
Abstract
Reticulated flatwoods salamander (Ambystoma bishopi) populations began decreasing dramatically in the 1900s. Contemporary populations are small, isolated, and may be susceptible to inbreeding and reduced adaptive potential because of low genetic variation. Genetic variation at immune genes is especially important as it influences disease susceptibility and adaptation to emerging infectious pathogens, a central conservation concern for declining amphibians. We collected samples from across the extant range of this salamander to examine genetic variation at major histocompatibility complex (MHC) class Iα and IIβ exons as well as the mitochondrial control region. We screened tail or toe tissue for ranavirus, a pathogen associated with amphibian declines worldwide. Overall, we found low MHC variation when compared to other amphibian species and did not detect ranavirus at any site. MHC class Iα sequencing revealed only three alleles with a nucleotide diversity of 0.001, while MHC class IIβ had five alleles with a with nucleotide diversity of 0.004. However, unique variation still exists across this species’ range with private alleles at three sites. Unlike MHC diversity, mitochondrial variation was comparable to levels estimated for other amphibians with nine haplotypes observed, including one haplotype shared across all sites. We hypothesize that a combination of a historic disease outbreak and a population bottleneck may have contributed to low MHC diversity while maintaining higher levels of mitochondrial DNA variation. Ultimately, MHC data indicated that the reticulated flatwoods salamander may be at an elevated risk from infectious diseases due to low levels of immunogenetic variation necessary to combat novel pathogens.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Immune genes are crucial for species survival, as immunogenetic diversity is necessary to effectively combat a broad range of infections; without it, complex organisms face a greater risk of disease and extinction. Moreover, disease threatens many amphibian species and is a key factor in some recent amphibian extinctions (McCallum 2007; Richmond et al. 2009). Ranaviruses (frog virus 3 and Ambystoma tigrinum virus) are a type of emerging amphibian pathogen that is increasing in prevalence and becoming more virulent through recombination of viral strains, threatening species worldwide (Chinchar 2002; Gray and Chinchar 2015; Claytor et al. 2017; Peace et al. 2019) while chytrid fungi (Batrachochytrium dendrobatidis and B. salamandrivorans) have devastated populations of amphibians around the world (Berger et al. 1998; Martel et al. 2013; O’Hanlon et al. 2018).
In North America, ranavirus accounts for a large percentage of disease-related amphibian deaths each year (Green et al. 2002), affecting both captive and wild populations (Johnson et al. 2008; Claytor et al. 2017). Ranavirus kills its host through a combination of lesions, abdominal swelling, and hemorrhaging (Gray and Chinchar 2015). Larval amphibians are the most susceptible to fatal infection, and mass die-offs can devastate entire larval cohorts resulting in complete recruitment failure (Teacher et al. 2009). Some ambystomatid species, like the tiger salamander (Ambystoma tigrinum), can tolerate ranavirus infection (Greer et al. 2009) while others like the California tiger salamander (Ambystoma californiense) exhibit high mortality rates following experimental infection (Picco et al. 2007). This disease is spreading, and in recent years, amphibian deaths on five continents have been attributed to ranavirus (Marsh et al. 2002; Fox et al. 2006; Kik et al. 2011; Miller et al. 2011; Price et al. 2014). The incipient threat of disease in an interconnected world underscores the importance of disease screening, biosecurity, and immune gene research for vulnerable species.
Major histocompatibility complex (MHC) immune genes are among the most important determinants of disease resistance in jawed vertebrates (Sommer 2005). This region is well studied and considered to be one of the most genetically diverse regions in the genome (Sommer 2005). MHC genes code for transmembrane proteins with cell surface domains, which bind peptides derived from antigens and then present them for inspection to T cells that in turn activate other components of the immune response (Bernatchez and Landry 2003). The MHC consists of two main classes: class I, which monitors intracellular fluid, and class II, which interacts with extracellular fluid. Class I proteins bind antigens found inside the cell and present them on the cell’s surface to cytotoxic T cells, which destroy infected cells (Alberts et al. 2015). These antigens usually derive from viruses or intracellular bacteria. Class II proteins present to and activate helper T cells, which in turn activate B cells, macrophages, and other immune cells. Class II proteins typically present antigens from extracellular bacteria and fungi (Alberts et al. 2015). Together, these two classes of proteins combine to protect vertebrates against a diverse array of pathogens.
Genetic variation at MHC genes plays an important role in fighting infectious diseases. Variation is vital to the long-term persistence of a species, and high MHC diversity allows a species to adapt to, and survive, a broad range of pathogens. This diversity is especially important when a species is faced with a novel pathogen because higher diversity can increase the chance a population survives. To date, most MHC-related amphibian research has focused on anurans with less attention to caudates, but research has shown associations between diseases and MHC diversity in amphibians. For instance, specific MHC class Iα and class IIβ alleles have been associated with increased survival following exposure to chytrid fungus and ranavirus (Teacher et al. 2009; Savage et al. 2016; Savage and Zamudio 2016; Fu and Waldman 2017; Savage et al. 2018). In other species, MHC class IIβ heterozygosity correlates with increased survival in Chiricahua leopard frogs (Lithobates chiricahuensis) and lowland leopard frogs (L. yavapaiensis) infected with chytrid fungus (Savage and Zamudio 2011; Savage et al. 2018). Similarly, wood frogs (Rana sylvatica) that are heterozygous at MHC class IIβ had a lower ranavirus infection intensity when compared to homozygotes (Savage et al. 2019). Because MHC diversity is important in combating pathogens, conserving that diversity will be a key component in countering global amphibian species declines (Elbers and Taylor 2016; Savage et al. 2018).
The reticulated flatwoods salamander (RFS, Ambystoma bishopi) is a federally endangered species (USFWS 2009) that has experienced severe declines in population size and the number of breeding sites. These declines may have caused a genetic bottleneck, leading to inbreeding and reduced genetic diversity. Inbreeding increases genome-wide homozygosity and may lead to inbreeding depression, which can manifest as reduced fecundity and decreased survival, including that caused by increased disease susceptibility (Allendorf et al. 2013). RFSs occur in fire-maintained longleaf pine (Pinus palustris) ecosystems (Palis 1996; Petranka 2010). Once a wide-ranging species, the RFS was locally abundant throughout the coastal plain of the southeastern USA and could be found in southern Alabama, western Georgia, and the panhandle of Florida west of the Apalachicola River (Palis 1996; Pauly et al. 2007; IUCN 2008). Over the last century, however, fire suppression, extensive land conversion, extended droughts, and loss of longleaf pine habitat have severely reduced and fragmented RFS populations (Frost 1993; Palis 1997; Bishop and Haas 2005; IUCN 2008; Chandler et al. 2016; McIntyre et al. 2018). As a result, only twenty-two breeding sites could be identified when the RFS was listed as endangered in 2009 (USFWS 2009, Farmer et al. 2016). All known breeding sites are restricted to Florida (n = 20) and Georgia (n = 2), with an estimated adult population size of just 10,000 individuals in 2009 (Pauly et al. 2007; IUCN 2008). Since 2009, the number of known active breeding sites has declined from twenty-two to six, although part of this decline is because many known sites on private land have not been surveyed in the intervening years (Farmer et al. 2016; O’Donnell et al. 2017; Semlitsch et al. 2017). A loss of genetic diversity in RFS may affect its long-term persistence because genetic variation is the foundation upon which natural selection acts, enabling populations to adapt to changing environmental conditions (Frankham et al. 2002).
In this study, we estimated MHC diversity in the RFS to investigate range-wide genetic variation at breeding sites defined by USFWS. The control region of mitochondrial DNA was also sequenced to provide an additional estimate of genetic variation unrelated to immune gene variation and so unlikely to be affected by selection driven by disease. We also screened RFS for ranavirus infection to assess its occurrence across the range and to examine associations between immunogenetic variation and ranavirus, if present. Understanding the prevalence of ranavirus and the extent of immunogenetic diversity following bottlenecks will inform conservation efforts and management strategies for the RFS.
Methods
Sample collection
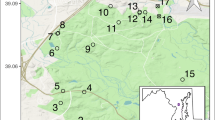
Sampling occurred at five breeding sites across the extant range of the RFS (Fig. 1). We identified “breeding sites” as defined by the US Fish and Wildlife Service in 2015, which considers any grouping of RFS ponds within 3.2 km (2 miles) of each other as a single breeding site (USFWS 2015, data by pond available in Supplemental 1). Between 2011 and 2019, we collected samples from: East and West Eglin Air Force Base (AFB), Florida (East: n = 147, West: n = 41); Escribano Point Wildlife Management Area (WMA), Florida (n = 48); Garcon Point Water Management Area, Florida (n = 4); and Mayhaw WMA, Georgia (n = 5). Sample sizes were unbalanced across sites, reflecting salamander abundance as well as sampling effort. Tissue was taken with sterilized scissors from the toes of adult salamanders caught in drift-fence funnel traps or from the tails of larvae captured during dipnet surveys. Both tissue types were stored in 95% ethanol at 4 °C, and DNA was extracted using a DNEasy Blood and Tissue Kit (Qiagen) following the manufacturer’s protocol.

Map of breeding sites sampled for RFS from 2011 to 2019
DNA amplification and sequencing
We sequenced three loci: MHC class Iα exon 3, MHC class IIβ exon 2, and the mitochondrial control region (D-loop). MHC class Iα was amplified using primers P3S and P3AS as described in Sammut et al. (1999), but with different amplification conditions. PCRs optimized for RFS were performed at a final volume of 20 μl with concentrations of 1X PCR buffer, 3 mM of MgCl2, 4 mM dNTPs, 6 μM of each primer, 0.1 μl of Taq polymerase (New England Labs, Ipswich MA), and 1.0 μl of DNA template. The thermal profile consisted of an initial denaturation step of 30 s at 95 °C followed by 35 cycles of 95 °C for 30 s, 63 °C for 30 s, 68 °C for 30 s, and a final extension step of 68 °C for 5 min. Despite multiple attempts to optimize conditions, previously published primers (Bos and DeWoody 2005) did not amplify the MHC class IIβ locus in RFS, so we designed new primers targeting MHC class IIβ using program Geneious v11.1.2 (Kearse et al. 2012). Primers were designed by aligning published sequences of all ambystomatid species available in GenBank (AF209115, AF209117, DQ125478-80, KP408179-209) and initially creating degenerate primers to target a conserved region across all sequences, an exonic portion of MHC class IIβ. After obtaining sequences from the degenerate primers, we redesigned new primers to target conserved regions of the RFS’s MHC class IIβ exon (forward 5′ GGATCTCCTTCTGGCTGTTC 3′, reverse 5′ CGAGTGCCGCWTTCTGAACG 3′). PCRs were performed as above except with a final concentration of 1 mM of MgCl2 and an annealing temperature of 60 °C. We also sequenced the mitochondrial D-loop, which has been used previously for genetic studies in the RFS (Pauly et al. 2007). We amplified this region using primers THR and DL1 following Shaffer and McKnight (1996) and Pauly et al. (2007).
All purified PCR products were cycle sequenced in forward and reverse directions using 5× BigDye buffer (Applied Biosystems Inc), Big Dye v3.1, 10 μM primer, 1.5 μl purified PCR product, and nanopure water for a total reaction volume of 7.0 μl. The thermal profile followed an initial step of 60 s at 96 °C followed by 24 cycles of 96 °C for 10 s, 50 °C for 5 s, and 60 °C for 4 min. Cycle-sequenced product was purified with Sephadex G50 Fine (Sigma-Aldrich) and sequenced on an ABI 3130xl DNA analyzer at the LSU Genomics Facility (Baton Rouge, LA).
Pathogen screening
RFS samples (n = 249) were screened for ranavirus using primers 4 and 5, and the PCR protocol was described in Mao et al. (1997). Primers 4 and 5 amplify a region of the major capsid protein and have reliably detected ranavirus infection in many amphibian species including the closely related tiger salamander (Ambystoma tigrinum: Picco et al. 2007; Greer et al. 2009) as well as more distantly related plethodontid salamanders (Blackburn et al. 2015). To confirm results, a random subsample (n = 119) was re-tested with primers M151 and M152 (which amplify a different region of the major capsid protein gene) and the PCR protocol described in Marsh et al. (2002). Negative and positive controls were included with every PCR run. The negative control consisted of nanopure water, whereas the positive control was a 521-bp fragment of linearized ranavirus plasmid (Allender et al. 2013). All PCR product was run on 2% agarose gels, where the presence or absence of bands equal to the size of the positive control indicated the presence or absence of ranavirus.
Data analysis
MHC variation was examined both by using sequence data and by assigning each unique sequence an allele number (e.g., allele 01) to create genotype data. Before analysis, all sequences were quality checked, low-quality reads were trimmed, and aligned using Geneious, then phased using DnaSP v6 (Rozas et al. 2017). All nucleotide and protein sequences were checked against sequences from other ambystomatid species using the NCBI BLAST algorithms blastx and blastp (https://blast.ncbi.nlm.nih. gov/Blast.cgi). Estimates of nucleotide diversity, or the average number of nucleotide differences per site between sequences, were calculated in DnaSP v6 (Rozas et al. 2017). Observed and expected heterozygosities (HO and HE) for MHC loci (Weir and Cockerham 1984) were estimated in GenePop v4.6 (Raymond and Rousset 1995; Rousset 2008). Each MHC locus was tested for Hardy-Weinberg Equilibrium (HWE) within each breeding site using GenePop v4.6. The p.adjust function in R (Core Team 2018) was then used to correct for multiple tests (n = 5) using the false discovery rate (FDR, Benjamini and Hochberg 1995) as well as both sequential and regular Bonferroni-adjusted of 0.05. Allelic richness (AR) was estimated with FSTAT v2.9.4 (Goudet 2001). This calculation uses a rarefaction technique to estimate the expected number of alleles in a subsample and is standardized to the smallest number of individuals typed at a location (Garcon Point n = 4). In order to translate DNA to amino acid sequences and determine synonymous and non-synonymous nucleotide substitutions, the reading frame was selected after sequence processing by comparing the translation starting at nucleotide 1, 2, or 3. Each translation was inspected for stop codons (indicating an improper reading frame) in Geneious and compared to sequences of similar species published in GenBank. The non-synonymous to synonymous (dN/dS) substitution ratio was calculated and examined for evidence of selection using two separate tests. First, MEGA X v10.0.5 was used to conduct a Z test with 5000 bootstraps to determine if dN = dS using the Nei and Gojobori method with a Jukes-Cantor correction (Kumar et al. 2018). The dN/dS ratio was measured using the entire sequence, as well as the codons predicted to be within the peptide binding region (Lillie et al. 2014; Bondinas et al. 2006). Second, HyPhy was used to test for selection on a per site basis with a fixed effects likelihood (FEL) method implemented in DataMonkey 2.0 (Kosakovsky et al. 2005; Weaver et al. 2018). For both MHC regions and mitochondrial DNA, a haplotype network was created using a minimum joining network in Popart 1.7 (Fig. 2; Bandelt et al. 1999). Finally, to compare MHC diversity in the RFS with other amphibians, a literature search was conducted reviewing 23 other species for MHC class Iα and class IIβ diversity. Data on sample size, number of alleles, and nucleotide diversity were summarized and compared to results from the RFS.

Haplotype network of MHC class Iα (a), MHC class IIβ (b), and mitochondrial D-loop (c) sequences. Circle size is proportional to the number of individuals per haplotype, and each dash represents a single base substitution
Results
At the MHC class Iα locus, we sequenced 190 individuals and amplified DNA fragments between 243 and 248 bp. Those fragments were trimmed to 243 bp each and after phasing three alleles were observed (Tables 1 and 2). The nucleotide sequences of these alleles matched the MHC class Iα chain of the Mexican axolotl (Ambystoma mexicanum) with more than 94% similarity and e-values between 6e−93 and 3e−96 (NCBI blastn algorithm, https://blast.ncbi.nlm.nih.gov/Blast.cgi, GenBank accession numbers U88185.1, U83137.1, and U83138.1). Protein sequences for MHC class Iα matched the Mexican axolotl with more than 91% similarity and e-values between 7e−44 and 7e−46 (NCBI blastp algorithm, https://blast.ncbi.nlm.nih.gov/Blast.cgi, GenBank accession numbers AAC60111.1, AAC60108.1, and AAC60109.1). At the MHC class IIβ locus, we sequenced 93 individuals and amplified DNA fragments between 160 and 163 bp. Those fragments were trimmed to 160 bp each and after phasing five alleles were observed (Tables 1 and 3). These sequences matched with more than 92% similarity to the MHC class IIβ chain of both the Mexican axolotl and tiger salamander with e-values between 1e−53 and 2e−55 (NCBI blastn algorithm, GenBank accession numbers KP408205.1, DQ125478.1, and DQ071905.1). Protein sequences for MHC class IIβ matched Mexican axolotl and tiger salamander with 78–81% similarity and e-values between 1e−22 and 3e−23 (NCBI blastp algorithm, https://blast.ncbi.nlm.nih.gov/Blast.cgi, GenBank accession numbers AKC35261.1 and AAY99198.1). After correcting for multiple tests, MHC loci at all breeding sites were in Hardy-Weinberg equilibrium except for the MHC class IIβ locus at Escribano Point (X2 = 16.9, p = 0.03). We sequenced 682 bp of the mitochondrial D-loop in 238 individuals and observed nine haplotypes: seven were previously undescribed while two were 100% matches to haplotypes recovered from other study sites sampled by Pauly et al. (2007; Genbank accession numbers H2: EU517607.1 and H3: EU517606.1). One haplotype, H3, was found at all five breeding sites, whereas haplotypes H2 and H9 were shared among two sites each, and seven haplotypes were private to a single breeding site (Fig. 2).
For each MHC locus, a maximum of two alleles was recovered in every individual, indicating that pseudogenes and gene duplication were absent, as previously demonstrated for other ambystomatids with MHC primers (Sammut et al. 1997; Bos and DeWoody 2005; Tracy et al. 2015). For MHC class Iα, only a single reading frame (beginning with nucleotide 2) produced an amino acid sequence with no stop codons. MHC class IIβ had two reading frames that did not produce stop codons (starting with nucleotide 2 or 3), so we compared the sequence to the same region of the tiger salamander (DQ125478.1 and DQ071905.1) and used the reading frame of the tiger salamander (reading frame 3). All further analyses were conducted using reading frame 2 for MHC class Iα and reading frame 3 for MHC class IIβ. For the analysis in MEGA, the dN/dS ratio did not differ from neutral expectations at either MHC locus, indicating no evidence for selection (MHC class Iα Z = − 0.24, p = 0.81; MHC class IIβ Z = 0.55, p = 0.60). For MHC class Iα, no amino acids in our sequences were predicted to be part of the peptide binding region (Lillie et al. 2014). For MHC class IIβ, seven amino acids were associated with peptide binding (Bondinas et al. 2006), although this region did not appear to be under selection as the dN/dS ratio did not differ from neutral expectations (Z = 0.38, p = 0.88). Using HyPhy, no selection was detected for MHC class Iα but for MHC class IIβ, purifying selection was detected at amino acid 33 in the peptide binding region (α = 84.5, p = 0.019).
Eglin AFB, as a whole (combining the East and West sites), had the highest levels of allelic richness, heterozygosity, and nucleotide diversity for the MHC class IIβ (Table 1), as well as the greatest number of haplotypes, highest nucleotide diversity, and greatest number of private haplotypes at the mtDNA D-loop (Table 1). In contrast, Eglin had lower MHC class Iα nucleotide diversity and allelic richness than all other sites except Garcon Point. Escribano Point exhibited the highest MHC class Iα nucleotide diversity and allelic richness. Escribano Point also was the only site to exhibit private MHC alleles, at both loci. Mayhaw WMA had intermediate levels of nucleotide diversity at both MHC loci. It also had very low levels of mtDNA haplotype and nucleotide diversity. Garcon Point was the least diverse site and exhibited low nucleotide diversity and heterozygosity at both MHC loci and mtDNA. Although Garcon Point and Mayhaw WMA did not have any private alleles, they did share a unique MHC class IIβ allele that was not found on Eglin AFB or Escribano Point (Table 1).
When these results were compared to other amphibian species, RFS had the fewest alleles and lowest nucleotide diversity at MHC class Iα, and only one species, the Mountain Stream Salamander (Ambystoma altamirani), had lower MHC class IIβ diversity (Table 4). Though MHC diversity was lower than almost all surveyed amphibian species, mitochondrial variation was similar to other ambystomatid species with RFS exhibiting comparable haplotype diversity (Table 1; Church et al. 2003; Zamudio and Savage 2003).
We did not detect ranavirus in any RFS samples, during any year, with either protocol. This result is in accordance with records from the US Geological Service wildlife health center database (https://www.usgs.gov/centers/nwhc/data-tools), which had no reports of ranavirus over the last 100 years for either the RFS or the closely related frosted flatwoods salamander (Ambystoma cingulatum).
Discussion
MHC class Iα and IIβ diversity in RFS was lower than levels observed in many other amphibians (Table 4), including species that have experienced severe population declines, such as the southern corroboree frog (Pseudophryne corroboree), Mexican axolotl, and Chinese giant salamander (Andrias davidianus, Contreras et al. 2009; Zhu et al. 2014; Kosch et al. 2017). It is, however, difficult to make direct comparisons of MHC diversity among amphibian species because different selective pressures are probably acting on each species, and many frogs and some salamanders have MHC pseudogenes and gene duplications (i.e., multiple loci) at MHC genes (Sammut et al. 1997; Bos and DeWoody 2005; Kiemnec-Tyburczy et al. 2012; Zhao et al. 2013; Tracy et al. 2015; Kosch et al. 2017). However, a few species did have low levels of MHC class IIβ diversity similar to RFS: the mountain stream salamander (Ambystoma altamirani), plateau tiger salamander (Ambystoma velasci), and Chiricahua leopard frog all have five or fewer alleles. Also, northern (post-glacial) populations of Great Crested Newt (Triturus cristatus), an abundant and widespread species, had the lowest MCH class IIβ diversity with only 2 alleles (Babik et al. 2009). These three species have all experienced recent and dramatic population declines, caused mostly by extensive habitat loss for the salamanders and chytridiomycosis for the Chiricahua leopard frog. Although these species had similarly low levels of diversity at MHC class IIβ, the RFS also exhibited depauperate MHC class Iα diversity with few alleles and low nucleotide diversity when compared to all other species examined (Table 4).
Low levels of MHC diversity in RFS may have been caused by many factors, such as the documented population declines, neutral processes like drift, or perhaps selection caused by previous exposure to disease. Microsatellite M-ratios are consistently < 0.5 in all Eglin breeding ponds, indicating that a severe bottleneck has occurred in this area (James Roberts, unpublished data). Yet population declines alone do not typically reduce MHC diversity to the extent observed in RFS. For example, the San Nicolas island fox (Urocyon littoralis dickeyi), Peary caribou (Rangifer tarandus pearyi), Chinese giant salamander, and the Lake Patzcuaro salamander (Ambystoma dumerilii) all retained more MHC alleles than observed in RFS despite large population bottlenecks (Aguilar et al. 2004; Taylor et al. 2012; Tracy et al. 2015). Although these species may have had historically larger effective population sizes (and therefore greater diversity), comparing the relative loss of diversity among marker types is suggestive of a bottleneck for the RFS. For example, genetic diversity as estimated with microsatellite data (HE, HO, AR) for RFS on Eglin AFB was comparable to other amphibian species (Wendt 2017), but MHC diversity was lower in RFS than in eighteen other amphibian species (Table 4). This pattern also holds for mitochondrial diversity: RFS mitochondrial diversity was similar to other ambystomatid species (Church et al. 2003; Zamudio and Savage 2003), whereas MHC diversity was lower. Similarly, the Chiricahua leopard frog, which experienced a bottleneck caused by a combination of habitat loss and disease pressure, apparently lost much of its MHC diversity, but retained mitochondrial diversity (Savage et al. 2018). Thus, a population bottleneck could reduce genetic diversity of MHC, microsatellite and mitochondrial markers evenly, but if that bottleneck was in combination with a historical disease outbreak, RFS could have lost MHC diversity to a greater extent. Low MHC diversity suggests that the RFS could be at an elevated risk from infectious diseases, as it lacks the broad spectrum of alleles useful in combating pathogens (Savage et al. 2019). Ranavirus is spread easily, and if the few remaining RFS breeding sites were exposed to this virus or any other amphibian pathogen, it could negatively impact population size, cause local extirpations, or even drive the species to extinction.
Biosecurity measures to prevent the spread of disease, such as washing all equipment in a 5% bleach solution (Gold et al. 2013; Gray and Chinchar 2015), are currently in place at most breeding sites; however, application is not uniform and disease screening is irregular. Biosecurity is vital as new, more virulent, chimeric ranaviruses are occurring as a result of recombination among different strains (Claytor et al. 2017; Peace et al. 2019). Although we did not detect ranavirus in RFS, it is a highly virulent disease and, consequently, is one of only two amphibian pathogens that must be reported to the US Geological Service wildlife health center (Schloegel et al. 2010). Lack of detection in our samples was probably not the result of amplification issues: during every screening, the positive control amplified ranavirus DNA while the negative control never produced amplified product. However, other factors could have negatively impacted our ability to identify virus, as ranavirus has been detected at low concentrations on Eglin with qPCR in other amphibian species (Dr. Debra Miller, University of Tennessee, personal communication). Tissue type may have been a factor: although Greer et al. (2009) successfully used tail clips to detect ranavirus, other tissues like spleen harbor more viral DNA and may have returned different results. Moreover, tail tissue tests positive for ranavirus later in the infection timeline when compared to organ tissue, thus using tail tissue can underestimate ranavirus prevalence (Greer and Collins 2007). Ranavirus can also occur in pulse events; therefore, its occurrence can vary widely from year to year (Gray and Chinchar 2015), and it is possible that samples were collected during a period of little ranavirus activity. However, it is also possible that ranavirus really was absent as the nature of RFS wetlands might reduce the presence of disease. RFS ponds are dry for much of the year, and the basins are burned with some regularity. If dry conditions and fire are inhospitable to ranavirus, it may not be surprising that we did not detect this disease. However, given the elevated risk of disease due to low immunogenetic variation, continued disease monitoring and proper biosecurity measures should be implemented at all sites to minimize future exposure to novel pathogens. Specifically, sampling multiple taxa across several sites on a bi-weekly or monthly basis, while host species are present, should detect most outbreaks. Non-lethal samples like toe clips can be used, but if available, organ samples should result in better detection (Gray and Chinchar 2015).
Despite low levels of genetic diversity, private alleles still exist across the extant range of the RFS. In particular, MHC IIβ allele 5 was found only at Garcon Point and Mayhaw (Fig. 2). In spite of their smaller population sizes, these sites still harbor unique genetic variants, which contribute meaningfully to the overall diversity at that locus. In contrast, mitochondrial diversity was unevenly distributed across the breeding sites. Eglin AFB as a whole accounts for more than half of the recovered haplotypes, five of which were private, although the large sample size at Eglin AFB may explain higher levels of diversity than that observed at other sites. Nevertheless, not one location contains all remaining diversity and thus each extant breeding site retains high conservation value.
We have demonstrated low levels of immune gene diversity, a result that emphasizes the urgency for further conservation and the need to consider genetic diversity as a valuable asset alongside other restoration goals (Bonin et al. 2007). Because the RFS is at an elevated risk from disease, biosecurity should be a priority at all breeding sites. Management should include considerations to preserve unique genetic variants, increase allelic richness, and reduce the loss of genetic diversity generally across the RFS range (Radwan et al. 2010). Genetic variation takes thousands of generations to replace; therefore, preserving genetic diversity of the reticulated flatwoods salamander will be crucial for conserving this vulnerable species.
References
Aguilar A, Roemer G, Debenham S et al (2004) High MHC diversity maintained by balancing selection in an otherwise genetically monomorphic mammal. Proc Natl Acad Sci 101:3490–3494
Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P (2015) Molecular biology of the cell, 6th edn. Chapter 24. Garland Science, New York
Allender MC, Bunick D, Mitchell MA (2013) Development and validation of TaqMan quantitative PCR for detection of frog virus 3-like virus in eastern box turtles (Terrapene carolina carolina). J Virol Methods 188:121–125
Allendorf FW, Luikart G, Aitken S (2013) Conservation and the genetics of populations. Blackwell publishing
Babik W, Pabijan M, Arntzen JW, Cogâlniceanu D, Durka W, Radwan J (2009) Long-term survival of a urodele amphibian despite depleted major histocompatibility complex variation. Mol Ecol 18:769–781
Bandelt HJ, Forster P, Rohl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16:37–48
Bataille A, Cashins SD, Grogan L et al (2015) Susceptibility of amphibians to chytridiomycosis is associated with MHC class II conformation. Proc R Soc B Biol Sci 282:20143127–20143127
Belasen AM, Bletz MC, Leite D et al (2019) Long-term habitat fragmentation is associated with reduced MHC IIB diversity and increased infections in amphibian hosts. Front Ecol Evol 6:236
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B 57:289–300
Berger L, Speare R, Daszak P et al (1998) Chytridiomycosis causes amphibian mortality associated with population declines in the rain forests of Australia and Central America. Proc Natl Acad Sci 95:9031–9036
Bernatchez L, Landry C (2003) MHC studies in nonmodel vertebrates: what have we learned about natural selection in 15 years? J Evol Biol 16:363–377
Bishop DC, Haas CA (2005) Burning trends and potential negative effects of suppressing wetland fires on flatwoods salamanders. Nat Areas J 25:290–294
Blackburn M, Wayland J, Smith WH et al (2015) First report of ranavirus and Batrachochytrium dendrobatidis in green salamanders (Aneides aeneus) from Virginia, USA. Herpetol Rev 46:357–361
Bondinas GP, Moustakas AK, Papadopoulos GK (2006) The spectrum of HLA-DQ and HLA-DR alleles: a listing correlating sequence and structure with function. Immunogenetics 59:539–553
Bonin A, Nicole F, Pompanon F, Miaud C, Taberlet P (2007) Population adaptive index: a new method to help measure intraspecific genetic diversity and prioritize populations for conservation. Conserv Biol 21:697–708
Bos DH, DeWoody JA (2005) Molecular characterization of major histocompatibility complex class II alleles in wild tiger salamanders (Ambystoma tigrinum). Immunogenetics 57:775–781
Chandler HC, Rypel AL, Jiao Y et al (2016) Hindcasting historical breeding conditions for an endangered salamander in ephemeral wetlands of the southeastern USA: implications of climate change. PLoS One 11:e0150169
Chinchar VG (2002) Ranaviruses (family Iridoviridae): emerging cold-blooded killers. Arch Virol 147:447–470
Church SA, Kraus JM, Mitchell JC, Church DR, Taylor DR (2003) Evidence for multiple Pleistocene refugia in the postglacial expansion of the eastern tiger salamander, Ambystoma tigrinum tigrinum. Evolution 57:372–383
Claytor SC, Subramaniam K, Landrau-Giovannetti N, Chinchar VG, Gray MJ, Miller DL, Mavian C, Salemi M, Wisely S, Waltzek TB (2017) Ranavirus phylogenomics: signatures of recombination and inversions among bullfrog ranaculture isolates. Virology 511:330–343
Contreras V, Martínez-Meyer E, Valiente E et al (2009) Recent decline and potential distribution in the last remnant area of the microendemic Mexican axolotl (Ambystoma mexicanum). Biol Conserv 142:2881–2885
Core Team R (2018) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Elbers JP, Taylor SS (2016) Major histocompatibility complex polymorphism in reptile conservation. Herpetol Conserv Biol 11:1–12
Farmer AL, Walls SC, Haas C et al (2016) A statewide species and habitat assessment for the reticulated flatwoods salamander, frosted flatwoods salamander, and striped newt. Florida Fish and Wildlife Annual Report
Fox S, Greer A, Torres-Cervantes R, Collins J (2006) First case of ranavirus-associated morbidity and mortality in natural populations of the South American frog Atelognathus patagonicus. Dis Aquat Org 72:87–92
Frankham R, Ballou JD, Briscoe DA (2002) Introduction to conservation genetics. Cambridge University Press
Frost CC (1993) Four centuries of changing landscape patterns in the longleaf pine ecosystem. In: Proceedings of the Tall Timbers Fire Ecology Conference. pp 17–43
Fu M, Waldman B (2017) Major histocompatibility complex variation and the evolution of resistance to amphibian chytridiomycosis. Immunogenetics 69:529–536
Gold K, Reed P, Bemis D, Miller DL, Gray MJ, Souza MJ (2013) Efficacy of common disinfectants and terbinafine in inactivating the growth of Batrachochytrium dendrobatidis in culture. Dis Aquat Org 107:77–81
Goudet J (2001) FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3). http://www2.unil.ch/popgen/softwares/fstat.htm
Gray MJ, Chinchar VG (eds) (2015) Ranaviruses. Springer International Publishing, Cham
Green DE, Converse KA, Schrader AK (2002) Epizootiology of sixty-four amphibian morbidity and mortality events in the USA, 1996-2001. Ann N Y Acad Sci 969:323–339
Greer AL, Collins JP (2007) Sensitivity of a diagnostic test for amphibian Ranavirus varies with sampling protocol. J Wildl Dis 43:525–532
Greer AL, Brunner J, Collins JP (2009) Spatial and temporal patterns of Ambystoma tigrinum virus (ATV) prevalence in tiger salamanders Ambystoma tigrinum nebulosum. Dis Aquat Org 85:1–6
IUCN [International Union for Conservation of Nature] (2008) Ambystoma bishopi: John Palis, Geoffrey Hammerson: the IUCN Red List of Threatened Species. International Union for Conservation of Nature
Johnson AJ, Pessier AP, Wellehan JFX, Childress A, Norton TM, Stedman NL, Bloom DC, Belzer W, Titus VR, Wagner R, Brooks JW, Spratt J, Jacobson ER (2008) Ranavirus infection of free-ranging and captive box turtles and tortoises in the United States. J Wildl Dis 44:851–863
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A (2012) Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649
Kiemnec-Tyburczy KM, Richmond JQ, Savage AE et al (2012) Genetic diversity of MHC class I loci in six non-model frogs is shaped by positive selection and gene duplication. Heredity 109:146
Kik M, Martel A, der Sluijs AS et al (2011) Ranavirus-associated mass mortality in wild amphibians, the Netherlands, 2010: a first report. Vet J 190:284–286
Kosakovsky P, Sergei L, Frost SDW (2005) Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol Biol Evol 22:1208–1222
Kosch TA, Eimes JA, Didinger C, Brannelly LA, Waldman B, Berger L, Skerratt LF (2017) Characterization of MHC class IA in the endangered southern corroboree frog. Immunogenetics 69:165–174
Kumar S, Stecher G, Li M, Knyaz C, Tamura K, Battistuzzi FU (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549
Lillie M, Shine R, Belov K (2014) Characterisation of major histocompatibility complex class I in the Australian cane toad, Rhinella marina. PLoS One 9:e102824
Mao J, Hedrick RP, Chinchar VG (1997) Molecular characterization, sequence analysis, and taxonomic position of newly isolated fish iridoviruses. Virology 229:212–220
Marsh IB, Whittington RJ, O’Rourke B et al (2002) Rapid differentiation of Australian, European and American ranaviruses based on variation in major capsid protein gene sequence. Mol Cell Probes 16:137–151
Martel A, Spitzen-van der Sluijs A, Blooi M et al (2013) Batrachochytrium salamandrivorans sp. nov. causes lethal chytridiomycosis in amphibians. Proc Natl Acad Sci 110:15325–15329
McCallum ML (2007) Amphibian decline or extinction? Current declines dwarf background extinction rate. J Herpetol 41:483–491
McIntyre RK, Guldin JM, Ettel T et al (2018) Restoration of longleaf pine in the southern United States: a status report 6
Miller D, Gray M, Storfer A (2011) Ecopathology of ranaviruses infecting amphibians. Viruses 3:2351–2373
O’Donnell KM, Messerman AF, Barichivich WJ et al (2017) Structured decision making as a conservation tool for recovery planning of two endangered salamanders. J Nat Conserv 37:66–72
O’Hanlon SJ, Rieux A, Farrer RA et al (2018) Recent Asian origin of chytrid fungi causing global amphibian declines. Science 360:621–627
Palis J (1996) Flatwoods salamander (Ambystoma cingulatum Cope). Nat Areas J 16:49–54
Palis JG (1997) Breeding migration of Ambystoma cingulatum in Florida. J Herpetol 31:71
Pauly GB, Piskurek O, Shaffer HB (2007) Phylogeographic concordance in the southeastern United States: the flatwoods salamander, Ambystoma cingulatum, as a test case: flatwoods salamander phylogeography. Mol Ecol 16:415–429
Peace A, O’Regan SM, Spatz JA et al (2019) A highly invasive chimeric ranavirus can decimate tadpole populations rapidly through multiple transmission pathways. Ecol Model 410:108777
Petranka J (2010) Salamanders of the United States and Canada. Smithsonian Institution Press, Washington
Picco AM, Brunner JL, Collins JP (2007) Susceptibility of the endangered California tiger salamander, Ambystoma californiense, to ranavirus infection. J Wildl Dis 43:286–290
Price SJ, Garner TWJ, Nichols RA, Balloux F, Ayres C, Mora-Cabello de Alba A, Bosch J (2014) Collapse of amphibian communities due to an introduced ranavirus. Curr Biol 24:2586–2591
Radwan J, Biedryzycka A, Babik W (2010) Does reduced MHC diversity decrease viability of vertebrate populations. Biol Conserv 143:537–544
Raymond M, Rousset F (1995) GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86:248–249
Richman AD, Herrera G, Reynoso VH, Méndez G, Zambrano L (2007) Evidence for balancing selection at the DAB locus in the axolotl, Ambystoma mexicanum. Int J Immunogenet 34:475–478
Richmond JQ, Savage AE, Zamudio KR, Rosenblum EB (2009) Toward immunogenetic studies of amphibian chytridiomycosis: linking innate and acquired immunity. BioScience 59:311–320
Rousset F (2008) Genepop’007: a complete reimplementation of the Genepop software for Windows and Linux. Mol Ecol Resour 8:103–106
Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, Sánchez-Gracia A (2017) DnaSP 6: DNA sequence polymorphism analysis of large datasets. Mol Biol Evol 34:3299–3302
Sammut B, Laurens V, Tournefier A (1997) Isolation of MHC class I cDNAs from the axolotl Ambystoma mexicanum. Immunogenetics 45:285–294
Sammut B, Du Pasquier L, Ducoroy P et al (1999) Axolotl MHC architecture and polymorphism. Eur J Immunol 29:2897–2907
Savage AE, Zamudio KR (2011) MHC genotypes associate with resistance to a frog-killing fungus. Proc Natl Acad Sci 108:16705–16710
Savage AE, Zamudio KR (2016) Adaptive tolerance to a pathogenic fungus drives major histocompatibility complex evolution in natural amphibian populations. Proc R Soc B Biol Sci 283:20153115
Savage AE, Terrell KA, Gratwicke B et al (2016) Reduced immune function predicts disease susceptibility in frogs infected with a deadly fungal pathogen. Conservation Physiology
Savage AE, Mulder KP, Torres T, Wells S (2018) Lost but not forgotten: MHC genotypes predict overwinter survival despite depauperate MHC diversity in a declining frog. Conserv Genet 19:309–322
Savage AE, Muletz-Wolz CR, Campbell Grant EH, Fleischer RC, Mulder KP (2019) Functional variation at an expressed MHC class IIβ locus associates with ranavirus infection intensity in larval anuran populations. Immunogenetics 71:335–346
Schloegel L, Daszak P, Cunningham A, Speare R, Hill B (2010) Two amphibian diseases, chytridiomycosis and ranaviral disease, are now globally notifiable to the World Organization for Animal Health (OIE): an assessment. Dis Aquat Org 92:101–108
Semlitsch RD, Walls SC, Barichivich WJ, O’Donnell KM (2017) Extinction debt as a driver of amphibian declines: an example with imperiled flatwoods salamanders. J Herpetol 51:12–18
Shaffer HB, McKnight ML (1996) The polytypic species revisited: genetic differentiation and molecular phylogenetics of the tiger salamander Ambystoma tigrinum (Amphibia: Caudata) complex. Evolution 50:417–433
Sommer S (2005) The importance of immune gene variability (MHC) in evolutionary ecology and conservation. Front Zool 2:16–34
Taylor SS, Jenkins DA, Arcese P (2012) Loss of MHC and neutral variation in Peary Caribou: genetic drift is not mitigated by balancing selection or exacerbated by MHC allele distributions. PLoS One
Teacher AGF, Garner TWJ, Nichols RA (2009) Evidence for directional selection at a novel major histocompatibility class I marker in wild common frogs (Rana temporaria) exposed to a viral pathogen (ranavirus). PLoS One
Tracy KE, Kiemnec-Tyburczy KM, DeWoody JA, Parra-Olea G, Zamudio KR (2015) Positive selection drives the evolution of a major histocompatibility complex gene in an endangered Mexican salamander species complex. Immunogenetics 67:323–335
USFWS [U.S. Fish and Wildlife Service] (2009) Endangered and threatened wildlife and plants; determination of endangered status for reticulated flatwoods salamander; designation of critical habitat for frosted flatwoods salamander and reticulated flatwoods salamander. Federal Register. 74 FR 6700
USFWS [U.S. Fish and Wildlife Service] (2015) Reticulated flatwoods salamander (Ambystoma bishopi) 5-year review: summary and evaluation. Federal Register. 79 FR 56821
Weaver S, Shank SD, Spielman SJ, Li M, Muse SV, Kosakovsky Pond SL (2018) Datamonkey 2.0: a modern web application for characterizing selective and other evolutionary processes. Mol Biol Evol 35:773–777
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Wendt AS (2017) A population genetic investigation of the reticulated flatwoods salamander (Ambystoma bishopi) on Eglin Air Force Base (MS thesis). Georgia Southern University, Statesboro
Zamudio KR, Savage WK (2003) Historical isolation, range expansion, and secondary contact of two highly divergent mitochondrial lineages in spotted salamanders (Ambystoma maculatum). Evolution 57:1631–1652
Zhao M, Wang Y, Shen H et al (2013) Evolution by selection, recombination, and gene duplication in MHC class I genes of two Rhacophoridae species. BMC Evol Biol 13:113
Zhu R, Chen Z, Wang J, Yuan JD, Liao XY, Gui JF, Zhang QY (2014) Extensive diversification of MHC in Chinese giant salamanders Andrias davidianus (Anda-MHC) reveals novel splice variants. Dev Comp Immunol 42:311–322
Acknowledgments
We are thankful to A. Perez-Umphrey, C. Rutt, A. Snider, A. Settlecowski, A. Bresnan, and T. Turner for extensive edits to this manuscript as well as Dr. Kim Terrell and Dr. Chris Austin, for their support and helpful comments. Thank you to everyone who helped in sample acquisition: K. Jones, B. Rincon, V. Porter, C. Abeles, A. Farmer, P. Hill, L. Smith, R. Bilbow, J. de Silva, K. Erwin, B. Moore, J. Sandoval, and E. Browning. We are grateful to A. Wendt who extracted all of the samples collected on Eglin and to Dr. M. Allender for providing a linearized ranavirus plasmid. Portions of this research were conducted with high-performance computational resources provided by the Louisiana Optical Network Infrastructure (http://www.loni.org). This material is based upon work that is supported by the National Institute of Food and Agriculture, U.S. Department of Agriculture, McIntire Stennis program. Collection of tissue samples and extraction of DNA from salamanders on Eglin Air Force Base was supported by Eglin Air Force Base Jackson Guard, the US Air Force Civil Engineer Center, and the US Fish and Wildlife Service. Salamander samples from Eglin Air Force Base were collected under US Fish and Wildlife Service Threatened and Endangered Species Permit #TE049502 and Virginia Tech IACUC protocols #13-129 and #16-100. Samples from Mayhaw WMA were collected under Georgia Department of Natural Resources scientific collection permit #029, and samples at Garcon Point and Escribano Point were collected under the section 6 agreement between USFWS and Florida Fish and Wildlife Conservation Commission.
Funding
This work would not be possible without the funding provided by Audubon Center for Research of Endangered Species (ACRES) grant. This project/work used Genomics core facilities that are supported in part by COBRE (NIH8 1P30GM118430-02) and NORC (NIH 2P30DK072476) center grants from the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(XLSX 35.1 kb)
Rights and permissions
About this article

Cite this article
Williams, S.T., Haas, C.A., Roberts, J.H. et al. Depauperate major histocompatibility complex variation in the endangered reticulated flatwoods salamander (Ambystoma bishopi). Immunogenetics 72, 263–274 (2020). https://doi.org/10.1007/s00251-020-01160-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00251-020-01160-y