
Abstract
Natural killer T (NKT) cells are a unique subset of lymphocytes that bridge the innate and adaptive immune system. NKT cells possess a classic αβ T cell receptor (TCR) that is able to recognize self and foreign glycolipid antigens presented by the nonclassical class I major histocompatibility complex (MHC) molecule, CD1d. Type I NKT cells (referred to as invariant NKT cells) express a semi-invariant Vα14Jα18 TCR in mice and Vα24Jα18 TCR in humans. Type II NKT cells are CD1d-restricted T cells that express a more diverse set of TCR α chains. The two types of NKT cells often exert opposing effects especially in tumor immunity, where type II cells generally suppress tumor immunity while type I NKT cells can enhance anti-tumor immune responses. In this review, we focus on the role of NKT cells in cancer. We discuss their effector and suppressive functions, as well as describe preclinical and clinical studies utilizing therapeutic strategies focused on harnessing their potent anti-tumor effector functions, and conclude with a discussion on potential next steps for the utilization of NKT cell-targeted therapies for the treatment of cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The initial discovery of conserved Vα14 chains across several hybridoma lines was quite unusual (Imai et al. 1986) because T cell receptors (TCRs) are highly diverse, with 70–80 α chain genes giving an enormous diversity to TCRα. Then a population of thymocytes possessing a single Vβ chain (Vβ) was described and posited to “represent a distinct lineage” (Fowlkes et al. 1987). It was this discovery that prompted the search for a subtype characterized by an invariant Vα14 chain with Vβ chains of limited diversity. Koseki et al. discovered that the frequency of the Vα14+ cells was much higher than expected, with Vα14+ cells comprising as much as 10–20 % of T cells in the livers of mice (Koseki et al. 1990). Almost a decade after the discovery of this remarkable population of Vα14+ cells, Adachi et al. noted a significant reduction in β2-microglobulin-deficient mice (Adachi et al. 1995). Later, Lantz and Bendelac ascertained that the Vα14+ cells were CD1d restricted and it became clear that this was a distinct population of T cells (Lantz and Bendelac 1994).
NKT cell subsets and cytokine profiles
NKT cells have been characterized according to their expression of CD4 or lack thereof, particularly the distinct Th1 and Th2 cytokine profiles of these subsets. In humans, CD4+ NKT cells produce Th1 and Th2 cytokines, whereas the CD4− subset, which includes both CD8+ and double negative (DN), primarily produces Th1 cytokines (Godfrey et al. 2010). In addition, NKT cells are phenotypically similar to effector T cells because they express non-lymphoid tissue homing chemokine receptors such as CCR2, CCR5, and CXCR3.
Mouse NKT cells also include CD4+ and CD4− subsets, with different subsets possessing diverse functional profiles. There is heterogeneity within CD4+ NKT cell subset in regard to the expression of the IL-17 receptor B (IL-17RB), a receptor for IL-25. CD4+ IL-17RB+ NKT cells are reported to produce high levels of IL-13 and IL-4, but little IFN-γ in response to IL-25 stimulation. Another subset of NKT cells within the NK1.1− CD4− subset is the retinoic acid receptor-related orphan receptor (ROR)γt+ NKT cells, which produces IL-17A and IL-22 (Michel et al. 2007; Terashima et al. 2008; Watarai et al. 2012).
Coquet et al. demonstrated that mouse NKT cell populations exhibit diversity in their ability to produce cytokines, depending on the organ and subsets. NKT cell subpopulations from thymus, spleen, and liver produced 19 cytokines with differences of 10- to 100-fold in response to anti-CD3/CD28 stimulation, depending on the organ (Coquet et al. 2008). Liver-derived NKT cells produced equal or higher amounts of IFN-γ, as compared to thymic NKT cells in response to CD3/CD28 stimulation (Coquet et al. 2008). The majority of NKT cells found in the periphery express both IL-4 and IFN-γ mRNA (Stetson et al. 2003). Thus, NKT cells are primed to exert their effector functions could thus provide immediate protection at sites of pathogen entry, while conventional T cells are undergoing antigen-specific expansion (Stetson et al. 2003). Moreover, production of these cytokines may facilitate conventional T cell differentiation, with NKT cells serving as primary initiators of antigen-specific responses.
Depending on the lipid presented to NKT cells in the context of CD1d, differential cytokine profiles can be induced (Zajonc and Girardi 2015). The prototypical NKT cell agonist is α-galactosylceramide (α-GalCer), a glycolipid originally isolated from a marine sponge, Agelas mauritianus (Kawano et al. 1997). α-GalCer induces rapid cytokine production and proliferation and has been extensively studied as an adjuvant in cancer. For example, α-GalCer induces IL-4, IL-13, and IFN-γ, but β-GalCer is a poor inducer of IFN-γ, TNF-α, GM-CSF, and IL-4 gene expression (Ortaldo et al. 2004).
IL-12p70 and IL-23 are members of a small family of heterodimeric cytokines predominantly produced by dendritic cells (DCs) and macrophages. IL-12p70 is involved in the induction and amplification of the Th1 response, while IL-23 mediates inflammatory responses, through induction of expansion of Th17 cells (Ortaldo et al. 2004). Uemura et al. demonstrated that when NKT cells are co-cultured with DCs, NKT cells enhance the IL-12p70 production while downregulating IL-23 production by DCs (Uemura et al. 2009).
Effects of cytokines produced by NKT cells
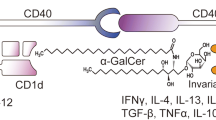
NKT cells can mediate anti-tumor activity via multiple mechanisms (Fig. 1). First, they can directly kill tumor cells. Second, they can induce maturation of dendritic cells in a CD40-CD40L-dependent manner (Fujii et al. 2007), thus initiating adaptive anti-tumor immunity. Finally, they activate NK cells and T cells by producing pro-inflammatory cytokines, such as IFN-γ and TNF-α. Using mouse tumor models of FBL-3 erythroleukemia and B16 melanoma, it was shown that in the absence of NKT cells, NK and T cells could not mediate tumor rejection (Cui et al. 1997).

NKT cells bridge innate and adaptive immune response. NKT cells have been shown to augment anti-tumor responses due, in part, to their capacity for rapid production of large amounts of IFN-γ, which acts on NK cells to target MHC negative tumors, and also, to target CD8 cytotoxic T cells to promote killing of MHC-positive tumors
In vivo administration of α-GalCer rapidly activates NKT cells to release Th1 and Th2 cytokines, which contribute to the activation of NK cells, dendritic cells, and T lymphocytes. Immature DCs can present antigens to NKT cells, which induce DC maturation, which in turn provides the necessary co-stimulation for NKT cell activation (Zaini et al. 2007).
Co-stimulatory requirements
NKT cells constitutively express cytokine mRNA and can synthesize cytokines in the absence of CD28 signaling, unlike conventional T cells, which require CD28 for cytokine gene transcription (Wang et al. 2009). Cytokine production by NKT cells is independent of CD28/CD40 co-stimulatory pathways. While CD28−/− mice have NKT cells, CD28 and CD40 signaling has been shown to be required for expansion of the NKT cells in vivo. Thus, NKT cells can be activated without CD28 co-stimulation, while the production of downstream factors such as IL-12 by DCs may be necessary to mount a full NKT cell response (Uldrich et al. 2005).
NKT cells express CD28 and CD28-CD80/CD86 co-stimulation is necessary for IL-4 production, which is drastically inhibited in presence of anti-CD80 and anti-CD86 antibodies (Hayakawa et al. 2001). Moreover, blockade of CD80 and CD86 resulted in a marked but partial inhibition of IFN-γ production. CD28- and CD40-deficient B6 mice have impaired IFN-γ production, but IL-4 production is impaired in CD28-deficient mice only. In addition, NKT cells from CD28 or CD40-deficient mice have impaired cytotoxic activity. Thus, CD154-CD40 blockade polarizes NKT cells towards a Th2 phenotype, with Th2 cytokine production being dependent on CD28 co-stimulation. On the other hand, IFN-γ production is dependent on IL-12 production by DCs and requires CD154-CD40 interaction, while CD28 is able to regulate IFN-γ production independently of IL-12, suggesting that IFN-γ production is dependent on both CD28 and CD40 co-stimulation in C57BL/6 mice (Hayakawa et al. 2001).
α-GalCer has been shown to promote DC maturation and subsequent antigen-specific T cell responses, with CD40-CD40L necessary for optimal DC-NKT cell interaction (Croudace et al. 2008). Moreover, TNF-α-induced maturation of DCs led to OX40L upregulation, which, through engagement of OX-40 on NKT cells, activated NKT cells (Zaini et al. 2007). IL-12 produced by DCs is critical for IFN-γ production by NKT cells and requires CD154-CD40 interaction. Due to their ability to rapidly produce a wide variety of cytokines, NKT cells can polarize the immune response towards a Th1 profile; while in other cases, NKT cells drive Th2 responses. In addition, NKT cells play an immunoregulatory or immunosuppressive role in some systems, usually through their production of the Th2-type cytokines; while in other systems, they appear to promote enhanced cell-mediated immunity via production of Th1-type cytokines. Work focused on delineating the factors that drive cytokine polarity of the NKT cell response and influence NKT cell responses in the context of initiating or regulating the host’s innate and adaptive immune system are ongoing.
Suppression of NKT cell activation
As a pillar of innate immunology and initiation of the adaptive immune response, methods of NKT cell suppression are of great interest in disease pathology. Decreased suppression results in inflammatory diseases, while increased suppression or dysfunction of the immune system is often a characteristic of exacerbated chronic infections, autoimmunity, and cancer. Cancers are the masters of immunosuppression. Some types of tumors can suppress NKT cells via shedding of inhibitory glycolipids (Sriram et al. 2002; Webb et al. 2012).
There have been three primary focuses of cellular suppression of NKT cell activation in anti-tumor immunity: regulatory T cells, immature myeloid cells, and NKT cells. Regulatory T cells (Tregs) are a well-known immunoregulatory component, and thus, the Treg:NKT interaction was one of the first to be examined closely. In vitro studies testing Vα24+ NKT proliferation, cytokine production, and direct cytotoxicity indicated that CD4+CD25+ Tregs can inhibit NKT responses in a dose-dependent, contact-dependent manner (Azuma et al. 2003). Yanagisawa et al. discovered that NKT cell responses are suppressed by the nitric oxide production of myeloid-derived suppressor cells (MDSCs) and that this suppression can be subverted by forced maturation of the MDSCs using all-trans-retinoic acid (Yanagisawa et al. 2006). In addition, it was shown that CCL20-producing tumor-associated macrophages (TAMs) can act as a hypoxic trap for tumor-infiltrating NKT cells, resulting in a loss of NKT cell function within the tumor microenvironment (Liu et al. 2012). Lastly, NKT cells can inhibit anti-tumor immunity (Osada et al. 2005). This phenomenon initially seemed very contradictory, but is explained by two concepts. First, the initial studies looked at bulk NKT cells. Type II NKT cells can be suppressive in the context of anti-tumor immunity by producing IL-13 that induces MDSCs (Ambrosino et al. 2008; Marrero et al. 2015; Terabe et al. 2005). Secondly, type I NKT cells are heterogeneous and the context of activation may affect their anti-tumor capacity through Th2 skewing (Renukaradhya et al. 2006). As shown by Izhak et al., there is an intricate network of suppression in the tumor microenvironment with multiple levels of suppression acting at once (Izhak et al. 2013). Collectively, these studies demonstrate that understanding NKT cell suppression within the tumor microenvironment mediated by tumors, Tregs, MDSCs, and type II NKT cells is extremely important in the context of developing effective cancer immunotherapy.
Utilization of NKT cells in vaccines for cancer treatment
Cancer therapies that incorporate NKT cell agonists have shown promise in improving vaccination strategies. Dendritic cell vaccines have been on the cusp of incorporation into standard cancer immunotherapy since their inception over two decades ago, but their complexity has slowed their progress. In an attempt to control all the variables, the first iterations of DC vaccines involved ex vivo antigen pulsing and maturation of monocyte-derived DCs (MoDCs) and suffered from low efficacy. In vivo maturation of DCs produces much better results, but the process must be carefully controlled to produce the desired Th1, CTL skewed anti-tumor immune response. DC maturation signals can include microbial products that trigger Toll-like receptors (TLRs) and co-stimulation provided by conventional T cells or NKT cells which occurs at a much higher frequency than antigen-specific conventional T cells at the start of an adaptive immune response (Vincent et al. 2002). The activated, memory phenotype of NKT cells makes them a natural choice for in vivo DC maturation. Upon reinfusion, DCs present α-GalCer to NKT cells via CD1d and the NKT cells in turn supply maturation signals to the DC (Toura et al. 1999).
The complexities of traditional DC-based vaccines have encouraged research into simpler methods such as using NKT cell activation as a type of adjuvant. NKT cells activated by α-GalCer stimulate anti-tumor immunity via IFN-γ that enhances the innate response through NK cell activity and the adaptive response via DC production of IL-12 and encouragement of a Th1, CTL response (Nakagawa et al. 2001). In mice, the ability of NK cells to be activated by IFN-γ from NKT cells and IL-12 from APCs has been well established (Eberl and MacDonald 2000). While the experiments to determine the ability of NKT cells to transactivate NK cells with the help of DCs have not been performed in humans, the interplay between NKT cells and the innate and adaptive immune systems is very important. It has been demonstrated in human cancer patients that NKT cell stimulation by α-GalCer-loaded mature DCs leads to increased IL-12 serum levels and a robust T cell response with the ability to produce memory CD8 T cells (Chang et al. 2005).
Protein antigen co-delivered with i.v. α-GalCer results in rapid DC maturation and induction of an effective adaptive immune response to challenge with tumors expressing that antigen (Fujii et al. 2003b; Hermans et al. 2003). This response is dependent on co-stimulatory molecules such as CD40 and CD80/86. DCs cross-present necrotic cell antigen more effectively than soluble antigen, therefore irradiated tumor cells administered α-GalCer (i.v.) as an adjuvant resulted in even more potent immune response (Liu et al. 2005). This concept was demonstrated in another study by induction of anti-tumor immunity to poorly immunogenic cancers such as plasmacytoma and myelomonocytic leukemia after i.v. administration of irradiated tumor cells loaded with α-GalCer (Shimizu et al. 2007). Other effective cell options for adjuvant loading have included antigen-loaded MDSCs and allogeneic, antigen mRNA-transfected fibroblasts (Fujii et al. 2009; Ko et al. 2009a).
Although α-GalCer has been shown to be relatively safe even at high doses, there has been a significant amount of research into alternative NKT-activating ligands for use as adjuvants. Tumors such as human melanoma can express NKT cell-activating ligands that enhance specific targeting (Wu et al. 2003). Because NKT cells can produce Th1, Th2, and immunosuppressive cytokines upon activation, the most pressing issue for this research is ensuring a Th1 phenotype of NKT cell activation. Indeed, many NKT cell antigens elicit differing cytokine and activation profiles (Wu et al. 2005). α-C-GalCer is a promising synthetic C-glycoside analog of α-GalCer because it shows much higher specificity for Th1 skewing in vivo when tested in anti-tumor models (Schmieg et al. 2003). While alpha-C-GalCer has been extremely effective in mice, it has shown reduced capacity in humans, leading to investigation of C-glycoside analogs (Li et al. 2009). Synthetic glycolipids that have shown potential for the strong Th1 skewing needed in cancer immunity include PyrC-αGalCer, 7DW8-5, NU-αGalCer, and DB06-1 and highlight the need to characterize how different glycolipid structures affect binding to CD1d and interaction with the NKT TCR (Aspeslagh et al. 2011; Aspeslagh et al. 2013; Birkholz et al. 2015; Li et al. 2010). Similarly, the NKT antigen β-mannosylceramide represents a complementary adjuvant to α-GalCer by inducing anti-tumor immunity that is dependent on TNF-α and NOS instead of IFN-γ (O’Konek et al. 2011). The phenotype of the DC interacting with the NKT cell can also impact its anti-tumor capacity. For instance, cooperative stimulation of TLR4 or TLR9 on DCs and NKT cells enhances the resulting adaptive immune response (Hermans et al. 2007; Paget et al. 2007). Administering anti-tumor antibodies with TLR agonists and NKT-activating ligands enhances NK-mediated ADCC (Moreno et al. 2008). CD1d fusion proteins allow a more physiologic stimulation of NKT cells that eliminates the risk of anergy. α-GalCer-loaded CD1d-anti-HER2 scFV has been shown to localize activated NKT cells to the tumor site and induce anti-tumor adaptive immunity (Corgnac et al. 2012; Stirnemann et al. 2008). In allergic airway inflammation models, α-GalCer-antigen conjugates have been used to induce tolerance to allergens (Anderson et al. 2014). The cumulative work of the past decade has made consideration of NKT cells in vaccine design second nature.
NKT cells in preclinical studies
One of the earliest preclinical studies that determined the significance of NKT cell-mediated anti-tumor responses was demonstrated by the IL-12-mediated rejection of tumors (Cui et al. 1997). It was initially hypothesized that IL-12-induced anti-tumor effects were mediated mainly by NK cells or CD8+ T cells (Tahara 1995). To elucidate the specific mechanism of IL-12, Jα18−/− mice (lacking the invariant TCR) were generated and injected with melanoma cell lines, which preferentially metastasize to the liver. Liver metastases were greater in Jα18−/− mice than WT and were similar to RAG−/− mice. This study confirmed that NKT cells are primarily responsible for IL-12-mediated tumor rejection and shed light on the functions of the various subsets of lymphocytes in vivo. Since this initial study, many advances have been made in our understanding of NKT cells, in general, and their anti-tumor properties, as reviewed in (McEwen-Smith et al. 2015; Robertson et al. 2014). To date, NKT cell responses to a variety of solid tumors and hematopoietic malignancies have shown preclinical efficacy.
Hematological malignancies
NKT cells are known to exert their effector functions by recognition of CD1d on target cells. Thus, expression of CD1d is important for tumor recognition and subsequent elimination (Haraguchi et al. 2006). Many hematological malignancies, including myelomonocytic leukemia (Metelitsa et al. 2003), chronic and acute lymphoblastic leukemia (CLL and ALL; Fais et al. 2005; Anastasiadis et al. 2014), and lymphoid neoplasms (Guo et al. 2014), express CD1d molecules and the co-stimulatory proteins needed to induce an anti-tumor immune response by NKT cells. Despite this, most of these cancers are poorly immunogenic, and CD1d expression correlates with a poor prognosis (Fais et al. 2005). However, these malignancies are primed for NKT cell-based immunotherapeutic interventions, and numerous preclinical studies demonstrate their potential. Studies by Dhodapker’s group have identified CD1d-binding ligands that are inflammation-associated lysophospholipids isolated from myeloma patients (Chang et al. 2008).
Our lab has assessed the NKT cell responses in spontaneous model of B cell lymphoma. It was found that treatment of these tumor-bearing mice with a single intravenous dose of α-GalCer resulted in delayed disease progression and prolonged survival compared to vehicle treated controls (Li et al. 2014a). Additional treatment methods involve α-GalCer incorporated into vaccination strategies. Consistent with our work, a single vaccination with α-GalCer-loaded A20 lymphoma cells elicited effective anti-tumor immunity against tumor challenge and tumor regression (Chung et al. 2007). Thus, these studies set the basis for the efficacy of more innovative strategies such as NKT cell adjuvant-based vaccination strategies, and adoptive immunotherapy.
Tumor cell vaccines are a more recent immunotherapeutic strategy. It involves loading of α-GalCer into autologous tumors administered as an irradiated cellular vaccine. The rationale for this approach is that the irradiated tumor cells will act as a source of tumor-specific neoantigens, in addition to the agonistic α-GalCer, thus allowing for a robust immune response mediated largely by NKT cells. Moreover, this method is relatively simple, inexpensive (in comparison to genetically modified T cells), and patient-specific (Mattarollo and Smyth 2013). These vaccines have shown potential in the preclinical setting of hematological malignancies. In a murine mouse model of acute leukemia, C1498, one such study demonstrated anti-cancer efficacy with i.v. administered, irradiated α-GalCer-loaded leukemic cells. The vaccine was highly effective as a prophylactic in preventing the onset of leukemia, but was unable to provide any protective benefit in established malignancies. However, if mice were treated with standard chemotherapy and the vaccine, the mice were protected upon rechallenge with second dose of leukemic cells. The lack of response to established cancers was found to be due to several immunosuppressive mechanisms including the presence of Tregs, and MDSCs, as well as tumor cell intrinsic means (Gibbins 2014). It would be interesting to determine if further modulation of NKT cells via adjuvants can render other cytotoxic cells resistant to immune suppression, as previously described in breast cancer (Kmieciak et al. 2011). Taken together, these findings have far-reaching clinical implications.
NKT cell adoptive immunotherapy in the preclinical setting has demonstrated potential in lymphoid neoplasms (Bagnara et al. 2009). Ex vivo expanded NKT cells were shown to limit the growth of a lymphoblastic CD1d+ cell line, but not CD1d− or CD1c−expressing negative controls. A significant reduction in tumor burden was observed after two sequential doses of α-GalCer and NKT cells. Moreover, these cells were present within the tumor after 24 h (Bagnara et al. 2009). These studies highlight a role for ex vivo expanded NKT cells as an effective therapeutic tool for treatment of CD1d-expressing hematologic malignancies.
Solid tumors
Preclinical studies examining NKT cell responses within solid tumors have been investigated extensively. Initial work assessing the role of NKT cells in immune regulation showed differential anti-tumor immunity of murine NKT cell subsets in methylcholanthrene (MCA)-induced sarcomas (Crowe et al. 2005). It was determined that liver-derived, but not spleen or thymus-derived, NKT cells are the predominant mediators of α-GalCer-induced rejection of established MCA tumors. Additionally, CD4− and CD4+ liver-derived NKT cells were administered separately to tumor-bearing TCR Jα18−/− mice. Tumor rejection was only observed in the CD4− population, while the CD4+ population only partially slowed growth compared to PBS-injected control. Further investigation in this study found that IL-4−/− mouse-derived NKT cells transferred to B16F10 (metastatic melanoma cell line)-inoculated mice conferred protection and decreased the number of lung metastases (Crowe et al. 2005). This investigation supports the notion that NKT cell subsets show varying levels of anti-tumor immune regulation and that the CD4+ subsets that produce IL-4 enhance tumor immune evasion.
Type II NKT cells have been shown to be the major contributor of NKT cell subsets to anti-cancer immune suppression (Renukaradhya et al. 2008; Terabe et al. 2005). Sulfatide has been established as an antigen specifically recognized by type II NKT cells, solely (Ambrosino et al. 2007). Therefore, the use of sulfatide should abrogate an immune response to cancer, and tolerate tumor growth and metastasis. Consistent with this, treatment of CT26 lung metastases with α-GalCer was hindered by the simultaneous injection of sulfatide (Kato et al. 2014). Thus, various NKT cell subsets play distinct roles against cancer, and these subsets are highly dependent, and responsive, to antigenic stimuli.
Improved characterization of NKT cell-specific antigens has revealed several agonists with anti-tumor potential. Unlike α-GalCer, which protects against cancers in a largely IFN-γ-dependent manner, β-mannosylceramide (β-ManCer) demonstrates anti-tumor potential by way of an IFN-γ-independent mechanism. One study has reported that NKT cell anti-tumor immunity against CT26 lung metastasis induced by β-ManCer is nitric oxide and TNF-α-dependent. Moreover, β-ManCer can activate type I NKT cells without inducing the long-term anergy associated with α-GalCer activation demonstrated by PD-1 expression. This lack of functional anergy allows for a second dose to provide therapeutic benefit against lung metastasis (O’Konek et al. 2013). It will be interesting to see how targeting co-stimulation influences NKT cell anti-tumor responses (Parekh et al. 2009; Zaini et al. 2007).
Lastly, combination therapy of three mAbs incorporating NKT cell-specific stimulation (termed 1DMab) demonstrated eradication of a variety of established tumor cell lines in mice (Teng et al. 2009). These results strongly support the rational combination NKT cell-based immunotherapeutics, in the form of mAbs, vaccines, or ex vivo expansion for adoptive immunotherapy.
NKT cell-based immunotherapy
Numerous studies have shown that cancer patients have a deficiency in both NKT cell number and function (Fujii et al. 2003a; Kawano et al. 1999; Tahir et al. 2001), suggesting that in vivo NKT cell modulation may be ineffective in patients with low NKT cell numbers. In 2005, Molling et al. found that NKT cell numbers were 47 % lower in cancer patients compared to age and gender matched healthy controls among a large cohort of patients (Molling et al. 2005). This reduction in NKT cell numbers was independent of tumor type or stage/grade. Chang et al. showed that multiple injections of α-GalCer-loaded mature dendritic cells lead to sustained expansion of NKT cells and antigen-specific T cells. However, these expanded NKT cells from cancer patients still exhibited reduced capacity for IFN-γ secretion compared to NKT cells from healthy controls (Chang et al. 2005). Importantly, it was shown that lenalidomide enhances antigen-specific expansion of NKT cells in response to α-GalCer in both healthy donors and patients with myeloma (Chang et al. 2006). Studies by this group later demonstrated that treatment of patients with asymptomatic myeloma with three cycles of combination of α-GalCer-loaded monocyte-derived dendritic cells and low-dose lenalidomide led to reduction in tumor-associated monoclonal immunoglobulin in three of four patients (Richter et al. 2013). Adoptive transfer of in vitro-activated NKT cells in patients with refractory lung cancer resulted in an increase in both the number of circulating NKT cells as well as the number of IFN-γ producing cells in the peripheral blood. The objective anti-tumor response rate, however, remained low. These studies indicate that cancer patients have a deficiency in both NKT cell number and function, suggesting that NKT cell modulation may be ineffective in patients with low NKT cells. Therefore, for these patients, adoptive immunotherapy using ex vivo-expanded NKT cells may be a more productive strategy (Motohashi et al. 2006).
Adoptive immunotherapy refers to the ex vivo stimulation and expansion of autologous T or NK cells, followed by reinfusion of the expanded lymphocyte population back into the patient. Thus far, the most successful efforts in adoptive immunotherapy have focused on the expansion of cancer-specific T cells isolated from tumors with a high mutation rate (i.e., melanoma) or ex vivo expansion of cancer-reactive T cells from the peripheral blood of patients. Rosenberg et al. were the first to demonstrate that ex vivo-expanded autologous tumor-specific cells are able to traffic to tumor sites and directly induce tumor shrinkage in vivo (Rosenberg et al. 1988). However, the efficacy of cancer immunotherapy using targeted T cells has been limited by tumor-induced immunosuppression mediated by myeloid-derived suppressor cells (MDSC) that interfere with T cell effector functions. To overcome MDSC-mediated immunosuppression, one strategy has focused on the conversion of MDSCs to APCs, which identified NKT cells as a key facilitator in the maturation of MDSCs into mature myeloid cells with anti-tumor immune stimulatory function (Ko et al. 2009b; Lee et al. 2012). A previous study using PBMCs of patients with early-stage breast cancer demonstrated that CD25+ NKT cells in combination with reprogrammed immune cells and cultured in the presence of MDSCs induced them to lose/downregulate CD11b, which was associated with HLA-DR up-regulation (Payne et al. 2013). More recent work by the same group demonstrated that concurrent administration of the epigenetic drug, decitabine, with adoptive transfer of tumor-sensitive T cells and CD25+ NKT cells rendered T cells resistant to remaining MDSCs and resulted in significantly prolonged survival of animals bearing metastatic tumor cells (Payne et al. 2016).
A limitation of adoptive immunotherapy is that patients must have preexisting tumor reactive cells, which are difficult to identify in non-melanoma malignancies. In addition, conventional T cells are limited by their HLA restriction (Vivier et al. 2012).To address this issue, methods have been developed to engineer T cells to express genes encoding tumor antigen specific T cell receptors or chimeric antigen receptors.
Chimeric antigen receptors (CAR)
The chimeric antigen receptor (CAR) T cell field has historically focused on using autologous T cells to derive the therapeutic dose. Currently used CARs typically consist of an extracellular single chain variable fragment of an antibody for antigen binding and an intracellular T cell receptor zeta chain that mimics TCR activation as well as 1 or 2 co-stimulatory signaling domains, such as CD28 or 4-1BB (Imai et al. 2004; Maher et al. 2002; Sadelain et al. 2013). A promising target for CAR-modified T cells is CD19 for B cell malignancies, since CD19 is only expressed on B cells and perhaps follicular dendritic cells (Uckun et al. 1988) and destruction of normal B cells is well tolerated. Indeed, recent clinical trials demonstrated that T cells redirected against the CD19 antigen induce sustained complete responses in patients with B cell malignancies (Brentjens et al. 2013; Grupp et al. 2013; Kochenderfer et al. 2012; Porter et al. 2011).
The use of polyclonal T cells for CAR therapy, however, is associated with a concomitant risk of toxicity and GVHD. An alternate approach would be to use nonconventional donor T cells, such as NKT cells, to generate CAR NKT cells. NKT cells are of particular interest since NKT-cell infiltration of primary tumors is associated with better outcomes in diverse tumors (Metelitsa et al. 2004; Tachibana et al. 2005). As outlined in Fig. 2, CAR NKT cells have distinct mechanistic advantages over CARs generated from bulk T cells. After activation, NKT cells exhibit NK-like MHC-independent cytotoxic activity against tumor cells through several mechanisms, including perforin and granzyme secretion, Fas ligand, or tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) (Kawano et al. 1999; Nieda et al. 2001). In addition, NKT cells can also rapidly produce large amounts of IFN-γ as well as other cytokines that can activate other immune cells such as NK cells, DCs, and CD8 T cells (Carnaud et al. 1999; Fujii et al. 2004; Fujii et al. 2003b), thereby serving as a bridge between innate and acquired immunity (Taniguchi et al. 2003). In addition, because NKT cells are CD1d restricted, they can be adoptively transferred to patients regardless of HLA allele expression. CD1d is expressed by only a few cell types (Brossay et al. 1998); thus, the potential toxicity of adoptively transferred NKT cells is limited. Several studies have reported that donor-derived NKT cells may suppress GVHD while still maintaining anti-tumor function (Morris et al. 2005; Pillai et al. 2007). In agreement with these reports, a recent study demonstrated that reconstitution of NKT cells in peripheral blood is associated with long-term remission of pediatric leukemia patients receiving haplo-identical transplantation (Casorati et al. 2012; de Lalla et al. 2011; Dellabona et al. 2011). Moreover, NKT cells have been shown to colocalize with tumor-associated macrophages (TAMs) and can kill or inhibit these growth-promoting cells in a CD1d-dependent manner (Song et al. 2009).

NKT cells can mount strong anti-tumor responses and have become a major focus in the development of effective cancer immunotherapy. Following activation, NKT cells rapidly secrete cytokines (i.e., IFN-γ, TNF-α, and GM-CSF) and can directly mediate cytotoxicity through FAS/FASL, perforin, and granzyme. Cytokine production by NKT cells can lead to the activation of NK cells, CD8+ T cells, and in combination with CD40/CD40L interactions lead to the maturation of dendritic cells (DC). NKT cells may be directed towards tumor cells expressing specific antigen through the transduction and expression of chimeric antigen receptors (CAR). CARs utilize an extracellular antigen-targeting domain (purple); thus, CARs are capable of binding their target antigen in an MHC-independent manner. Current research efforts are focused on harnessing the adaptability of CARs to enhance NKT cell targeting of tumors
Studies using CAR-modified NKT cells, however, have been relatively scant. Using a model of neuroblastoma, Heczey et al. demonstrated that the ex vivo expansion of human primary NKT modified with CARs specific for the GD2 ganglioside exhibited potent yet specific cytotoxicity against both GD2-positive tumor cells as well as CD1d-positive M2 macrophages in vitro (Heczey et al. 2014). Because the presence of TAMs with M2-like phenotype is associated with poor patient prognosis in neuroblastoma (Asgharzadeh et al. 2012), the elimination or inhibition of TAMs by CAR.GD2 NKT cells may sensitize tumor cells to CAR-mediated cytotoxicity and decrease the chances of tumor immune escape. CAR.GD2 NKT cells also effectively localized to the tumor site and had potent anti-tumor activity in a metastatic model of neuroblastoma without the induction of GVHD, successfully demonstrating the potential of NKT cells to serve as a safe and effective platform for CAR-redirected cancer immunotherapy (Heczey et al. 2014).
Artificial antigen-presenting cells
Effective adoptive T cell immunotherapy of cancer requires sufficient numbers of in vitro-generated, tumor-specific lymphocytes. The in vitro expanded T cells must also possess the capacity to engraft, proliferate, and persist in vivo after transfer. During and after the initial encounter with tumor antigens, the signals that T cell receive from antigen-presenting cells (APCs) can influence their programming and subsequent therapeutic efficacy (Harty and Badovinac 2008). Due to the inability to precisely regulate the signals and interactions provided during natural APC and T cell interactions, interest has increased in the use of artificial antigen-presenting cells (aAPC) in order to provide a greater degree of control to the generation of T cells for adoptive immunotherapy.
Originally, autologous antigen-presenting cells such as dendritic cells, monocytes, and activated B cells have been employed to generate tumor-specific T cells in vitro. However, the requirement to access cancer patients’ blood to prepare autologous APC from each patient in a timely manner is burdensome and the quantity and quality of prepared autologous APC varies between individuals. To overcome these issues, investigators have recently proposed the use of cell-based artificial APCs as a standardized reagent to reliably expand anti-tumor T cells in vitro. Cellular aAPCs derived from primary, transformed human, or xenogenic cells have been generated using retroviral or lentiviral transduction to introduce molecules that provide the necessary TCR, co-stimulatory, and adhesion molecules for synapse formation (Paulos et al. 2008).
Non-cellular aAPC can be utilized both for their potential clinical value in ex vivo T cell expansion as well as to investigate the basic requirements for T cell activation. Studies from Schneck’s group have shown that MHC-Ig-based aAPCs can be used to effectively expand CMV and MART-1-specific CTL (Oelke et al. 2003). Monocyte-derived DCs from cancer patients express reduced levels of co-stimulatory molecules and produce less inflammatory cytokines (Onishi et al. 2002). To determine the effectiveness of bead-based approaches on NKT cell propagation and activation, CD1d-Ig-based aAPCs have been generated (Shiratsuchi et al. 2009; Webb et al. 2009). CD1d-based aAPC can be modified to systematically evaluate the role of a panel of potential co-stimulatory molecules on NKT cell proliferation and function.
Bispecific antibodies
The concept of bispecific antibodies was first introduced in the 1980s as a method to target multiple antigens by a single antibody (Raso and Griffin 1981). They are composed of fragments of two different monoclonal antibodies that can bind two different antigens. Recombinant bispecific antibodies can be generated with or without an Fc region (Kontermann and Brinkmann 2015). Bispecific antibodies with anti-CD3 and anti-CD19 components have been utilized in numerous preclinical studies, resulting in rapid and effective cytotoxicity specific for CD19+ B cells (Loffler et al. 2000; Schlereth et al. 2006). Blinatumomab, a bi-specific T cell engager (BiTE®) antibody construct made by fusing an anti-CD3 scFv to an anti-CD19 scFv via a short five residue peptide linker has performed impressively in multiple clinical trials as a single agent for relapsed/refractory B cell ALL (Topp et al. 2015; Topp et al. 2014). Although not as common, bi-specific antibodies have also been used for NKT cells. Using CD8+ NKT cells redirected with a bi-specific antibody against HER2 and CD3, Scheffold et al. demonstrated that administration of these NKT cells resulted in rapid, and in most instances sustained, eradication of HER2-expressing tumor cells in a scid mouse model, highlighting a promising strategy for NKT-based adoptive immunotherapy of neoplastic diseases (Scheffold et al. 2002; Table 1).
Clinical studies using NKT cell-based therapies
Many studies have highlighted NKT cells as an ideal anti-tumor therapy (Fujii et al. 2013). Trials to date have included various types of solid and hematological malignancies (Dhodapkar and Richter 2011; Schneiders et al. 2011). Here, we will provide a brief overview of clinical trials using NKT cells in cancer patients, which reveal the potential of NKT cell-based therapeutics, refer to Tables 2 and 3.
Giaccone et al. described one of the first clinical trials utilizing α-GalCer (KRN7000) therapy (Giaccone et al. 2002). Although KRN7000 was well tolerated in cancer patients over a wide range of doses, no objective clinical response was observed. They observed a transient decrease in NKT cells following injection of α-GalCer. Importantly, increased serum cytokine levels (TNF-α and GM-CSF) were observed in patients with relatively normal NKT cell numbers, suggesting that efficacy depends on pretreatment NKT cell numbers.
Studies by Nicol’s group evaluated the clinical and immunologic effects of α-GalCer-pulsed monocyte-derived DCs administered at a 2-week interval to 12 human subjects with metastatic malignancy. It was found that activation of NKT cells results in increased serum IFN-γ and IL-12 (Nieda et al. 2004). In another study, this group examined the percentage, absolute number, and function (responsiveness to α-GalCer stimulation) of peripheral blood NKT cells in healthy donors and cancer patients (Crough et al. 2004). Similar to other studies, it was found that NKT cells were significantly reduced in melanoma and breast cancer patients. They performed a phase I clinical trial with α-GalCer-pulsed DC and found that the treatment could induce immune responses, and that the responses elicited are highly dependent on cell dose and administration route (Nicol et al. 2011).
Ishikawa et al. performed a phase 1 clinical trial to target endogenous NKT cell activation. Patients with advanced and recurrent non-small cell lung cancer were treated with α-GalCer (KRN7000)-pulsed dendritic cells (Ishikawa et al. 2005). Each patient received four doses of α-GalCer-pulsed DCs by intravenous injection. The DCs used in this study were prepared by stimulation with GM-CSF and IL-2. No severe adverse events were observed. After the first and second injection of α-GalCer-pulsed DCs, a dramatic increase in NKT cells (>20-fold) was observed in one case and significant responses were seen in two cases receiving the level 3 dose. This study indicates that α-GalCer-pulsed DC therapy was well tolerated and it can be done safely, even in patients with advanced disease. Motohashi et al. conducted a phase I study in advanced and recurrent non-small cell lung cancer patients using autologous in vitro activated NKT cells (Motohashi et al. 2006). Similar to their previous study, no severe adverse events were observed. While the number of IFN-γ-producing cells in peripheral blood mononuclear cells (PBMC) increased after the administration of activated NKT cells, no patient was found to meet the criteria for either a partial or a complete response.
Notably, Motohashi and colleagues have extended the study of α-GalCer–pulsed DC to test immunological safety and clinical responses (Motohashi et al. 2009). α-GalCer (KRN7000)-pulsed PBMC cultured in the presence of IL-2 and GM-CSF were given to patients with advanced and recurrent non-small cell lung cancer. Twenty-three patients were enrolled and 17 cases completed the study. After the injection of α-GalCer-pulsed IL-2/GM-CSF cultured PBMC, an increased number of IFN-γ-producing cells in the peripheral blood was detected in ten patients. Five cases remained as stable disease, and the remaining 12 cases were evaluated as progressive disease. Similar to the phase I trial, α-GalCer-pulsed DC injection was well tolerated and accompanied by the successful induction of NKT cells. Ten patients with increased IFN-γ-producing cells (>2-fold), following α-GalCer stimulation, showed prolonged median survival time (MST) of 31.9 months as compared to the control group (MST of 9.7 months). The authors proposed that the increased number of IFN-γ-producing cells in PBMCs may serve as a biological marker for predicting the clinical course in response to α-GalCer-pulsed IL-2/GM-CSF-cultured PBMC administration. There are other ongoing trials exploring NKT cell immunotherapy in cancer patients. Zhang and colleagues have initiated a phase I–II study of NKT cell infusion in patients with advanced solid tumor (non-small cell lung cancer, gastric cancer, hepatocellular carcinoma, and colorectal cancer; NCT02562963) to determine whether NKT cells are effective and safe in the treatment of patients with unresectable advanced solid tumor. Another ongoing phase I clinical trial of adoptive transfer with autologous NKT cells in metastatic melanoma patients (NCT02619058) is investigating the maximum tolerated dose (MTD) and dose limiting toxicity (DLT) of intravenous administration of autologous NKT cells in metastatic melanoma patients.
Future directions
While NKT cell-based immunotherapy for the treatment of cancer has shown promise, issues remain. Many immunotherapies work under the assumption of a normally functioning immune system. Yet it is known that cancer patients frequently have impaired immune responses, including exhausted NKT cells. We have developed qPCR-based diagnostic (Sohn et al. 2014) that can be used to rapidly determine which patients have functional NKT cells and would benefit from antibody or adjuvant-based immunotherapy and which patients have defective NKT cell responses and would need the more robust treatment of adoptive cell transfer (Fig. 3).

Future directions for NKT cell-mediated cancer therapy. While NKT cell immunotherapies have shown promise, it is unclear which patient populations are most likely to benefit from this treatment. We have developed a qPCR-based diagnostic to assess NKT cell function in cancer patients. This assay will allow one to determine which patients have functional NKT cells and may suggest the need for checkpoint inhibiting therapy or adoptive transfer of NKT cells in patients with dysfunctional or exhausted NKT cell populations. Studies from our group and others have shown that human NKT cells can be generated from stem cells. In contrast, patients with high IFN-γ producing NKT cells may benefit from treatment with NKT cell agonists, used as adjuvants and delivered using a variety of platforms
Current studies focused on inducing the activation of endogenous NKT cells are utilizing nanoparticle-based therapy. Nanoparticles, typically poly(lactic-co-glycolic acid) (PLGA)-based, are extremely versatile and can be modified to include adjuvants, tumor-derived antigens and targeting mechanisms. Like CD1d antibody fusion proteins, nanoparticle-delivered α-GalCer does not induce anergy (Thapa et al. 2009). Anti-DEC-205 coated nanoparticles loaded with α-GalCer and antigen were shown to specifically target DCs that are highly efficiently at cross-presentation and promote significant anti-tumor immunity (Macho-Fernandez et al. 2014). Recent work has shown that co-encapsulation of α-GalCer and antigen produces a stronger anti-tumor and CTL response than either co-encapsulated TLR agonists or antigen-nanoparticles mixed with adjuvant (Dölen et al. 2016). R8-peptide-coated liposomes selectively target APCs and when loaded with α-GalCer and administered with IL-12 demonstrate the benefits of combination therapy by allowing lower doses of liposome to produce a more potent result (Abdelmegeed et al. 2016). Finally, adipose-derived mesenchymal stem cell exosomes have been shown to stimulate anti-tumor immunity via NKT cells (Ko et al. 2015).
For patients with low immune function, adoptive cell transfer is an appealing remedy. Previous options involved ex vivo expansion of autologous NKT isolated from the blood or tumor. Current research is moving towards the generation of NKT cells from stem cells using various methods. CD4+CD8α+ NKT cells have been generated from CD34+ cells isolated from cord blood using a liquid culture system, dependent on IL-15 and flt-3 ligand or stem cell factor (SCF). While double positive NKT cells are not a standard population found in vivo, these cells produced both Th1 and Th2 cytokines (Woo et al. 2003). Another group has differentiated murine induced pluripotent stem cells (iPSCs) into NKT cells using the OP9/DL-1 culture system and has shown that replacing the OP9/DL-1 culture system at day 14 with a WT OP-9 culture system generated Th1 skewed NKT cells that would be more useful in cancer immunotherapy (Watarai et al. 2010a; Watarai et al. 2010b; Watarai et al. 2012). It was shown that human NKT cells generated from iPSCs could be redifferentiated in vitro using a IL-7/IL-15-based cytokine co-culture system (Kitayama et al. 2016) Human NKT cells have been generated from bone marrow-derived adult hematopoietic stem-progenitor cells using the OP-9/DL-1 culture system in the presence of IL-7 and flt-3 ligand for 21 days followed by expansion with CD1d-based aAPC. The resulting cells were shown to be functionally mature and responsive to α-GalCer stimulation (Sun et al. 2015).
Advancements in the study of NKT cells are necessary and important. Recent studies examining the efficacy of cancer therapeutics have utilized zebrafish models. Zebrafish are an attractive model for multiple reasons: short generation time, large progeny, ease of care, transparency for in vivo imaging, genetically tractable, and ease of chemical screening (Yen et al. 2014). A transgenic hepatocellular carcinoma zebrafish model has shown that their immune system plays a similarly prominent role in tumor progression (Li et al. 2014b). They possess innate and adaptive immune systems that are comparable to higher organisms (Rauta et al. 2012). While NKT cells have not yet been discovered in zebrafish, amphibians have a comparable subset called invariant T (iT) cells that play a conserved role in immunity (Robert and Edholm 2014). Collectively, these studies demonstrate the feasibility of targeting NKT cells for cancer immunotherapy and reveal innovative strategies that can be employed to increase our understanding of this important population of T cells.
References
Abdelmegeed H, Nakamura T, Harashima H (2016) In vivo inverse correlation in the activation of natural killer T cells through dual-signal stimulation via a combination of α-galactosylceramide-loaded liposomes and interleukin-12. J Pharm Sci 105:250–256
Adachi Y, Koseki H, Zijlstra M, Taniguchi M (1995) Positive selection of invariant V alpha 14+ T cells by non-major histocompatibility complex-encoded class I-like molecules expressed on bone marrow-derived cells. Proc Natl Acad Sci USA 92:1200–1204
Ambrosino E, Berzofsky JA, Terabe M (2008) Regulation of tumor immunity: the role of NKT cells. Expert Opin Biol Ther 8:725–734
Ambrosino E, Terabe M, Halder RC, Peng J, Takaku S, Miyake S, Yamamura T, Kumar V, Berzofsky JA (2007) Cross-regulation between type I and type II NKT cells in regulating tumor immunity: a new immunoregulatory axis. J Immunol 179:5126–5136
Anastasiadis A, Kotsianidis I, Papadopoulos V, Spanoudakis E, Margaritis D, Christoforidou A, Gouliamtzi S, Tsatalas C (2014) CD1d expression as a prognostic marker for chronic lymphocytic leukemia. Leuk Lymphoma 55:320–325
Anderson RJ, Tang C-W, Daniels NJ, Compton BJ, Hayman CM, Johnston KA, Knight DA, Gasser O, Poyntz HC, Ferguson PM, Larsen DS, Ronchese F, Painter GF, Hermans IF (2014) A self-adjuvanting vaccine induces cytotoxic T lymphocytes that suppress allergy. Nat Chem Biol 10:943–949
Asgharzadeh S, Salo JA, Ji L, Oberthuer A, Fischer M, Berthold F, Hadjidaniel M, Liu CW, Metelitsa LS, Pique-Regi R, Wakamatsu P, Villablanca JG, Kreissman SG, Matthay KK, Shimada H, London WB, Sposto R, Seeger RC (2012) Clinical significance of tumor-associated inflammatory cells in metastatic neuroblastoma. J Clin Oncol 30:3525–3532
Aspeslagh S, Li Y, Yu ED, Pauwels N, Trappeniers M, Girardi E, Decruy T, Van Beneden K, Venken K, Drennan M, Leybaert L, Wang J, Franck RW, Van Calenbergh S, Zajonc DM, Elewaut D (2011) Galactose-modified iNKT cell agonists stabilized by an induced fit of CD1d prevent tumour metastasis. EMBO J 30:2294–2305
Aspeslagh S, Nemčovič M, Pauwels N, Venken K, Wang J, Van Calenbergh S, Zajonc DM, Elewaut D (2013) Enhanced TCR footprint by a novel glycolipid increases NKT-dependent tumor protection. J Immunol 191:2916–2925
Azuma T, Takahashi T, Kunisato A, Kitamura T, Hirai H (2003) Human CD4+ CD25+ regulatory T cells suppress NKT cell functions. Cancer Res 63:4516–4520
Bagnara D, Ibatici A, Corselli M, Sessarego N, Tenca C, De Santanna A, Mazzarello A, Daga A, Corvo R, De Rossi G, Frassoni F, Ciccone E, Fais F (2009) Adoptive immunotherapy mediated by ex vivo expanded natural killer T cells against CD1d-expressing lymphoid neoplasms. Haematologica 94:967–974
Birkholz AM, Girardi E, Wingender G, Khurana A, Wang J, Zhao M, Zahner S, Illarionov PA, Wen X, Li M, Yuan W, Porcelli SA, Besra GS, Zajonc DM, Kronenberg M (2015) A novel glycolipid antigen for NKT cells that preferentially induces IFN-γ production. J Immunol 195:924–933
Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, Bartido S, Stefanski J, Taylor C, Olszewska M, Borquez-Ojeda O, Qu J, Wasielewska T, He Q, Bernal Y, Rijo IV, Hedvat C, Kobos R, Curran K, Steinherz P, Jurcic J, Rosenblat T, Maslak P, Frattini M, Sadelain M (2013) CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med 5:177ra38
Brossay L, Chioda M, Burdin N, Koezuka Y, Casorati G, Dellabona P, Kronenberg M (1998) CD1d-mediated recognition of an alpha-galactosylceramide by natural killer T cells is highly conserved through mammalian evolution. J Exp Med 188:1521–1528
Carnaud C, Lee D, Donnars O, Park SH, Beavis A, Koezuka Y, Bendelac A (1999) Cutting edge: cross-talk between cells of the innate immune system: NKT cells rapidly activate NK cells. J Immunol 163:4647–4650
Casorati G, de Lalla C, Dellabona P (2012) Invariant natural killer T cells reconstitution and the control of leukemia relapse in pediatric haploidentical hematopoietic stem cell transplantation. Oncoimmunology 1:355–357
Chang DH, Deng H, Matthews P, Krasovsky J, Ragupathi G, Spisek R, Mazumder A, Vesole DH, Jagannath S, Dhodapkar MV (2008) Inflammation-associated lysophospholipids as ligands for CD1d-restricted T cells in human cancer. Blood 112:1308–1316
Chang DH, Liu N, Klimek V, Hassoun H, Mazumder A, Nimer SD, Jagannath S, Dhodapkar MV (2006) Enhancement of ligand-dependent activation of human natural killer T cells by lenalidomide: therapeutic implications. Blood 108:618–621
Chang DH, Osman K, Connolly J, Kukreja A, Krasovsky J, Pack M, Hutchinson A, Geller M, Liu N, Annable R, Shay J, Kirchhoff K, Nishi N, Ando Y, Hayashi K, Hassoun H, Steinman RM, Dhodapkar MV (2005) Sustained expansion of NKT cells and antigen-specific T cells after injection of alpha-galactosyl-ceramide loaded mature dendritic cells in cancer patients. J Exp Med 201:1503–1517
Chung Y, Qin H, Kang CY, Kim S, Kwak LW, Dong C (2007) An NKT-mediated autologous vaccine generates CD4 T-cell dependent potent antilymphoma immunity. Blood 110:2013–2019
Coquet JM, Chakravarti S, Kyparissoudis K, McNab FW, Pitt LA, McKenzie BS, Berzins SP, Smyth MJ, Godfrey DI (2008) Diverse cytokine production by NKT cell subsets and identification of an IL-17-producing CD4–NK1.1–NKT cell population. Proc Natl Acad Sci 105:11287–11292
Corgnac S, Perret R, Derré L, Zhang L, Stirnemann K, Zauderer M, Speiser DE, Mach J-P, Romero P, Donda A (2012) CD1d-antibody fusion proteins target iNKT cells to the tumor and trigger long-term therapeutic responses. Cancer Immunol Immunother 62:747–760
Croudace JE, Curbishley SM, Mura M, Willcox CR, Illarionov PA, Besra GS, Adams DH, Lammas DA (2008) Identification of distinct human invariant natural killer T-cell response phenotypes to alpha-galactosylceramide. BMC Immunol 9:71
Crough T, Purdie DM, Okai M, Maksoud A, Nieda M, Nicol AJ (2004) Modulation of human V[alpha]24 + V[beta]11+ NKT cells by age, malignancy and conventional anticancer therapies. Br J Cancer 91:1880–1886
Crowe NY, Coquet JM, Berzins SP, Kyparissoudis K, Keating R, Pellicci DG, Hayakawa Y, Godfrey DI, Smyth MJ (2005) Differential antitumor immunity mediated by NKT cell subsets in vivo. J Exp Med 202:1279–1288
Cui J, Shin T, Kawano T, Sato H, Kondo E, Toura I, Kaneko Y, Koseki H, Kanno M, Taniguchi M (1997) Requirement for V14 NKT cells in IL-12-mediated rejection of tumors. Science 278
de Lalla C, Rinaldi A, Montagna D, Azzimonti L, Bernardo ME, Sangalli LM, Paganoni AM, Maccario R, Di Cesare-Merlone A, Zecca M, Locatelli F, Dellabona P, Casorati G (2011) Invariant NKT cell reconstitution in pediatric leukemia patients given HLA-haploidentical stem cell transplantation defines distinct CD4+ and CD4- subset dynamics and correlates with remission state. J Immunol 186:4490–4499
Dellabona P, Casorati G, de Lalla C, Montagna D, Locatelli F (2011) On the use of donor-derived iNKT cells for adoptive immunotherapy to prevent leukemia recurrence in pediatric recipients of HLA haploidentical HSCT for hematological malignancies. Clin Immunol 140:152–159
Dhodapkar MV, Richter J (2011) Harnessing natural killer T (NKT) cells in human myeloma: progress and challenges. Clin Immunol 140:160–166
Dölen Y, Kreutz M, Gileadi U, Tel J, Vasaturo A, van Dinther EAW, van Hout-Kuijer MA, Cerundolo V, Figdor CG (2016) Co-delivery of PLGA encapsulated invariant NKT cell agonist with antigenic protein induce strong T cell-mediated antitumor immune responses. OncoImmunology 5:e1068493
Eberl G, MacDonald HR (2000) Selective induction of NK cell proliferation and cytotoxicity by activated NKT cells. Eur J Immunol 30:985–992
Fais F, Tenca C, Cimino G, Coletti V, Zanardi S, Bagnara D, Saverino D, Zarcone D, De Rossi G, Ciccone E, Grossi CE (2005) CD1d expression on B-precursor acute lymphoblastic leukemia subsets with poor prognosis. Leukemia 19:551–556
Fowlkes BJ, Kruisbeek AM, Ton-That H, Weston MA, Coligan JE, Schwartz RH, Pardoll DM (1987) A novel population of T-cell receptor ab-bearing thymocytes which predominantly expresses a single Vb gene family. Nature 329:251–254
Fujii S, Liu K, Smith C, Bonito AJ, Steinman RM (2004) The linkage of innate to adaptive immunity via maturing dendritic cells in vivo requires CD40 ligation in addition to antigen presentation and CD80/86 costimulation. J Exp Med 199:1607–1618
Fujii S, Shimizu K, Klimek V, Geller MD, Nimer SD, Dhodapkar MV (2003a) Severe and selective deficiency of interferon-gamma-producing invariant natural killer T cells in patients with myelodysplastic syndromes. Br J Haematol 122:617–622
Fujii S, Shimizu K, Smith C, Bonifaz L, Steinman RM (2003b) Activation of natural killer T cells by alpha-galactosylceramide rapidly induces the full maturation of dendritic cells in vivo and thereby acts as an adjuvant for combined CD4 and CD8 T cell immunity to a coadministered protein. J Exp Med 198:267–279
Fujii S-I, Goto A, Shimizu K (2009) Antigen mRNA-transfected, allogeneic fibroblasts loaded with NKT-cell ligand confer antitumor immunity. Blood 113:4262–4272
Fujii S-I, Shimizu K, Hemmi H, Steinman RM (2007) Innate V14+ natural killer T cells mature dendritic cells, leading to strong adaptive immunity. Immunol Rev 220:183–198
Fujii S-I, Shimizu K, Okamoto Y, Kunii N, Nakayama T, Motohashi S, Taniguchi M (2013) NKT cells as an ideal anti-tumor immunotherapeutic. Front Immunol 4:409
Giaccone G, Punt CJ, Ando Y, Ruijter R, Nishi N, Peters M, Von Blomberg BM, Scheper RJ, Van Der Vliet HJ, Van Den Eertwegh AJ, Roelvink M, Beijnen J, Zwierzina H, Pinedo HM (2002) A phase I study of the natural killer T-cell ligand a- galactosylceramide (KRN7000) in patients with solid tumors. Clin Cancer Res 8:3702–3709
Gibbins JD (2014) An autologous leukemia cell vaccine prevents murine acute leukemia relapse after cytarabine treatment. Blood 124
Godfrey DI, Stankovic S, Baxter AG (2010) Raising the NKT cell family. Nat Immunol 11:197–206
Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, Milone MC, Levine BL, June CH (2013) Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med 368:1509–1518
Guo W, Dong A, Xing C, Lin X, Pan X, Lin Y, Zhu B, He M, Yao RX (2014) CD1d levels in peripheral blood of patients with acute myeloid leukemia and acute lymphoblastic leukemia. Oncol Lett 8:825–830
Haraguchi K, Takahashi T, Nakahara F, Matsumoto A, Kurokawa M, Ogawa S, Oda H, Hirai H, Chiba S (2006) CD1d expression level in tumor cells is an important determinant for anti-tumor immunity by natural killer T cells. Leuk Lymphoma 47:2218–2223
Harty JT, Badovinac VP (2008) Shaping and reshaping CD8+ T-cell memory. Nat Rev Immunol 8:107–119
Hayakawa Y, Takeda K, Yagita H, Van Kaer L, Saiki I, Okumura K (2001) Differential regulation of Th1 and Th2 functions of NKT cells by CD28 and CD40 costimulatory pathways. J Immunol 166:6012–6018
Heczey A, Liu D, Tian G, Courtney AN, Wei J, Marinova E, Gao X, Guo L, Yvon E, Hicks J, Liu H, Dotti G, Metelitsa LS (2014) Invariant NKT cells with chimeric antigen receptor provide a novel platform for safe and effective cancer immunotherapy. Blood 124:2824–2833
Hermans IF, Silk JD, Gileadi U, Masri SH, Shepherd D, Farrand KJ, Salio M, Cerundolo V (2007) Dendritic cell function can be modulated through cooperative actions of TLR ligands and invariant NKT cells. J Immunol 178:2721–2729
Hermans IF, Silk JD, Gileadi U, Salio M, Mathew B, Ritter G, Schmidt R, Harris AL, Old L, Cerundolo V (2003) NKT cells enhance CD4+ and CD8+ T cell responses to soluble antigen in vivo through direct interaction with dendritic cells. J Immunol 171:5140–5147
Imai C, Mihara K, Andreansky M, Nicholson IC, Pui CH, Geiger TL, Campana D (2004) Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 18:676–684
Imai K, Kanno M, Kimoto H, Shigemoto K, Yamamoto S, Taniguchi M (1986) Sequence and expression of transcripts of the T-cell antigen receptor alpha-chain gene in a functional, antigen-specific suppressor-T-cell hybridoma. Proc Natl Acad Sci USA 83:8708–8712
Ishikawa A, Motohashi S, Ishikawa E, Fuchida H, Higashino K, Otsuji M, Iizasa T, Nakayama T, Taniguchi M, Fujisawa T (2005) A phase I study of alpha-galactosylceramide (KRN7000)-pulsed dendritic cells in patients with advanced and recurrent non-small cell lung cancer. Clin Cancer Res 11:1910–1917
Izhak L, Ambrosino E, Kato S, Parish ST, O’Konek JJ, Weber H, Xia Z, Venzon D, Berzofsky JA, Terabe M (2013) Delicate balance among three types of T cells in concurrent regulation of tumor immunity. Cancer Res 73:1514–1523
Kato S, Terabe M, Berzofsky JA (2014) Liver sulfatide-reactive type II NKT cells recognize endogenous phospholipids. J ImmunoTher Cancer 2:P215
Kawano T, Cui J, Koezuka Y, Toura I, Kaneko Y, Motoki K, Ueno H, Nakagawa R, Sato H, Kondo E, Koseki H, Taniguchi M (1997) CD1d-restricted and TCR-mediated activation of valpha14 NKT cells by glycosylceramides. Science 278:1626–1629
Kawano T, Nakayama T, Kamada N, Kaneko Y, Harada M, Ogura N, Akutsu Y, Motohashi S, Iizasa T, Endo H, Fujisawa T, Shinkai H, Taniguchi M (1999) Antitumor cytotoxicity mediated by ligand-activated human V alpha24 NKT cells. Cancer Res 59:5102–5105
Kitayama S, Zhang R, Liu T-Y, Ueda N, Iriguchi S, Yasui Y, Kawai Y, Tatsumi M, Hirai N, Mizoro Y, Iwama T, Watanabe A, Nakanishi M, Kuzushima K, Uemura Y, Kaneko S (2016) Cellular adjuvant properties, direct cytotoxicity of Re-differentiated Vα24 invariant NKT-like cells from human induced pluripotent stem cells. Stem Cell Rep 6:213–227
Kmieciak M, Basu D, Payne KK, Toor A, Yacoub A, Wang XY, Smith L, Bear HD, Manjili MH (2011) Activated NKT cells and NK cells render T cells resistant to myeloid-derived suppressor cells and result in an effective adoptive cellular therapy against breast cancer in the FVBN202 transgenic mouse. J Immunol 187:708–717
Ko H-J, Lee J-M, Kim Y-J, Kim Y-S, Lee K-A, Kang C-Y (2009a) Immunosuppressive myeloid-derived suppressor cells can Be converted into immunogenic APCs with the help of activated NKT cells: an alternative cell-based antitumor vaccine. J Immunol 182:1818–1828
Ko HJ, Lee JM, Kim YJ, Kim YS, Lee KA, Kang CY (2009b) Immunosuppressive myeloid-derived suppressor cells can be converted into immunogenic APCs with the help of activated NKT cells: an alternative cell-based antitumor vaccine. J Immunol 182:1818–1828
Ko S-F, Yip H-K, Zhen Y-Y, Lee C-C, Lee C-C, Huang C-C, Ng S-H, Lin J-W (2015) Adipose-derived mesenchymal stem cell exosomes suppress hepatocellular carcinoma growth in a rat model: apparent diffusion coefficient, natural killer T-cell responses, and histopathological features. Stem Cells Int 2015:11
Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, Stetler-Stevenson M, Phan GQ, Hughes MS, Sherry RM, Yang JC, Kammula US, Devillier L, Carpenter R, Nathan DA, Morgan RA, Laurencot C, Rosenberg SA (2012) B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 119:2709–2720
Kontermann RE, Brinkmann U (2015) Bispecific antibodies. Drug Discov Today 20:838–847
Koseki H, Imai K, Nakayama F, Sado T, Moriwaki K, Taniguchi M (1990) Homogenous junctional sequence of the V14+ T-cell antigen receptor alpha chain expanded in unprimed mice. Proc Natl Acad Sci USA 87:5248–5252
Lantz O, Bendelac A (1994) An invariant T cell receptor a chain is used by a unique subset of major histocompatibility complex class I-specific CD4+ and CD4−8− T cells in mice and humans. J Exp Med 180:1097–1106
Lee JM, Seo JH, Kim YJ, Kim YS, Ko HJ, Kang CY (2012) The restoration of myeloid-derived suppressor cells as functional antigen-presenting cells by NKT cell help and all-trans-retinoic acid treatment. Int J Cancer 131:741–751
Li J, Sun W, Subrahmanyam PB, Page C, Younger KM, Tiper IV, Frieman M, Kimball AS, Webb TJ (2014a) NKT cell responses to B cell lymphoma. Med Sci (Basel) 2:82–97
Li X, Chen G, Garcia-Navarro R, Franck RW, Tsuji M (2009) Identification of C-glycoside analogues that display a potent biological activity against murine and human invariant natural killer T cells. Immunology 127:216–225
Li X, Fujio M, Imamura M, Wu D, Vasan S, Wong C-H, Ho DD, Tsuji M (2010) Design of a potent CD1d-binding NKT cell ligand as a vaccine adjuvant. Proc Natl Acad Sci 107:13010–13015
Li Z, Luo H, Li C, Huo X, Yan C, Huang X, Al-Haddawi M, Mathavan S, Gong Z (2014b) Transcriptomic analysis of a transgenic zebrafish hepatocellular carcinoma model reveals a prominent role of immune responses in tumour progression and regression. Int J Cancer 135:1564–1573
Liu D, Song L, Wei J, Courtney AN, Gao X, Marinova E, Guo L, Heczey A, Asgharzadeh S, Kim E, Dotti G, Metelitsa LS (2012) IL-15 protects NKT cells from inhibition by tumor-associated macrophages and enhances antimetastatic activity. J Clin Invest 122:2221–2233
Liu K, Idoyaga J, Charalambous A, S-I F, Bonito A, Mordoh J, Wainstok R, Bai X-F, Liu Y, Steinman RM (2005) Innate NKT lymphocytes confer superior adaptive immunity via tumor-capturing dendritic cells. J Exp Med 202:1507–1516
Loffler A, Kufer P, Lutterbuse R, Zettl F, Daniel PT, Schwenkenbecher JM, Riethmuller G, Dorken B, Bargou RC (2000) A recombinant bispecific single-chain antibody, CD19 x CD3, induces rapid and high lymphoma-directed cytotoxicity by unstimulated T lymphocytes. Blood 95:2098–2103
Macho-Fernandez E, Cruz LJ, Ghinnagow R, Fontaine J, Bialecki E, Frisch B, Trottein F, Faveeuw C (2014) Targeted delivery of α-galactosylceramide to CD8α + Dendritic cells optimizes type I NKT cell-based antitumor responses. J Immunol 193:961–969
Maher J, Brentjens RJ, Gunset G, Riviere I, Sadelain M (2002) Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta /CD28 receptor. Nat Biotechnol 20:70–75
Marrero I, Ware R, Kumar V (2015) Type II NKT cells in inflammation, autoimmunity, microbial immunity and cancer. Front Immunol 6
Mattarollo SR, Smyth MJ (2013) NKT cell adjuvants in therapeutic vaccines against hematological cancers. Oncoimmunology 2:e22615
McEwen-Smith RM, Salio M, Cerundolo V (2015) The regulatory role of invariant NKT cells in tumor immunity. Cancer Immunol Res 3:425–435
Metelitsa LS, Weinberg KI, Emanuel PD, Seeger RC (2003) Expression of CD1d by myelomonocytic leukemias provides a target for cytotoxic NKT cells. Leukemia 17:1068–1077
Metelitsa LS, Wu HW, Wang H, Yang Y, Warsi Z, Asgharzadeh S, Groshen S, Wilson SB, Seeger RC (2004) Natural killer T cells infiltrate neuroblastomas expressing the chemokine CCL2. J Exp Med 199:1213–1221
Michel ML, Keller AC, Paget C, Fujio M, Trottein F, Savage PB, Wong CH, Schneider E, Dy M, Leite-de-Moraes MC (2007) Identification of an IL-17-producing NK1.1(neg) iNKT cell population involved in airway neutrophilia. J Exp Med 204:995–1001
Molling JW, Kolgen W, van der Vliet HJ, Boomsma MF, Kruizenga H, Smorenburg CH, Molenkamp BG, Langendijk JA, Leemans CR, von Blomberg BM, Scheper RJ, van den Eertwegh AJ (2005) Peripheral blood IFN-gamma-secreting Valpha24 + Vbeta11+ NKT cell numbers are decreased in cancer patients independent of tumor type or tumor load. Int J Cancer 116:87–93
Moreno M, Mol BM, Mensdorff-Pouilly SV, Verheijen RHM, von Blomberg BME, van den Eertwegh AJM, Scheper RJ, Bontkes HJ (2008) Toll-like receptor agonists and invariant natural killer T-cells enhance antibody-dependent cell-mediated cytotoxicity (ADCC). Cancer Lett 272:70–76
Morris ES, MacDonald KP, Rowe V, Banovic T, Kuns RD, Don AL, Bofinger HM, Burman AC, Olver SD, Kienzle N, Porcelli SA, Pellicci DG, Godfrey DI, Smyth MJ, Hill GR (2005) NKT cell-dependent leukemia eradication following stem cell mobilization with potent G-CSF analogs. J Clin Invest 115:3093–3103
Motohashi S, Ishikawa A, Ishikawa E, Otsuji M, Iizasa T, Hanaoka H, Shimizu N, Horiguchi S, Okamoto Y, Fujii S, Taniguchi M, Fujisawa T, Nakayama T (2006) A phase I study of in vitro expanded natural killer T cells in patients with advanced and recurrent non-small cell lung cancer. Clin Cancer Res 12:6079–6086
Motohashi S, Nagato K, Kunii N, Yamamoto H, Yamasaki K, Okita K, Hanaoka H, Shimizu N, Suzuki M, Yoshino I, Taniguchi M, Fujisawa T, Nakayama T (2009) A phase I-II study of alpha-galactosylceramide-pulsed IL-2/GM-CSF-cultured peripheral blood mononuclear cells in patients with advanced and recurrent non-small cell lung cancer. J Immunol 182:2492–2501
Nakagawa R, Nagafune I, Tazunoki Y, Ehara H, Tomura H, Iijima R, Motoki K, Kamishohara M, Seki S (2001) Mechanisms of the antimetastatic effect in the liver and of the hepatocyte injury induced by a-galactosylceramide in mice. J Immunol 166:6578–6584
Nicol AJ, Tazbirkova A, Nieda M (2011) Comparison of clinical and immunological effects of intravenous and intradermal administration of α-GalactosylCeramide (KRN7000)-pulsed dendritic cells. Clin Cancer Res 17:5140–5151
Nieda M, Nicol A, Koezuka Y, Kikuchi A, Lapteva N, Tanaka Y, Tokunaga K, Suzuki K, Kayagaki N, Yagita H, Hirai H, Juji T (2001) TRAIL expression by activated human CD4(+)V alpha 24NKT cells induces in vitro and in vivo apoptosis of human acute myeloid leukemia cells. Blood 97:2067–2074
Nieda M, Okai M, Tazbirkova A, Lin H, Yamaura A, Ide K, Abraham R, Juji T, Macfarlane DJ, Nicol AJ (2004) Therapeutic activation of Valpha24 + Vbeta11+ NKT cells in human subjects results in highly coordinated secondary activation of acquired and innate immunity. Blood 103:383–389
O’Konek JJ, Illarionov P, Khursigara DS, Ambrosino E, Izhak L, Castillo BF II, Raju R, Khalili M, Kim H-Y, Howell AR, Besra GS, Porcelli SA, Berzofsky JA, Terabe M (2011) Mouse and human iNKT cell agonist β-mannosylceramide reveals a distinct mechanism of tumor immunity. J Clin Invest 121:683–694
O’Konek JJ, Kato S, Takao S, Izhak L, Xia Z, Illarionov P, Besra GS, Terabe M, Berzofsky JA (2013) Beta-mannosylceramide activates type I natural killer t cells to induce tumor immunity without inducing long-term functional anergy. Clin Cancer Res 19:4404–4411
Oelke M, Maus MV, Didiano D, June CH, Mackensen A, Schneck JP (2003) Ex vivo induction and expansion of antigen-specific cytotoxic T cells by HLA-Ig-coated artificial antigen-presenting cells. Nat Med 9:619–624
Onishi H, Morisaki T, Baba E, Kuga H, Kuroki H, Matsumoto K, Tanaka M, Katano M (2002) Dysfunctional and short-lived subsets in monocyte-derived dendritic cells from patients with advanced cancer. Clin Immunol 105:286–295
Ortaldo JR, Young HA, Winkler-Pickett RT, Bere EW Jr, Murphy WJ, Wiltrout RH (2004) Dissociation of NKT stimulation, cytokine induction, and NK activation in vivo by the use of distinct TCR-binding ceramides. J Immunol 172:943–953
Osada T, Morse MA, Lyerly HK, Clay TM (2005) Ex vivo expanded human CD4+ regulatory NKT cells suppress expansion of tumor antigen-specific CTLs. Int Immunol 17:1143–1155
Paget C, Mallevaey T, Speak AO, Torres D, Fontaine J, Sheehan KCF, Capron M, Ryffel B, Faveeuw C, Leite de Moraes M, Platt F, Trottein F (2007) Activation of invariant NKT cells by toll-like receptor 9-stimulated dendritic cells requires type I interferon and charged glycosphingolipids. Immunity 27:597–609
Parekh VV, Lalani S, Kim S, Halder R, Azuma M, Yagita H, Kumar V, Wu L, Kaer LV (2009) PD-1/PD-L blockade prevents anergy induction and enhances the anti-tumor activities of glycolipid-activated invariant NKT cells. J Immunol 182:2816–2826
Paulos CM, Suhoski MM, Plesa G, Jiang T, Basu S, Golovina TN, Jiang S, Aqui NA, Powell DJ Jr, Levine BL, Carroll RG, Riley JL, June CH (2008) Adoptive immunotherapy: good habits instilled at youth have long-term benefits. Immunol Res 42:182–196
Payne KK, Keim RC, Graham L, Idowu MO, Wan W, Wang XY, Toor AA, Bear HD, Manjili MH (2016) Tumor-reactive immune cells protect against metastatic tumor and induce immunoediting of indolent but not quiescent tumor cells. J Leukoc Biol
Payne KK, Zoon CK, Wan W, Marlar K, Keim RC, Kenari MN, Kazim AL, Bear HD, Manjili MH (2013) Peripheral blood mononuclear cells of patients with breast cancer can be reprogrammed to enhance anti-HER-2/neu reactivity and overcome myeloid-derived suppressor cells. Breast Cancer Res Treat 142:45–57
Pillai AB, George TI, Dutt S, Teo P, Strober S (2007) Host NKT cells can prevent graft-versus-host disease and permit graft antitumor activity after bone marrow transplantation. J Immunol 178:6242–6251
Porter DL, Levine BL, Kalos M, Bagg A, June CH (2011) Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med 365:725–733
Raso V, Griffin T (1981) Hybrid antibodies with dual specificity for the delivery of ricin to immunoglobulin-bearing target cells. Cancer Res 41:2073–2078
Rauta PR, Nayak B, Das S (2012) Immune system and immune responses in fish and their role in comparative immunity study: a model for higher organisms. Immunol Lett 148:23–33
Renukaradhya GJ, Khan MA, Vieira M, Du W, Gervay-Hague J, Brutkiewicz RR (2008) Type I NKT cells protect (and type II NKT cells suppress) the host’s innate antitumor immune response to a B-cell lymphoma. Blood 111:5637–5645
Renukaradhya GJ, Sriram V, Du W, Gervay-Hague J, Van Kaer L, Brutkiewicz RR (2006) Inhibition of antitumor immunity by invariant natural killer T cells in a T-cell lymphoma model in vivo. Int J Cancer 118:3045–3053
Richter J, Neparidze N, Zhang L, Nair S, Monesmith T, Sundaram R, Miesowicz F, Dhodapkar KM, Dhodapkar MV (2013) Clinical regressions and broad immune activation following combination therapy targeting human NKT cells in myeloma. Blood 121:423–430
Robert J, Edholm E-S (2014) A prominent role for invariant T cells in the amphibian Xenopus laevis tadpoles. Immunogenetics 66:513–523
Robertson FC, Berzofsky JA, Terabe M (2014) NKT cell networks in the regulation of tumor immunity. Front Immunol 5:543
Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, Simon P, Lotze MT, Yang JC, Seipp CA, et al. (1988) Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med 319:1676–1680
Sadelain M, Brentjens R, Riviere I (2013) The basic principles of chimeric antigen receptor design. Cancer Discov 3:388–398
Scheffold C, Kornacker M, Scheffold YC, Contag CH, Negrin RS (2002) Visualization of effective tumor targeting by CD8+ natural killer T cells redirected with bispecific antibody F(ab’)(2)HER2xCD3. Cancer Res 62:5785–5791
Schlereth B, Quadt C, Dreier T, Kufer P, Lorenczewski G, Prang N, Brandl C, Lippold S, Cobb K, Brasky K, Leo E, Bargou R, Murthy K, Baeuerle PA (2006) T-cell activation and B-cell depletion in chimpanzees treated with a bispecific anti-CD19/anti-CD3 single-chain antibody construct. Cancer Immunol Immunother 55:503–514
Schmieg J, Yang G, Franck RW, Tsuji M (2003) Superior protection against malaria and melanoma metastases by a C-glycoside analogue of the natural killer T cell ligand alpha-galactosylceramide. J Exp Med 198:1631–1641
Schneiders FL, Scheper RJ, von Blomberg BME, Woltman AM, Janssen HLA, van den Eertwegh AJM, Verheul HMW, de Gruijl TD, van der Vliet HJ (2011) Clinical experience with α-galactosylceramide (KRN7000) in patients with advanced cancer and chronic hepatitis B/C infection. Clin Immunol 140:130–141
Shimizu K, Kurosawa Y, Taniguchi M, Steinman RM, Fujii S (2007) Cross-presentation of glycolipid from tumor cells loaded with alpha-galactosylceramide leads to potent and long-lived T cell mediated immunity via dendritic cells. J Exp Med 204:2641–2653
Shiratsuchi T, Schneck J, Kawamura A, Tsuji M (2009) Human CD1 dimeric proteins as indispensable tools for research on CD1-binding lipids and CD1-restricted T cells. J Immunol Methods 345:49–59
Sohn S, Tiper I, Japp E, Sun W, Tkaczuk K, Webb TJ (2014) Development of a qPCR method to rapidly assess the function of NKT cells. J Immunol Methods 407:82–89
Song L, Asgharzadeh S, Salo J, Engell K, Wu HW, Sposto R, Ara T, Silverman AM, DeClerck YA, Seeger RC, Metelitsa LS (2009) Valpha24-invariant NKT cells mediate antitumor activity via killing of tumor-associated macrophages. J Clin Invest 119:1524–1536
Sriram V, Cho S, Li P, O’Donnell PW, Dunn C, Hayakawa K, Blum JS, Brutkiewicz RR (2002) Inhibition of glycolipid shedding rescues recognition of a CD1+ T cell lymphoma by natural killer T (NKT) cells. Proc Natl Acad Sci USA 99:8197–8202
Stetson DB, Mohrs M, Reinhardt RL, Baron JL, Wang ZE, Gapin L, Kronenberg M, Locksley RM (2003) Constitutive cytokine mRNAs mark natural killer (NK) and NK T cells poised for rapid effector function. J Exp Med 198:1069–1076
Stirnemann K, Romero JF, Baldi L, Robert B, Cesson V, Besra GS, Zauderer M, Wurm F, Corradin G, Mach J-P, MacDonald HR, Donda A (2008) Sustained activation and tumor targeting of NKT cells using a CD1d–anti-HER2–scFv fusion protein induce antitumor effects in mice. J Clin Invest 118:994–1005
Sun W, Wang Y, East JE, Kimball AS, Tkaczuk K, Kesmodel S, Strome SE, Webb TJ (2015) Invariant natural killer T cells generated from human adult hematopoietic stem-progenitor cells are poly-functional. Cytokine 72:48–57
Tachibana T, Onodera H, Tsuruyama T, Mori A, Nagayama S, Hiai H, Imamura M (2005) Increased intratumor Valpha24-positive natural killer T cells: a prognostic factor for primary colorectal carcinomas. Clin Cancer Res 11:7322–7327
Tahara H (1995) Effective eradication of established murine tumors with IL-12 gene therapy using a polycistronic retroviral vector. Immunology 6466–6474
Tahir SM, Cheng O, Shaulov A, Koezuka Y, Bubley GJ, Wilson SB, Balk SP, Exley MA (2001) Loss of IFN-gamma production by invariant NK T cells in advanced cancer. J Immunol 167:4046–4050
Taniguchi M, Seino K, Nakayama T (2003) The NKT cell system: bridging innate and acquired immunity. Nat Immunol 4:1164–1165
Teng MW, Sharkey J, McLaughlin NM, Exley MA, Smyth MJ (2009) CD1d-based combination therapy eradicates established tumors in mice. J Immunol 183:1911–1920
Terabe M, Swann J, Ambrosino E, Sinha P, Takaku S, Hayakawa Y, Godfrey DI, Ostrand-Rosenberg S, Smyth MJ, Berzofsky JA (2005) A nonclassical non-Valpha14Jalpha18 CD1d-restricted (type II) NKT cell is sufficient for down-regulation of tumor immunosurveillance. J Exp Med 202:1627–1633
Terashima A, Watarai H, Inoue S, Sekine E, Nakagawa R, Hase K, Iwamura C, Nakajima H, Nakayama T, Taniguchi M (2008) A novel subset of mouse NKT cells bearing the IL-17 receptor B responds to IL-25 and contributes to airway hyperreactivity. J Exp Med 205:2727–2733
Thapa P, Zhang G, Xia C, Gelbard A, Overwijk WW, Liu C, Hwu P, Chang DZ, Courtney A, Sastry JK, Wang PG, Li C, Zhou D (2009) Nanoparticle formulated alpha-galactosylceramide activates NKT cells without inducing anergy. Vaccine 27:3484–3488
Topp MS, Gokbuget N, Stein AS, Zugmaier G, O’Brien S, Bargou RC, Dombret H, Fielding AK, Heffner L, Larson RA, Neumann S, Foa R, Litzow M, Ribera JM, Rambaldi A, Schiller G, Bruggemann M, Horst HA, Holland C, Jia C, Maniar T, Huber B, Nagorsen D, Forman SJ, Kantarjian HM (2015) Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol 16:57–66
Topp MS, Gokbuget N, Zugmaier G, Klappers P, Stelljes M, Neumann S, Viardot A, Marks R, Diedrich H, Faul C, Reichle A, Horst HA, Bruggemann M, Wessiepe D, Holland C, Alekar S, Mergen N, Einsele H, Hoelzer D, Bargou RC (2014) Phase II trial of the anti-CD19 bispecific T cell-engager blinatumomab shows hematologic and molecular remissions in patients with relapsed or refractory B-precursor acute lymphoblastic leukemia. J Clin Oncol 32:4134–4140
Toura I, Kawano T, Akutsu Y, Nakayama T, Ochiai T, Taniguchi M (1999) Cutting edge: inhibition of experimental tumor metastasis by dendritic cells pulsed with a-galactosylceramide. J Immunol 163:2387–2391
Uckun FM, Jaszcz W, Ambrus JL, Fauci AS, Gajl-Peczalska K, Song CW, Wick MR, Myers DE, Waddick K, Ledbetter JA (1988) Detailed studies on expression and function of CD19 surface determinant by using B43 monoclonal antibody and the clinical potential of anti-CD19 immunotoxins. Blood 71:13–29
Uemura Y, Liu TY, Narita Y, Suzuki M, Nakatsuka R, Araki T, Matsumoto M, Iwai LK, Hirosawa N, Matsuoka Y, Murakami M, Kimura T, Hase M, Kohno H, Sasaki Y, Ichihara Y, Ishihara O, Kikuchi H, Sakamoto Y, Jiao SC, Senju S, Sonoda Y (2009) Cytokine-dependent modification of IL-12p70 and IL-23 balance in dendritic cells by ligand activation of Valpha24 invariant NKT cells. J Immunol 183:201–208
Uldrich AP, Crowe NY, Kyparissoudis K, Pellicci DG, Zhan Y, Lew AM, Bouillet P, Strasser A, Smyth MJ, Godfrey DI (2005) NKT cell stimulation with glycolipid antigen in vivo: costimulation-dependent expansion, Bim-dependent contraction, and Hyporesponsiveness to further antigenic challenge. J Immunol 175:3092–3101
Vincent MS, Leslie DS, Gumperz JE, Xiong X, Grant EP, Brenner MB (2002) CD1-dependent dendritic cell instruction. Nat Immunol 3:1163–1168
Vivier E, Ugolini S, Blaise D, Chabannon C, Brossay L (2012) Targeting natural killer cells and natural killer T cells in cancer. Nat Rev Immunol 12:239–252
Wang J, Cheng L, Wondimu Z, Swain M, Santamaria P, Yang Y (2009) Cutting edge: CD28 engagement releases antigen-activated invariant NKT cells from the inhibitory effects of PD-1. J Immunol 182:6644–6647
Watarai H, Fujii S-i, Yamada D, Rybouchkin A, Sakata S, Nagata Y, Iida-Kobayashi M, Sekine-Kondo E, Shimizu K, Shozaki Y, Sharif J, Matsuda M, Mochiduki S, Hasegawa T, Kitahara G, Endo TA, Toyoda T, Ohara O, Harigaya K-i, Koseki H, Taniguchi M (2010a) Murine induced pluripotent stem cells can be derived from and differentiate into natural killer T cells. J Clin Invest 120:2610–2618
Watarai H, Rybouchkin A, Hongo N, Nagata Y, Sakata S, Sekine E, Dashtsoodol N, Tashiro T, Fujii S-I, Shimizu K, Mori K, Masuda K, Kawamoto H, Koseki H, Taniguchi M (2010b) Generation of functional NKT cells in vitro from embryonic stem cells bearing rearranged invariant Vα14-Jα18 TCRα gene. Blood 115:230–237
Watarai H, Sekine-Kondo E, Shigeura T, Motomura Y, Yasuda T, Satoh R, Yoshida H, Kubo M, Kawamoto H, Koseki H, Taniguchi M (2012) Development and function of invariant natural killer T cells producing T(h)2- and T(h)17-cytokines. PLoS Biol 10:e1001255
Webb TJ, Bieler JG, Schneck JP, Oelke M (2009) Ex vivo induction and expansion of natural killer T cells by CD1d1-Ig coated artificial antigen presenting cells. J Immunol Methods 346:38–44
Webb TJ, Li X, Giuntoli RL 2nd, Lopez PH, Heuser C, Schnaar RL, Tsuji M, Kurts C, Oelke M, Schneck JP (2012) Molecular identification of GD3 as a suppressor of the innate immune response in ovarian cancer. Cancer Res 72:3744–3752
Woo S-Y, Jung Y-J, Ryu K-H, Park H-Y, Kie J-H, Im S-A, Chung W-S, Han H-S, Seoh J-Y (2003) In vitro differentiation of natural killer T cells from human cord blood CD34+ cells. Br J Haematol 121:148–156
Wu D, Xing GW, Poles MA, Horowitz A, Kinjo Y, Sullivan B, Bodmer-Narkevitch V, Plettenburg O, Kronenberg M, Tsuji M, Ho DD, Wong CH (2005) Bacterial glycolipids and analogs as antigens for CD1d-restricted NKT cells. Proc Natl Acad Sci USA 102:1351–1356
Wu DY, Segal NH, Sidobre S, Kronenberg M, Chapman PB (2003) Cross-presentation of disialoganglioside GD3 to natural killer T cells. J Exp Med 198:173–181
Yanagisawa K, Exley MA, Jiang X, Ohkochi N, Taniguchi M, Seino K (2006) Hyporesponsiveness to natural killer T-cell ligand alpha-galactosylceramide in cancer-bearing state mediated by CD11b + Gr-1+ cells producing nitric oxide. Cancer Res 66:11441–11446
Yen J, White RM, Stemple DL (2014) Zebrafish models of cancer: progress and future challenges. Curr Opin Genet Dev 24:38–45
Zaini J, Andarini S, Tahara M, Saijo Y, Ishii N, Kawakami K, Taniguchi M, Sugamura K, Nukiwa T, Kikuchi T (2007) OX40 ligand expressed by DCs costimulates NKT and CD4+ Th cell antitumor immunity in mice. J Clin Invest 117:3330–3338
Zajonc DM, Girardi E (2015) Recognition of microbial glycolipids by Natural Killer T cells. Front Immunol 6
Acknowledgments
Given the wide breadth of studies examining the role of NKT cells in cancer, only recent reviews and closely related articles have been cited and we apologize to those whose work may have been omitted due to space considerations. The authors have no competing financial interest. This work was supported by grants from the NIH/NCI to TJW.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is published in the Special Issue CD1, MR1, NKT, and MAIT: Evolution and Origins of Non-peptidic Antigen Recognition by T lymphocytes with Guest Editor Dr. Dirk Zajonc
Rights and permissions
About this article

Cite this article
Shissler, S.C., Bollino, D.R., Tiper, I.V. et al. Immunotherapeutic strategies targeting natural killer T cell responses in cancer. Immunogenetics 68, 623–638 (2016). https://doi.org/10.1007/s00251-016-0928-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00251-016-0928-8