
Abstract
The interaction of plants and root-associated bacteria encourage the removal of soil contaminants. Engineers and scientists have looked at this phenomenon as a possible means of soil treatment (rhizodegradation). In this study, root-associated bacteria were isolated and selected for growth on a model soil contaminant: polycyclic aromatic hydrocarbons. Isolates were compared genetically to see how plant–bacteria interactions change with soil contamination levels. Characterization of root-associated bacteria was performed using REP-PCR genetic fingerprinting and 16s rRNA gene alignments for identification. Genomic fingerprinting indicated that the composition of PAH-metabolizing bacteria (“guild”) was similar among plant species at each treatment level. However, guild composition changed with contamination level and differed from that of bulk soils, suggesting a common rhizosphere effect among plant species related to PAH contamination. PAH-metabolizing bacteria were identified through 16s rRNA gene alignment as members of the α-, β-, and γ-proteobacteria, Actinobacteria, and Bacilli classes. Burkholderia and Pseudomonas spp. were the only genera of bacteria isolated from all plant types in uncontaminated controls. Bacterial species found at the highest treatment included Achromobacter xylosoxidans, Rhodococcus spp., members of the Microbacteriae, Stenotrophomonas rhizophilia, as well as other members of the alpha-proteobacteria. Given their ability to grow on PAHs and inhabit highly contaminated rhizospheres, these bacteria appear good candidates for the promotion of rhizodegradation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Although the mechanisms which plants employ to remove soil contamination are not yet fully understood, rhizosphere microorganisms clearly play a major role in the process. In this regard, beneficial bacteria in the rhizosphere have been shown to reduce contaminant-related stress by decreasing the concentration of ethylene below toxic levels [1]. In addition, plants release exudates to induce metabolic responses by rhizobacteria which lead to contaminant degradation and a subsequent reduction of phytotoxicity [2, 3]. However, it is not clear how soil contamination affects bacterial populations in the rhizosphere and what influence plant species has on population composition. A better understanding of plant–microbe interactions under contamination stress may benefit phytoremediation applications designed to enhance biological degradation of soil contaminants.
The rhizosphere is the soil region surrounding plant roots that is influenced chemically and physically by plant activity as roots grow [4, 5]. Plants alter the soil by adding nutrients and organic carbon, detaching and translocating soil-bound compounds (biosurfactants, chelating agents), and promoting air exchange [4]. The addition of this plant material into the soil encourages microbial activity, leading to increased numbers and altered population composition when compared to bulk soil [6]. This increase in population numbers and activity is termed the rhizosphere effect [7].
The composition of the rhizosphere bacterial population (RBP) can vary between plant species due to the complexity of plant–microbe interactions. These interactions can be mutualistic, as is the case for nitrogen-fixing symbionts of legumes and plant growth-promoting rhizobacteria. The host plant sloughs root cells and mucilages into the soil, and actively exudes lysates, amino acids, proteins, sugars, complex carbohydrates, alcohols, vitamins, and hormones that bacteria can assimilate [4, 5]. Under uncontaminated conditions, plants and bacterial populations establish feedback loops involving heterotrophic and autotrophic bacteria [8]. Beneficial bacteria may ensure this mutualistic relationship by synthesizing plant hormones and producing antibiotics that suppress pathogens [4, 5]. In the case of high soil contamination, it would be expected that normal root exchange would be disrupted and microbial feedback loops altered. Presumably, the plant’s ability to respond to this disruption will lead to either an increase in beneficial bacteria to correct the imbalance or the growth of opportunistic plant pathogens that can withstand the host plant’s antimicrobial defenses.
Residues from historic petroleum industries are prevalent soil contaminants that contain polycyclic aromatic hydrocarbons (PAHs). PAH soil contamination is difficult to remediate as these aromatic compounds are hydrophobic and adhere strongly to the soil matrix [9, 10]. These compounds also inhibit soil microbial activity and deter seed germination [11]. However, bioremediation studies have found that bacteria inhabiting the rhizosphere and bulk soils can degrade PAHs [12,13,14,15]. In addition, some plant types, namely, Cucurbits, can translocate significant amounts of PAHs towards the root zone [16]. Evidence suggests that plants promote bacterial degradation of these compounds in the rhizosphere [13, 14, 17, 18]. This method of plant-mediated contaminant removal is termed rhizodegradation. Characterization of rhizobacteria that grow on PAHs (“guild”) may shed light on the host plant’s response to PAH soil toxicity and the effect on root-associated bacterial populations. By identifying members of this guild, bacteria-plant combinations can be tested for the improvement of rhizodegradation.
Plant responses to phytotoxic stress in the soil are difficult to ascertain, since the root zone is highly populated with a diverse array of microorganisms and cannot be easily observed in situ. Yet, various microscopic and molecular techniques have been developed to examine the microbial inhabitants of the rhizosphere. For example, the effect of environmental conditions on rhizosphere bacterial activity has been monitored through respiration rates and DNA/protein synthesis [19]. Direct microscopic observation of rhizosphere habitats has been achieved through the use of fluorescent probes and the inoculation of gfp-tagged strains onto developing roots [19, 20]. Molecular techniques such as denaturing gradient gel electrophoresis (DGGE) of 16S rRNA gene PCR amplicons and terminal restriction fragment length polymorphism (T-RFLP) have also allowed for examination of bacterial communities from the rhizosphere [6, 21,22,23]. In one example, community analysis has shown that bacterial populations change with exudate variations that occur with root activity as plants grow [24].
This study was conducted with the notion of an augmented rhizodegradation system that promotes PAH removal and decontamination of soil with petroleum residues. This system would involve the inoculation of a particular bacterial type that has shown the ability to metabolize PAHs onto the roots of a host plant to increase the potential for soil decontamination. In order to select which bacterial types should be candidates for rhizosphere augmentation, there are important questions to answer: (1) How do bacterial communities in the rhizosphere respond to soil contamination levels and host plant species? And (2) What bacteria are the most prevalent in the rhizosphere when plants are subjected to higher levels of PAH contamination stress? In order to answer these questions we compared bacteria that metabolize PAHs from four different plant types. Population changes due to contamination level and plant variety were considered. Genomic fingerprints of bacterial isolates were also identified to learn what bacteria primarily make up the communities isolated from the root zone.
Materials and Methods
Obtaining PAH-Utilizing Bacteria from Plant Rhizospheres
The methods used to grow plants have been previously described [23]. In brief, four test plants—zucchini (Cucurbita pepo ssp. pepo “Black Beauty”), pumpkin (Cucurbita pepo ssp. pepo “Howden”), lettuce (Lactuca sativa “Tango”), and wheat (Triticum aestivum “Cavalier”)—were grown in a soil mixture including soil from a former manufactured gas plant (“MGP”) in Winsted, CT. The MGP soil was highly contaminated with a suite of PAHs (760 ppm in total). The soil mixture included uncontaminated soil (“Pristine”) which was obtained from the Lockwood Farm in Hamden, CT, which is part of the Connecticut Agricultural Experiment Station. In addition, a small amount of peat moss was added for water retention. MGP soil levels in the mixture were 0% (control), 3%, or 30% (v/v). Plants were grown at room temperature (24 °C) with a 16-h photoperiod and supplied with sterile water and fertilizer.
Plants were harvested after 2 months of growth. Roots were withdrawn from the potting media and sonicated in a buffered solution to obtain rhizosphere bacteria. Five grams of the soil was used to obtain bacteria from MGP and pristine bulk soils. Cell suspensions were collected and subjected to PAH selection plating. Rhizosphere cell suspensions were spread on mineral salts minimal media (MSMM) plates prepared with noble agar. The plates were then sprayed with PAH–solvent solutions (500 mg/L anthracene in hexanes and 500 mg/L chrysene in hexanes). These plates were allowed to incubate for 30 days at room temperature (24 °C). PAH-metabolizing bacteria were selected based on the formation of a clearing zone on the PAH layer around the colonies. Single colonies were then purified by T-streaking on 0.1 R2A media.
Genome Fingerprinting
In order to distinguish between the bacterial colonies selected from PAH-amended plates, whole-genome fingerprints were developed for each isolate using REP-PCR. Repetitive Element Sequence PCR (REP-PCR) targets sequences which are repeated in the chromosome, called “palindromic units.” Because these sequences will occur in different locations for various types of bacteria, a specific band pattern will occur due to the different sizes of PCR amplicons. These band patterns are then employed as a genome fingerprint to compare types of bacteria.
All steps of the REP-PCR protocol were performed in 96-well plates to efficiently evaluate a large number of isolated bacteria. Crude DNA extracts were prepared by growing bacteria at room temperature in 96-well microtiter plates with 200 μL 0.25 Tryptic soy broth (TSB). After 2 days, 50 μL aliquots were transferred to a 96-well PCR plate. These cell suspensions were placed in a boiling water bath for 1 min and then cooled in an ice bath for 1 min. This step was repeated four times to lyse the cells. The 96-well PCR plate was then spun at 3700 rpm in an Eppendorf centrifuge. A 0.3 μL aliquot of the supernatant was then used as the template for the 20 μL REP-PCR reaction. The primer, BOXA1R (5’-CTACGGCAAGGCGACGCTGACG-3′), was at a final concentration of 0.9 μM for each reaction. Final concentrations for the rest of the PCR reagents were 1× PCR buffer, 2.5 mM MgCl2, 200 μM dNTPs and 0.6 U of Taq polymerase (Applied Biosystems). The thermocycler program was 94 °C for 2 min, 36 cycles of [94 °C for 30 s, 55 °C for 1 min, and 65 °C for 8 min], and then a final extension of 65 °C for an additional 8 min. Amplification products were separated on 1.5% agarose gels containing 0.5 μg/μL ethidium bromide and run at 70 V for 3.5 h in 1 × TBE. Figure 1 displays a gel with band patterns generated from REP-PCR amplicons. These band patterns were used to create whole-genome fingerprints for each isolate. Curve-based analysis was performed on the gel images using Bionumerics (Applied Maths, St-Martens-Latem, Belgium). Clustering for the band patterns was calculated using Pearson’s product-moment correlation coefficient and the unweighted pair group method with arithmetic mean (UPGMA). The cutoff percentage for clusters was determined by comparing ladder standards through cluster analysis to quantify gel variation. Genome fingerprints with similarities above the cutoff percentage were then grouped into unique clusters. The number of isolates in each cluster (Table 1) was then subjected to canonical correspondence analysis (CCA) to determine the influence of contamination level and plant species on the diversity of the bacterial populations. This ordination technique extracts continuous axes of variation from species occurrence or abundance data. CCA is represented by an ordination diagram which shows the patterns of variation in community composition best explained by environmental variables.

Image of a representative gel with band patterns produced through REP-PCR (genome fingerprints). Lanes 2–9 and 11–18 are band patterns for bacteria isolated from rhizospheres in 3% MGP soil mixture. Lanes 1, 10, and 19 are 1 Kb Plus ladder used as the DNA mass standard
16s rRNA Gene PCR Sequencing and Alignment
Groups of genomic fingerprints were then subjected to partial 16S rRNA gene sequencing and subsequent alignment with reference sequences. Representative bacteria from each group were grown in 0.25 TSB at room temperature. Chromosomal DNA from these cell suspensions was extracted using the UltraClean DNA extraction kit (MoBio). Eubacterial 16S rRNA genes were amplified with forward S-D-Bact-49-a-s-20 (5′- TNANACATGCAAGTCGAICG-3′) and reverse S-D-Bact-1492-a-A-19 (5′- GGYTACCTTGTTACGACTT-3′) primers, based on the Oligonucleotide Probe Database. The Qiagen Taq PCR core kit with Q solution (Qiagen, Valencia, CA) was used for all PCR reactions. Each 100-μL reaction mixture consisted of 1× PCR buffer, 1× Q solution, 5 mM Mg 2+, 0.4 μM of each primer, 200 μM dNTPs, and 2.5 U Taq polymerase. The thermocycler program was 94 °C for 2 min, 33 cycles of [94 °C for 30 s, 60 °C for 45 s, and 72 °C for 1 min], and a final extension of 72 °C for 1 min (Perkin Elmer Lifesciences Inc., Boston, MA, models 2400 and 9700). Six replicate PCR reactions were performed simultaneously in 96-well microtiter plates to obtain enough amplified product for sequencing. The six replicate reactions were combined and purified using the MoBio Purification kit. Partial 16S rRNA gene products were sequenced at the University of Connecticut Biotechnology Center or at Macrogen, Inc. (Seoul, South Korea). Sequences and chromatographs were edited using Chromas v. 2.01 freeware and aligned in the Ribosomal Database Project 2 (RDP). A phylogenetic tree was then created using the Treebuilder function to illustrate the similarity between the PAH-metabolizing rhizosphere isolates and the nearest relative in the RDP.
Results
PAH-utilizing bacterial isolates from the rhizosphere were characterized through genomic fingerprint clustering. REP-PCR band patterns as pictured in Fig. 1 were used as genomic fingerprints for bacterial isolates and clustered through curve-based analysis using Bionumerics software. Figure 2 shows a representative dendrogram displaying the clustering of isolates from the 30% MGP soil mixture. These fingerprints are ≥ 60% similar and identified as a single “cluster.” The 60% similarity was chosen as the cutoff point given the similarity of DNA mass standards used in the gels.

Representative cluster analysis of REP-PCR genetic fingerprints for 30% MGP rhizospheres isolates. The figure shows different fingerprints that are > 60% similar and identified as a single “cluster.” Breakdown of isolates by host plant species: Wheat = 16, Pumpkin = 20, Zucchini = 10, Lettuce = 23
In Fig. 3, all clusters are shown which contained 5 or more genomic fingerprints. The two enlargements of clusters 15 and 31 are provided in Fig. 3 to illustrate the curves resulting from REP-PCR band patterns. Both peak magnitude and position are considered in the clustering analysis. Figure 3 shows that 46 clusters with five or more isolates were found among the pool of genomic fingerprints. This pool includes the four plant types (wheat, lettuce, zucchini, and pumpkin) at each MGP soil level (0%, 3%, and 30%) and the two bulk soil samples (pristine and 100% MGP soil). Table 1 provides a breakdown of each cluster from Fig. 3 based on either treatment level, host plant type, or bulk soil. Treatment level is the amount of MGP soil included in the potting mixture to which plant rhizospheres were subjected. These numbers were subjected to canonical correspondence analysis (CCA) to see if plant type or treatment level were the major determining factor in the clustering of genome fingerprints. Figure 4 shows how the samples align in relation to treatment level or plant species. In total, the CCA method indicates that MGP concentration was more determinant of the guild composition of PAH-metabolizing bacteria than the plant species. Samples W0, W3, W30, L0, L30, Z0, Z3, Z30, P0, and P30 all align more along the MGP concentration variable. Samples L3 and P3 appear to align more along the plant species variable. In addition, the two bulk soils B0 and B100 align along the MGP concentration variable, which makes sense as these samples had no influence from plant roots and therefore, should not align along the plant species variable.

Curve-based analysis of genomic fingerprints for all clusters with five or more isolates. Fingerprints with a percentage similarity greater than 60% are considered a cluster. The two enlargements of cluster 15 and cluster 31 show curves developed from the band patterns used for analysis. Symbols are W = wheat, L = lettuce, Z = zucchini, P = pumpkin, 0 = 0% contamination, and 3 = 3% contamination

Canonical correspondence analysis (CCA) of the cluster values in Table 1. All samples are presented with the plant type (Z = Zucchini, P = Pumpkin, W = Wheat, and L = Lettuce), the rhizosphere treatment level (0 = 0% MGP, 3 = 3% MGP, and 30 = 30% MGP), and the bulk soil (B0 = Pristine soil, and B100 = MGP soil)
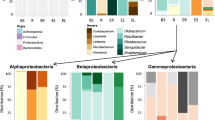
Based on the 16s rRNA gene sequencing, two phyla and five classes of bacteria are present among the PAH-metabolizing isolates (Fig. 5). We found three genera of β-proteobacteria and γ-proteobacteria, five genera of α-proteobacteria, one genera of Bacilli, and six genera of Actinobacteria, for a total of 18 genera. In total, 21 species of PAH-metabolizing bacteria were identified for all treatments.

Alignment of 16s RNA gene sequences for bacterial isolates with sequences in the Ribosomal Database Project (RDP). The phylogenetic tree shows bacteria from the three rhizosphere treatments (0%, 3%, and 30% MGP soil) and the bulk soil controls (Pristine and MGP). The scale indicates sequence dissimilarity (10%). Streptococcus infantarius (Class: Bacilli) was used as the outgroup
Two bacterial types, Burkholderia sp.1 and Pseudomonas sp.2, were present in the rhizospheres of all plant varieties grown in the 0% MGP soil mixture. 16s rRNA gene alignments for these two types aligned closest to Burkholderia fungorum and Pseudomonas rhodesiae (Fig. 5). Genome fingerprinting showed that 16 clusters made up these bacterial types.
Four classes of bacteria—β-proteobacteria, γ-proteobacteria, Actinobacteria, and Bacilli—were found among the guild for the 3% treatment level. Pseudomonas and Burkholderia were present among the PAH-metabolizing bacteria but are not found at the highest treatment level. An additional eight more genera and 34 total genome fingerprint clusters were found at the 3% level. One fingerprint cluster, “3% Rhizo10,” was unidentified though 16srRNA gene alignment.
At 30% MGP soil contamination, three of the same classes of bacteria are found, and several new genera were present (Table 2). One of these is Stenotrophomonas, of the γ-proteobacteria. A few genomic fingerprints of this bacterial species were found and this bacteria was only present at the 30% level. This condition indicates that this bacterial species can tolerate higher contamination levels and effectively colonize the root zone. One of the primary differences at this treatment level was the presence of α-proteobacteria and Actinobacteria. These classes were found almost exclusively at higher contamination levels. Interestingly, they do not necessarily correlate with the bacteria found in the bulk MGP soil. This prevalence would indicate that the populations of these types of bacteria are promoted and that these bacteria are relatively good root-colonizers (i.e., prevalence is not based on the fact that more bacteria from the MGP soil would be present given the higher amount of MGP soil mixed into the pots). No Pseudomonas or Burkholderia appeared at the 30% level which is in sharp contrast to the 0% MGP treatment where these were the only types of bacteria identified. The largest number of genera for a given pool of isolates occurred at the 30% MGP level (Table 3).
Discussion
In uncontaminated soils, plant root activity promotes rapidly growing heterotrophs. These bacteria feed on root turnover and exuded compounds. With high amounts of soil contamination, it would be expected that the plant would need to respond to the phytotoxicity, and that this response could change the microbial populations in the rhizosphere. Plants respond to environmental stress through the production and subsequent exudation of particular compounds, such as flavonoids [25]. Secondary metabolites, such as phenols and their oxygen-substituted derivatives—terpenoids, lactones, coumarins, simple phenolics, tannins, alkaloids, saponins, and quinones—can also protect plants from predatory microbes, insects, and herbivores [26]. These compounds can be actively exuded from roots [27]. These interactions result in the development of a pool of pollutant-degrading enzymes which aid in the detoxification of contaminated soil [28]. By controlling the flow of root exudates, plants encourage microbial breakdown of harmful substances in the soil [13].
In this study, rhizobacteria were characterized under PAH stress conditions to observe how guild composition changes with contamination level and host plant species. Comparison of cultured bacterial isolates was performed through genetic fingerprinting. 16s rRNA gene sequences were aligned with known species to identify bacterial species that inhabit the rhizosphere and show the potential to promote soil decontamination. Identification of bacterial species can be applied by the use of an augmented rhizodegradation system where known PAH-metabolizing bacteria that display PAH-metabolizing activity and root-colonization are introduced to host plant species. In addition, further understanding of the community dynamics in the rhizosphere can shed light on the way in which the local soil ecology is restored given a toxic pollution event. Genomic fingerprinting showed that PAH-metabolizing guild members were not specific to host plant species. The four plants generally showed the same genera of PAH-metabolizing bacteria; however, slight differences were evident in the abundance of these bacterial types. While the similarity in guild composition among plant varieties continued through the three contamination levels, the isolated bacterial types did change noticeably as the level of MGP soil contamination increased. These findings suggest that a commonly shared rhizosphere effect has occurred in response to soil toxicity.
In another previous study, a phytotoxicity assay confirmed that the presence of plant rhizospheres improved the soil conditions over the growth period by reducing the toxicity to plants [29]. This finding suggests that the interaction of the bacteria and the plant roots are able to clean and restore the soil habitat. In that same study, it was also found that the isolated bacteria can not only metabolize PAHs but also a number of aromatic plant exudates (simple phenols and flavonoids). The high rate of metabolism found for these potentially antimicrobial exudates suggests that active exudations of such compounds may be the mechanism by which host plants promote the presence of a guild of PAH-metabolizing bacteria to aid contaminant breakdown, the reduction of phytotoxicity, and the eventual restoration of the soil health.
Bacterial communities from both bulk soils are different from those associated with plant roots indicating that PAH-metabolizers found in the rhizosphere are better root colonizers. The ability of bacteria to colonize roots is another important consideration for an augmented rhizodegradation system. Survival of bacterial samples introduced onto plant roots would require a beneficial relationship with the host plant.
The two types of PAH-utilizing bacteria found in rhizospheres uncontaminated by PAHs were Burkholderia spp. and Pseudomonas spp. These bacterial types have special relationships with plants and have even been shown to be endophytic, living within the plant tissue [30]. Burkholderia species are found in a wide range of soil and rhizosphere environments [31, 32], and multiple strains are present in the rhizospheres of a wide variety of cultivated crop species including maize, yellow lupine, citrus plants, banana, pineapple, grape, and rice plants [33, 34]. Likewise, Pseudomonas sp. has been identified among isolates of marigold, carrot, soybean, and Scots pine [33]. Pseudomonas strains isolated in this study are identified as Pseudomonas rhodesiae, a species which has previously demonstrated strong PAH-degrading ability [35]. Pseudomonad soil isolates have also been shown to increase PAH solubility threefold through the production of biosurfactants [36]. The fact that these bacteria are present in such varied host plants and promote rhizodegradation of PAHs suggests that Pseudomonas and Burkholderia strains may benefit phytoremedial applications. In this study, though, Pseudomonas and Burkholderia spp. were not found at the higher contamination levels indicating that these species may not be able tolerate high PAH levels and still colonize the root zone.
Gram-positive bacteria and α-proteobacteria (Table 2) were found exclusively in the contaminated rhizospheres and the α-proteobacteria were only present at the 30% treatment level. Another study found that α-proteobacteria were found in oil-contaminated soils but not in uncontaminated soils [37]. Unlike the Burkholderia and Pseudomonas species mentioned above, the appearance of these bacterial types seems to be directly related to the presence of contamination in the soil.
Several bacterial genera previously unreported for having activity towards PAHs were identified in this study through community analysis: Microbacterium, Afipia, and Methylobacterium. All of these three genera are found only in rhizosphere environments, not in bulk soils. The bacterial species found exclusively in pristine bulk soil environments included Mesorhizobium loti, Bacillus niacini, and Sphingopyxis macrogoltabidus. In contrast to the bacteria found only in rhizosphere treatments, bacteria found only in the bulk soil appear to be poor root colonizers.
Many of the bacterial genera found in this study have previously shown the ability to utilize PAHs as a growth substrate. Pseudomonas stutzeri P16 and Pseudomonas saccharophila P15, have been grown on phenanthrene, naphthalene, 1-methylnaphthalene, and 2-methylnaphthalene as sole carbon and energy sources [38, 39]. These strains, as well as Sphingomonas yanoikuyae R1, and Bacillus cereus P21, also degrade pyrene [40]. Strains of Burkholderia, Rhodococcus, and Variovorax have also shown degradative abilities towards PAHs [12, 14, 41,42,43,44]. Degradation of chrysene is especially relevant to this study as this compound is one of the two PAHs used for bacteria selection. A Sphingomonas strain has been shown previously to mineralize radiolabeled chrysene [45].
Sphingomonas strains isolated in this study and selected for PAH-metabolism are possible candidates for rhizodegradation applications. Likewise, Stenotrophomonas species are absent from the 0% and 3% treatments, yet these bacteria were prevalent at the highest contamination level. The fact that this bacterium can utilize PAHs and is present in such abundance in the highly contaminated rhizospheres indicates that this bacteria also has good potential for rhizodegradation.
Conclusion
This study demonstrates that although guild compositions differ between plants at the various levels of contamination, the same genera of bacteria are generally found in all four plant types. However, the guild of bacteria that utilize PAHs changes significantly at the three contamination levels (Fig. 4). This finding is similar to a finding in a previous paper which showed that the whole rhizosphere community (not just the PAH-degrading guild) also was determined more by contamination level than plant type [23]. In combination, these findings indicate that the microbial community associated with the plant roots is changing due to the soil contamination in a similar way for different plant species. This change in microbial community suggests that there is a common mechanism by which the plants are stimulating a response in the rhizosphere.
This mechanism of population stimulation through aromatic exudates signaling is not a far-fetched idea. In fact, a very similar mechanism has been elucidated for the phenomenon of nodulation by root-associated bacteria. In this system, legume plants which require greater concentrations of nitrogen in the root zone, actively recruit rhizobacteria to infect the plant root, form nodules, and fix nitrogen to improve plant nutrition and growth. Plants stimulate nodulation by exuding isoflavonoids from their roots. These aromatic exudates then bind to the promoter of the nod operon in the bacterial chromosome [46]. The genes in the operon are then transcribed and translated into proteins which enable the infection of the root hair, the development of the infection thread, and the eventual formation of the nodule.
It appears, then, that plants also employ a similar mechanism in promoting rhizodegradation. As stated earlier, almost all PAH-metabolizing isolates also showed the ability to metabolize flavonoid compounds like morin, quercetin, and naringenin in a previous study. This method of signaling may then stimulate the change in rhizosphere population and PAH-metabolizing guild which has also been shown. A greater understanding of this mechanism may allow for more effective treatment applications in phytoremediation and faster soil rehabilitation.
References
Glick BR (2003) Phytoremediation, synergistic use of plants and bacteria to clean up the environment. Biotechnol Adv 21:383–393
Miya RK, Firestone MK (2001) Enhanced phenanthrene biodegradation in soil by slender oat root exudates and root debris. J Environ Qual 30(6):1911–1918
Kamath R, Schnoor J, Alvarez P (2004) Effect of root-derived substrates on the expression of nah-lux genes in Pseudomonas fluorescens hk44: implications for PAH biodegradation in the rhizosphere. Environ Sci Technol 38(1740):1740–1745
Knueffel DA (1993) The behavior and tracking of bacteria in the rhizosphere. Annu Rev Phytopathol 31:441–472
Dandurand LM and G.R. Knudsen (2001) Sampling Microbes from the Rhizosphere and Phyllosphere. In: Hurst CJ, et al. (eds) Manual of Environmental Microbiology. American Society for Microbiology
Duineveld BM, Kowalchuk GA, Keijzer A, van Elsas JD, van Veen JA (2001) Analysis of bacterial communities in the rhizosphere of chrysanthemum via denaturing gradient gel electrophoresis of PCR-amplified 16S rRNA as well as DNA fragments coding for 16S rRNA. Appl Environ Microbiol 67(1):172–178
Whipps JM (2001) Microbial interactions and biocontrol in the rhizosphere. J Exp Bot 52(suppl_1):487–511
Whipps JM (1990) Carbon Economy. In: Lynch JM (ed) The Rhizosphere. John Wiley and Sons, Chichester
Sims RC, Overcash MR (1983) Fate of polynuclear aromatic compounds (PNAs) in soil-plant systems. Residue Reviews 88:1–68
Shuttleworth KL, Cerniglia CE (1995) Environmental aspects of PAH biodegradation. Appl Biochem Biotechnol 54(1–3):291–302
Lepsis J, Blanke MM Environmental Effects, Phytotoxicity and Breakdown of Tar Oil From Impregnated Tree Stakes, in VIII International Symposium on Canopy, Rootstocks, and Environmental Physiology in Orchard Systems, ISHS Acta Horticulturae
Aitken MD, Stringfellow WT, Nagel RD, Kazunga C, Chen SH (1998) Characteristics of phenanthrene-degrading bacteria isolated from soils contaminated with polycyclic aromatic hydrocarbons. Can J Microbiol 44(8):743–752
Siciliano SD et al (2001) Selection of specific endophytic bacterial genotypes by plants in response to soil contamination. Appl Environ Microbiol 67(6):2469–2475
Daane LL et al (2001) Isolation and characterization of polycyclic aromatic hydrocarbon-degrading bacteria associated with the rhizosphere of salt marsh plants. Appl Environ Microbiol 67(6):2683–2691
Bastiaens L et al (2000) Isolation of adherent polycyclic aromatic hydrocarbon (PAH)-degrading bacteria using PAH-sorbing carriers. Appl Environ Microbiol 66(5):1834–1843
Parrish ZD, White JC, Isleyen M, Gent MPN, Iannucci-Berger W, Eitzer BD, Kelsey JW, Mattina MI (2006) Accumulation of weathered polycyclic aromatic hydrocarbons (PAHs) by plant and earthworm species. Chemosphere 64(4):609–618
Corgie SC, Beguiristain T, Leyval C (2004) Spatial distribution of bacterial communities and phenanthrene degradation in the rhizosphere of Lolium perenne L. Appl Environ Microbiol 70(6):3552–3557
Juhasz AL, Naidu R (2000) Enrichment and isolation of non-specific aromatic degraders from unique uncontaminated (plant and faecal material) sources and contaminated soils. J Appl Microbiol 89(4):642–650
Ramos C, Molbak L, Molin S (2000) Bacterial activity in the rhizosphere analyzed at the single-cell level by monitoring ribosome contents and synthesis rates. Appl Environ Microbiol 66(2):801–809
Bringhurst RM, Cardon ZG, Gage DJ (2001) Galactosides in the rhizosphere: utilization by Sinorhizobium meliloti and development of a biosensor. Proc Natl Acad Sci U S A 98:4540–4545
Niemi MR et al (2001) Extraction and purification of DNA in rhizosphere soil samples for PCR-DGGE analysis of bacterial consortia. J Microbiol Methods 45(3):155–165
Nunan N et al (2005) Links between plant and rhizoplane bacterial communities in grassland soils, characterized using molecular techniques. Appl Environ Microbiol 71(11):6784–6792
Pritchina O, Ely C, Smets B (2011) Effects of PAH-contaminated soil on rhizosphere microbial communities. Water Air Soil Pollut:1–9
Baudoin E, Benizri E, Guckert A (2002) Impact of growth stage on the bacterial community structure along maize roots, as determined by metabolic and genetic fingerprinting. Appl Soil Ecol 19(2):135–145
Winkel-Shirley B (2002) Biosynthesis of flavonoids and effects of stress. Curr Opin Plant Biol 5(3):218–223
Cowan MM (1999) Plant products as antimicrobial agents. Clin Microbiol Rev 12(4):564–582
Pillai BVS, Swarup S (2002) Elucidation of the flavonoid catabolism pathway in Pseudomonas putida PML2 by comparative metabolic profiling. Appl Environ Microbiol 68(1):143–151
Adams N et al (2000) Introduction to phytoremediation. Environmental Protection Agency, Washington, D.C.
Ely CS, Smets BF (2017) Bacteria from wheat and cucurbit plant roots metabolize PAHs and aromatic root exudates: implications for rhizodegradation. Int J Phytoremediat 19(10):877–883
Taghavi S et al (2005) Horizontal gene transfer to endogenous endophytic bacteria from poplar improves phytoremediation of toluene. Appl Environ Microbiol 71(12):8500–8505
Ramette A, LiPuma JJ, Tiedje JM (2005) Species abundance and diversity of Burkholderia cepacia complex in the environment. Appl Environ Microbiol 71(3):1193–1201
Salles JF, van Veen JA, van Elsas JD (2004) Multivariate analyses of Burkholderia species in soil: effect of crop and land use history. Appl Environ Microbiol 70(7):4012–4020
Rosenblueth M, Martinez-Romero E (2006) Bacterial endophytes and their interactions with hosts. Mol Plant-Microbe Interact 19(8):827–837
Compant S et al (2005) Endophytic colonization of Vitis vinifera L. by plant growth-promoting bacterium Burkholderia sp. strain PsJN. Appl Environ Microbiol 71(4):1685–1693
Kahng, et al. (2002) PAH utilization by Pseudomonas rhodesiae KK1 isolated from a former manufactured-gas plant site. Appl Microbiol Biotechnol 60(4):475–480
Deziel E et al (1996) Biosurfactant production by a soil Pseudomonas strain growing on polycyclic aromatic hydrocarbons. Appl Environ Microbiol 62(6):1908–1912
MacNaughton SJ et al (1999) Microbial population changes during bioremediation of an experimental oil spill. Appl Environ Microbiol 65(8):3566–3574
Stringfellow WT, Aitken MD (1994) Comparative physiology of phenanthrene degradation by 2 dissimilar pseudomonads isolated from a creosote-contaminated soil. Can J Microbiol 40(6):432–438
Stringfellow W, Aitken M (1995) Competitive metabolism of naphthalene, methylnaphthalenes, and fluorene by phenanthrene-degrading pseudomonads. Appl Environ Microbiol 61(1):357–2392
Kazunga C, Aitken MD (2000) Products from the incomplete metabolism of pyrene by polycyclic aromatic hydrocarbon-degrading bacteria. Appl Environ Microbiol 66(5):1917–1922
Eriksson M et al (2003) Degradation of polycyclic aromatic hydrocarbons at low temperature under aerobic and nitrate-reducing conditions in enrichment cultures from northern soils. Appl Environ Microbiol 69(1):275–284
Moody JD et al (2001) Degradation of phenanthrene and anthracene by cell suspensions of Mycobacterium sp. strain PYR-1. Appl Environ Microbiol 67(4):1476–1483
Brezna B, Khan A, Cerniglia CE (2003) Molecular characterization of dioxygenases from polycyclic aromatic hydrocarbon-degrading Mycobacterium spp. FEMS Microbiol Ecol 223:177–183
Larkin MJ et al (1999) Purification and characterization of a novel naphthalene dioxygenase from Rhodococcus sp. strain NCIMB12038. J Bacteriol 181(19):6200–6204
Demaneche S et al (2004) Identification and functional analysis of two aromatic-ring-hydroxylating dioxygenases from a Sphingomonas strain that degrades various polycyclic aromatic hydrocarbons. Appl Environ Microbiol 70(11):6714–6725
Masson-Boivin M et al (2009) Establishing nitrogen-fixing symbioses with legumes: how many rhizobium recipes? Trends Microbiol 17(10):458–466
Funding
This research was performed by a funding grant from the Environmental Protection Agency Star Program, R829405.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ely, C.S., Smets, B.F. Guild Composition of Root-Associated Bacteria Changes with Increased Soil Contamination. Microb Ecol 78, 416–427 (2019). https://doi.org/10.1007/s00248-019-01326-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-019-01326-6