Zhao Y, Zhao G, Tan Q (2013) Sequencing and comparative analysis of the straw mushroom (Volvariella volvacea) genome. PLoS One 8(3):e58294
Benoit I, Coutinho PM, Schols HA, Gerlach JP, Henrissat B, de Vries RP (2012) Degradation of different pectins by fungi: correlations and contrasts between the pectinolytic enzyme sets identified in genomes and the growth on pectins of different origin. BMC Genomics 13:321
Berensmeier S, Singh S, Meens J, Buchholz K (2004) Cloning of the pelA gene from Bacillus licheniformis 14A and biochemical characterization of recombinant, thermostable, high-alkaline pectate lyase. Appl Microbiol Biotechnol 64:560–567
Boland WE, Henriksen ED, Doran-Peterson J (2010) Characterization of two Paenibacillus amylolyticus strain 27C64 pectate lyases with activity on highly methylated pectin. Appl Environ Microbiol 76:6006–6009
Chen B, Gui F, Xie B, Deng Y, Sun X, Lin M, Tao Y, Li S (2013) Composition and expression of genes encoding carbohydrate-active enzymes in the straw-degrading mushroom Volvariella volvacea. PLoS One 8(3):e58780
Damak N, Hadj-Taieb N, Bonnin E, Bacha AB, Gargouri A (2011) Purification and biochemical characterization of a novel thermoactive fungal pectatelyase from Penicillium occitanis. Process Biochem 46:888–893
de Vries RP, Ferreira P, Findley K, Foster B, Gaskell J, Glotzer D, Górecki P, Heitman J, Hesse C, Hori C, Igarashi K, Jurgens JA, Kallen N, Kersten P, Kohler A, Kües U, Kumar TKA, Kuo A, LaButti K, Larrondo LF, Lindquist E, Ling A, Lombard V, Lucas S, Lundell T, Martin R, McLaughlin DJ, Morgenstern I, Morin E, Murat C, Nagy LG, Nolan M, Ohm RA, Patyshakuliyeva A, Rokas A, Ruiz-Dueñas FJ, Sabat G, Salamov A, Samejima M, Schmutz J, Slot JC, St John F, Stenlid J, Sun H, Sun S, Syed K, Tsang A, Wiebenga A, Young D, Pisabarro A, Eastwood DC, Martin F, Cullen D, Grigoriev IV, Hibbett DS (2012) The paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Sci 336:1715–1719
Di Pietro A, Roncero MIG (1996) Purification and characterization of a pectate lyase from Fusarium oxysporum f. sp. lycopersici produced on tomato vascular tissue. Physiol Mol Plant Pathol 49:177–185
Ding S, Ge W, Buswell JA (2006) Cloning of multiple cellulase cDNAs from Volvariella volvacea and their differential expression during substrate colonization and fruiting. FEMS Microbiol Lett 263:207–213
Gang L, Rao L, Xue Y, Zhou C, Zhang Y, Ma Y (2010) Cloning, expression, and characterization of a highly active alkaline pectate lyase from alkaliphilic Bacillus sp. N16-5. J Microbiol Biotechnol 20:670–677
Hatada Y, Kobayashi T, Ito S (2001) Enzymatic properties of the highly thermophilic and alkaline pectate lyase Pel-4B from alkaliphilic Bacillus sp. strain P-4-N and the entire nucleotide and amino acid sequences. Extremophiles 5:127–133
Hoondal G, Tiwari R, Tewari R, Dahiya N, Beg Q (2002) Microbial alkaline pectinases and their industrial applications: a review. Appl Microbiol Biotechnol 59:409–418
Jenkins J, Shevchik VE, Hugouvieux-Cotte-Pattat N, Pickersgill RW (2004) The crystal structure of pectate lyase Pel9A from Erwinia chrysanthemi. J Biol Chem 279:9139–9145
Kamen DE, Griko Y, Woody RW (2000) The stability, structural organization, and denaturation of pectate lyase C, a parallel β-helix protein. Biochem 39:15932–15943
Klug-Santner BG, Schnitzhofer W, Vrsanská M, Weber J, Agrawal PB, Nierstrasz VA, Guebitz GM (2006) Purification and characterization of a new bioscouring pectate lyase from Bacillus pumilus BK2. J Biotechnol 121:390–401
Li X, Wang H, Zhou C, Ma Y, Li J, Song J (2014) Cloning, expression and characterization of a pectate lyase from Paenibacillus sp. 0602 in recombinant Escherichia coli. BMC Biotechnol 14:18
Macauley-Patrick S, Fazenda ML, McNeil B, Harvey LM (2005) Heterologous protein production using the Pichia pastoris expression system. Yeast 22:249–270
Marín-Rodríguez MC, Orchard J, Seymour GB (2002) Pectate lyases, cell wall degradation and fruit softening. J Exp Bot 53:2115–2119
Martinez D, Larrondo LF, Putnam N, Gelpke MD, Huang K, Chapman J, Helfenbein KG, Ramaiya P, Detter JC, Larimer F, Coutinho PM, Henrissat B, Berka R, Cullen D, Rokhsar D (2004) Genome sequence of the lignocellulose degrading fungus Phanerochaete chrysosporium strain RP78. Nat Biotechnol 22:695–700
Morozova VV, Semenova MV, Salanovich TN, Okunev ON, Koshelev AV, Bubnova TV, Krichevskiĭ GE, Timatkov AG, Barysheva NV, Sinitsyn AP (2006) Application of neutral-alkaline pectate lyases to cotton fabric boil off. Prikl Biokhim Mikrobiol 42:686–691
Ohm RA, de Jong JF, Lugones LG, Aerts A, Kothe E, Stajich JE, de Vries RP, Record E, Levasseur A, Baker SE, Bartholomew KA, Coutinho PM, Erdmann S, Fowler TJ, Gathman AC, Lombard V, Henrissat B, Knabe N, Kües U, Lilly WW, Lindquist E, Lucas S, Magnuson JK, Piumi F, Raudaskoski M, Salamov A, Schmutz J, Schwarze FW, vanKuyk PA, Horton JS, Grigoriev IV, Wösten HA (2010) Genome sequence of the model mushroom Schizophyllum commune. Nat Biotechnol 28:957–963
Payasi A, Sanwal R, Sanwal GG (2009) Microbial pectate lyases: characterization and enzymological properties. World J Microbiol Biotechnol 25:1–14
Pokhrel S, Joo JC, Kim YH, Yoo YJ (2012) Rational design of a Bacillus circulans xylanase by introducing charged residue to shift the pH optimum. Process Biochem 47:2487–2493
Shevchik VE, Condemine G, Robert-Baudouy J, Hugouvieux-Cotte-Pattat N (1999) The exopolygalacturonate lyase PelW and oligogalacturonatelyase Ogl, two cytoplasmic enzymes of pectin catabolism in Erwinia chrysanthemi 3937. J Bacteriol 181:3912–3919
Skropeta D (2009) The effect of individual N-glycans on enzyme activity. Bioorgan Med Chem 17:2645–2653
Solbak AI, Richardson TH, McCann RT, Kline KA, Bartnek F, Tomlinson G, Tan X, Parra-Gessert L, Frey GJ, Podar M, Luginbühl P, Gray KA, Mathur EJ, Robertson DE, Burk MJ, Hazlewood GP, Short JM, Kerovuo J (2005) Discovery of pectin-degrading enzymes and directed evolution of a novel pectate lyase for processing cotton fabric. J Biol Chem 280:9431–9438
Soriano M, Diaz P, Pastor FIJ (2006) Pectate lyase C from Bacillus subtilis: a novel endo-cleaving enzyme with activity on highly methylated pectin. Microbiol 152:617–625
Starr ME, Moran P (1962) Eliminative split of pectic substances by phytopathogenic soft-rot bacteria. Sci 135:920–921
Tardy F, Nasser R-BJ, Hugouvieux-Cotte-Pattat N (1997) Comparative analysis of the five major Erwinia chrysanthemi pectate lyases: enzyme characteristics and potential inhibitors. J Bacteriol 179:2503–2511
Trombetta ES (2003) The contribution of N-glycans and their processing in the endoplasmic reticulum to glycoprotein biosynthesis. Glycobiology 13:77R–91R
Willats WG, McCartney L, Mackie W, Knox JP (2001) Pectin: cell biology and prospects for functional analysis. Plant Mol Biol 47:9–27
Yuan P, Meng K, Luo H, Shi P, Huang H, BaiY YP, Yao B (2011) A novel low-temperature active alkaline pectate lyase from Klebsiella sp. Y1 with potential in textile industry. Process Biochem 46:1921–1926
Yuan P, Meng K, Shi P, Luo H, Huang H, Tu T, Yang P, Yao B (2012) An alkaline-active and alkali-stable pectate lyase from Streptomyces sp. S27 with potential in textile industry. Process Biochem 39:909–915
Zhang J, Kang Z, Ling Z, Cao W, Liu L, Wang M, Du G, Chen J (2013) High-level extracellular production of alkaline polygalacturonate lyase in Bacillus subtilis with optimized regulatory elements. Bioresour Technol 146:543–548
Zhao QX, Yuan S, Zhang YL, Zhu H, Dai CC, Yang F, Han FM (2007) Expression, purification and characterization of pectate lyase A from Aspergillus nidulans in Escherichia coli. World J Microbiol Biotechnol 23:1057–1064
Zheng F, Ding S (2013) Processivity and enzymatic mode of a glycoside hydrolase family 5 endoglucanase from Volvariella volvacea. Appl Environ Microbiol 79:989–996
Zheng Y, Huang CH, Liu W, Ko TP, Xue Y, Zhou C, Guo RT, Ma Y (2012) Crystal structure and substrate-binding mode of a novel pectate lyase from alkaliphilic Bacillus sp. N16-5. Biochem Biophys Res Commun 420:269–274
Zou M, Guo F, Li X, Zhao J, Qu Y (2014) Enhancing production of alkaline polygalacturonate lyase from Bacillus subtilis by fed-batch fermentation. PLoS One 9(3):e90392
Acknowledgments
We thank Dr. Sandra Chapman, National Institute of Agricultural Botany in Cambridge, UK, for linguistic revision of the manuscript. This work was supported by a research grant (no. 31270628) from the National Natural Science Foundation of China, a Project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions, and the Doctorate Fellowship Foundation of Nanjing Forestry University.
Author information
Authors and Affiliations
Corresponding author
Additional information
Aiqin Shi and Hang Hu contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 191 kb)
Rights and permissions
About this article

Cite this article
Shi, A., Hu, H., Zheng, F. et al. Biochemical characteristics of an alkaline pectate lyase PelA from Volvariella volvacea: roles of the highly conserved N-glycosylation site in its secretion and activity. Appl Microbiol Biotechnol 99, 3447–3458 (2015). https://doi.org/10.1007/s00253-014-6146-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-014-6146-0