
Abstract
The organisms inhabiting the deep-seafloor are known to play a crucial role in global biogeochemical cycles. Chemolithoautotrophic prokaryotes, which produce biomass from single carbon molecules, constitute the primary source of nutrition for the higher organisms, being critical for the sustainability of food webs and overall life in the deep-sea hydrothermal ecosystems. The present study investigates the metabolic profiles of chemolithoautotrophs inhabiting the sediments of Menez Gwen and Rainbow deep-sea vent fields, in the Mid-Atlantic Ridge. Differences in the microbial community structure might be reflecting the distinct depth, geology, and distance from vent of the studied sediments. A metagenomic sequencing approach was conducted to characterize the microbiome of the deep-sea hydrothermal sediments and the relevant metabolic pathways used by microbes. Both Menez Gwen and Rainbow metagenomes contained a significant number of genes involved in carbon fixation, revealing the largely autotrophic communities thriving in both sites. Carbon fixation at Menez Gwen site was predicted to occur mainly via the reductive tricarboxylic acid cycle, likely reflecting the dominance of sulfur-oxidizing Epsilonproteobacteria at this site, while different autotrophic pathways were identified at Rainbow site, in particular the Calvin–Benson–Bassham cycle. Chemolithotrophy appeared to be primarily driven by the oxidation of reduced sulfur compounds, whether through the SOX-dependent pathway at Menez Gwen site or through reverse sulfate reduction at Rainbow site. Other energy-yielding processes, such as methane, nitrite, or ammonia oxidation, were also detected but presumably contributing less to chemolithoautotrophy. This work furthers our knowledge of the microbial ecology of deep-sea hydrothermal sediments and represents an important repository of novel genes with potential biotechnological interest.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Since the discovery of deep-sea hydrothermal vents 40 years ago, numerous studies on natural communities of Bacteria, Archaea, and highly specialized fauna, such as novel species of mussels, annelids, and crabs, have been undertaken in several vent habitats, distributed on the global ocean. Chemolithoautotrophic microorganisms at these ecosystems attracted a large body of research, given their relevant role at the basis of food chains and their intricate chemosynthetic symbioses with both micro- and macroorganisms [1, 2]. Those microorganisms are the primary producers of organic matter through their ability to fix inorganic carbon from the geothermal source and transfer of energy up to the higher trophic levels [3]. Considerable knowledge has already been gained about the diversity and the geographic distribution of microorganisms in distinct hydrothermal habitats, such as in sediments [4, 5], diffuse vent fluids [6,7,8,9], black smoker chimneys [10,11,12], hydrothermal plumes [13, 14], and deposits [15, 16].
Recent advances in metagenomic sequencing approaches have expanded our knowledge of the microbial ecology in these ecosystems, simultaneously exploring the taxonomic diversity and the metabolic potential and functions of microbial communities [6, 15, 17,18,19]. Possible scenarios for carbon fixation and energy generation in the deep-sea have been suggested by metagenome-derived information. The type of microorganisms mediating these reactions and the metabolic strategies employed in the different vent systems are still to be fully understood as many hydrothermal vent environments remain largely understudied.
This work reports on the investigation of two vent fields located on the northern Mid-Atlantic Ridge (MAR), one of the most slow-spreading ridge systems on the Earth. The MAR supports a series of high-temperature (250–400 °C) hydrothermal vent fields, some of which are magmatically heated and hosted by basaltic rocks while others are hosted by ultramafic rocks, undergoing reactions with peridotite [20, 21]. The Menez Gwen and the Rainbow hydrothermal fields are examples of ecosystems hosted by basaltic and ultramafic rocks, respectively, and were the chosen sites for this work. These two geochemically distinct vent systems are located on two different segments of the southeastern branch of the Azores Triple Junction (ATJ), at 800 and 2300 m deep, and within 2° of latitude (Fig. 1).

Location of the sampling sites in the hydrothermal vent fields Menez Gwen and Rainbow, southwest the Azores archipelago, in the Mid-Atlantic Ridge, at 850 and 2300 m deep, respectively
The Rainbow vent field is a vigorous ultramafic-hosted hydrothermal system that discharges hot fluids over a 20,000-m2 area from about 10 major groups of extremely active black smokers located on an isolated sulfide mound [22] (Fig. 2a). The hydrothermal fluids have the highest temperature reported for MAR fluids (~ 360 °C), very high rates of fluid flow [22], the lowest end-member pH (2.8), and the highest Fe, Cu, and Zn metal contents [23]. The fluids emitted by the active chimneys of the Rainbow, as a result of serpentinization processes, have elevated concentrations of H2 and CH4 and low levels of H2S, relative to fluids from basalt-hosted systems [24].

In contrast, the Menez Gwen vent field is the shallowest and youngest (~ 100 years) among all the MAR hydrothermal vent fields [21] (Fig. 2b). Its active area occupies approximately 200 m2 being composed by small and white anhydrite chimneys, typically less than 5 m high. Diffuse low-temperature (10–50 °C) hydrothermal vents dominate over focused ones, the black smokers are rare, and there are no plumes [21]. The Menez Gwen fluids are characterized by particularly high concentrations of CH4 and CO2, moderate concentrations of H2S, and very low H2 concentrations [25]. Moreover, Menez Gwen hydrothermal fluids reported the highest value ever of dissolved organic carbon (DOC) concentration for a diffusive vent system [26].
Both hydrothermal vent fields exhibit therefore contrasting environmental conditions, including different depths, geologic settings, and underlying rocks (Table 1). Consequently, the hydrothermal sediments formed during hydrothermal activity [27] presumably harbor distinct microbial communities, sustained by the energy and carbon sources available [28].
Until recently, very few surveys conducted in Menez Gwen and Rainbow vent fields had reported on the sediment-associated microbiota owing to the limited accessibility and critical sampling techniques [11, 29,30,31]. This lack of understanding has encouraged the authors to carry out a 16S rRNA gene sequencing approach to assess the microbial community diversity associated with sediments retrieved in both vent fields [32]. The outcome of this work showed sulfur-oxidizing Epsilonproteobacteria, archaeal methanogens, and thermoacidophiles from the archaeal DHVE group 2 together with some thermophilic microorganisms as the more abundant taxa associated with the Menez Gwen hydrothermal sediments. These results contrasted with the higher microbial diversity associated with Rainbow sediments, which were found to be mainly composed of sulfur-oxidizing Gammaproteobacteria, sulfate-reducing Deltaproteobacteria, acidophilic Actinobacteria, and some thermophilic and methanogenic archaeal members. However, the metabolic pathways used by the sediment-associated communities could not be inferred from the phylogenetic information provided by the amplicon sequencing tools.
In the present work, these studies were followed up by a shotgun metagenomic sequencing procedure to characterize the metabolic potential and functions of microorganisms inhabiting the sediments of basalt-hosted Menez Gwen and peridotite-hosted Rainbow fields. The comparative genomic analysis gave us a better understanding of the diversity and functional potential of microbial communities inhabiting sediments of these two deep-sea hydrothermal fields of the Azores.
Materials and Methods
Sampling and Total Community DNA Extraction
The sediment samples used in this study were retrieved from the Menez Gwen (MG) (37° 51′ N, 31° 31′ W) and Rainbow (RB) (36° 14′ N, 33° 54′ W) deep-sea hydrothermal fields in the MAR, during the BioBaz cruise, on August 2013, aboard the research vessel “Pourquoi Pas?” (Table 1). The more detailed sampling process, geochemical characteristics of sampling sites, as well as the DNA extraction procedures from the sediment samples have been described previously [32].
Briefly, sampling sites in each hydrothermal field were chosen from an area as close as possible to an active vent chimney, where the sediment surface was suitable for corer deployment. MG sediments were collected approximately at 4 m of the active vent “Woody,” where diffuse fluids are regularly discharged. However, the same sampling scheme could not be applied to RB site, since core sampling was hampered by the unsedimented nature of the field. Here, the black smokers are dispersed and built on massive hydrothermal structures hosted by ultramafic rocks with very scarce sedimentary cover. Thus, the RB sediments could only be retrieved at approximately 100 m from an active vent. Despite the circumstances, the very high flow rates of fluids emitted by the active chimneys of the Rainbow, together with the homogeneity of both the chemistry and temperature of the vent fluids [22], led us to assume that RB sampling site was under the influence of hydrothermal emissions from the nearby black smokers.
The outcome of our previous 16S rRNA gene sequencing approach [32] showed that MG and RB sites presented the most contrasting microbial communities from the studied hydrothermal sediments. This prompted us to further explore the functional potential of the sediment-associated communities by means of metagenomic analyzes. From the environmental DNA samples obtained in this previous work, a representative sample of each hydrothermal vent site, MG and RB, was selected for metagenomic shotgun sequencing. Based on the DNA integrity and absence of inhibitors, MG35B and RB35B were selected. Both samples were obtained from the same depth in the sediment, from 3 to 5 cm below the sediment surface, at each sampling site, and from the subcore B [32].
Metagenomic Sequencing, Assembly, and Annotation
Sequencing libraries were prepared with the Nextera XT DNA Library Prep Kit (Illumina, San Diego, USA). Shotgun sequencing of metagenomic DNA was performed with Illumina MiSeq platform using 2 × 250 bp cycles and the MiSeq Reagent Kit v2 (Illumina, San Diego, USA), according to manufacturer’s instructions, at the Next Generation Sequencing Unit in UC-Biotech (Cantanhede, Portugal). Assembly of the Illumina high-quality sequencing reads was performed by MetaSPAdes [33] for the two metagenomes (MG and RB). Final assemblies were submitted to the Joint Genome Institute (JGI) Integrated Microbial Genomes/Metagenomics Expert Review (IMG/MER) [34] for annotation and analysis.
A total of 398,414 and 1,382,338 protein-coding genes were identified in MG and RB metagenomes, respectively, using the IMG annotation. Genes were assigned the taxonomic classification of their “top” hits against the microbial reference genomes of NCBI (including Bacteria, Archaea, Eukaryota, and viruses). Genes having no identified homologue with an E value ≤ 0.01, a percent sequence identity ≥ 60%, and a percent alignment length ≥ 50% were designated as unclassified.
Unassembled raw sequencing reads from the two datasets (MG and RB) were also submitted to the MG-RAST server (version 4.0) provided by Argonne National Laboratory, for gene annotation and analyses [35]. For simplicity, we report only on results of assembled metagenomes in IMG/M.
Comparative Metagenomic Analyses
Of the identified protein-coding genes, 32.1 and 33.4% were assigned against Kyoto Encyclopedia of Genes and Genomes (KEGG) Orthology (KO) database for MG and RB metagenomes. To enable comparison, gene counts were normalized against the total number of gene assignments in the KO database for each metagenome.
Data Availability
Assembled sequences annotated using the IMG/M metagenome analysis system [34] are available in the IMG/M system (https://img.jgi.doe.gov/cgi-bin/mer/) with accession numbers 3300017525 and 3300017530 for Menez Gwen and Rainbow metagenomes, respectively.
The metagenomic sequence data is publicly available in the MG-RAST v4 database (http://metagenomics.anl.gov/) [35], with accession numbers 4718937.3 for the Menez Gwen metagenome and 4718936.3 for the Rainbow metagenome.
Results and Discussion
Two sediment cores were sampled from the Menez Gwen and Rainbow hydrothermal vent fields in the northern MAR, 278 km apart from each other (Figs. 1 and 2 and Table 1). A representative sample from each hydrothermal vent site was selected for metagenomic shotgun sequencing. A total of 4,550,463 and 12,720,510 high-quality sequences with an average length of 265 and 228 bp/read were obtained for MG and RB, respectively. Assembly and annotation resulted in 393,124 and 1,112,629 scaffolds for MG and RB metagenomes, respectively. Gene counts for each metagenome were retrieved from the IMG/M version 4.0 management system [34] (Table 2).
Microbial Community Composition
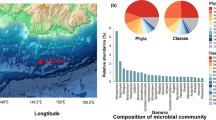
Taxonomic annotation of metagenomic sequencing data showed that the MG and RB hydrothermal sediments carry different microbial assemblages (Fig. 3). Bacteria prevailed relatively to Archaea and viruses in both metagenomes. Archaeal representatives abounded at Rainbow over Menez Gwen, while viral reads had similar relative abundances. Eukaryotic reads were also detected, more abundant in the Menez Gwen metagenome (Table 2 and Fig. 3).

Taxonomic distribution and relative abundance of the sediment communities in the two different metagenomes, Menez Gwen (MG) and Rainbow (RB), based on protein-encoding genes. Sunburst charts show the Phylum-Class-Genus taxonomic distribution of assigned reads. Alpha, Beta, Delta, Gamma, and Epsilon correspond to the related Proteobacterial classes. Poorly represented taxa are grouped into Other Phyla
Epsilonproteobacteria members accounted for nearly 40% of the total abundance found at Menez Gwen site, followed by Gammaproteobacteria (19%) and Deltaproteobacteria (10%). The epsilonproteobacterial genera Sulfurovum and Sulfurimonas were dominant, comprising 17.8 and 10.0% of the total MG sequences, respectively, which is in accordance with the taxonomic data obtained from 16S amplicon sequencing [32]. Less represented proteobacterial members belonged to the Alpha- (3.8%) and Betaproteobacteria (1.2%) classes (Fig. 3). The remaining bacterial sequences affiliated to Bacteroidetes (12.0%), Spirochaetes (1.3%), Firmicutes (1.1%), and also to thermophilic lineages of the Thermotogae (1.7%) and Caldiserica (0.5%) phyla.
MG dataset accounted for a substantial amount of eukaryotic hits (5.4%). The majority was associated to Mollusca which is in line with the presence of vent-associated macrofauna in Menez Gwen hydrothermal environments, mainly colonized by Bathymodiolus azoricus mussels. Previous studies already reported eukaryotic sequences related to mollusks in hydrothermal sediments of Menez Gwen vent field [29, 32].
Gammaproteobacteria was the most abundant class found at Rainbow site, accounting for 21.7% of the total sequences, while Alpha- (9.6%), Delta- (9.1%), and Betaproteobacteria (4.7%) were also present in relatively high contents (Fig. 3). Particularly Epsilon- and Gamma- subdivisions of Proteobacteria are known to play significant roles in carbon, nitrogen, and sulfur cycles in deep-sea hydrothermal environments, owing to their remarkably metabolic versatility in energy generation [38,39,40].
Planctomycetes (10.3%), Chloroflexi (8.0%), Actinobacteria (6.3%), Bacteroidetes (5.1%), and Firmicutes (4.4%) members were also present in the RB metagenome (Fig. 3), where Nitrospina (1.4%), Nitrospira (1.3%), and Thioalkalivibrio (1.3%) were the dominant genera found. Sequences of the RB metagenome were assigned to a larger number of genera relatively to the ones detected in MG metagenome, which suggest a higher level of taxonomic diversity in the microbial communities of the RB site.
A detailed taxonomic analysis of the archaeal representatives found in both metagenomes is presented in Fig. 4. Whereas MG archaeal community was predominantly represented by Euryarchaeota, the archaeal community at the RB site was equally dominated by Euryarchaeota and Thaumarchaeota members. Methanomicrobia was the prevalent class at both MG and RB sites. The thaumarchaeotal genus Nitrosopumilus dominated the archaeal community at RB.

Taxonomic distribution and relative abundance of the archaeal communities in the two different metagenomes, Menez Gwen (MG) and Rainbow (RB), based on protein-encoding genes. Sunburst charts show the Phylum-Class-Genus taxonomic distribution of the assigned archaeal reads
Overall, these findings are in agreement with the microbial diversity pattern previously reported using 16S rRNA gene amplicon sequencing [32].
As reflected by the DOC concentration of fluids emitted from the nearest vents (Table 1), it is reasonable to consider that the vent fluids represent an important carbon source fueling microbial communities at the MG site, while at RB site, the availability of DOC from hot hydrothermal vent fluids is considerably lower. For this reason, it would seem plausible that MG sediments were specifically enriched in heterotrophic microorganisms likely feeding on the available organic carbon.
However, the detected MG community was found to be particularly enriched in the epsilonproteobacterial genera Sulfurovum and Sulfurimonas (Fig. 3). The dominance of these groups, known to harbor chemoautotrophic species, suggests that the oxic mixing zone where MG site is located is likely a niche for Sulfurovum and Sulfurimonas, which is in agreement with previous studies that reported the abundance of Epsilonproteobacteria members at diffuse venting sites [30, 41, 42]. Moreover, MG site is likely exposed to particularly high rates of H2S and CO2 (Table 1) that may favor the growth of especially adapted microbes while limiting the diversity of other microbial taxa in the sediments.
On the other hand, the broader taxonomic diversity found at RB site might reflect a suitable combination of environmental conditions, including temperature, pH, and appropriate hydrothermal vents’ exposure, that favor the growth of distinct autotrophic and heterotrophic microbes, for which the available energy and nutrient supplies are sufficient.
Functional Composition
Functional properties of the MG and RB metagenomes were investigated and compared using KO. Similar distributions of annotated genes between the Menez Gwen and Rainbow metagenomes were observed for broad categories (data not shown). As deep-sea hydrothermal vents are typically dominated by chemolithoautotrophic communities known for fixing inorganic carbon, and using energy derived from the oxidation of reduced inorganic compounds that arise with hydrothermal fluids (such as reduced sulfur compounds, hydrogen, and methane), we analyzed these processes in detail by evaluating marker genes for key enzymatic steps for CO2 fixation, sulfur, nitrogen, and methane metabolism.
Carbon Fixation
Carbon fixation is a fundamental biosynthetic process used by hydrothermal vent microbes to assimilate inorganic carbon (i.e., CO2) into organic matter. Presently available data suggest that the Calvin–Benson–Bassham (CBB) cycle and the reductive tricarboxylic acid (rTCA) cycle are the predominant carbon fixation pathways at hydrothermal vent environments [38, 41, 43].
Gene sequences related to the key enzymes of CBB cycle, ribulose-bisphosphate carboxylase (RuBisCO) (rbc EC:4.1.1.39), and phosphoribulokinase (prk EC:2.7.1.19), were found in both metagenomic datasets (Fig. 5). The most abundant sequences were related to members of the gammaproteobacterial order Thiotrichales, represented by Beggiatoa, Thiomicrospira, Thiothrix, and Sedimenticola. Other affiliating with members of the betaproteobacterial genera Nitrosomonas and Nitrosospira and with the gammaproteobacterial genus Thioalkalivibrio were found only across the RB metagenome (Table S1). These findings are in line with previous studies that report the existence of many sulfur-oxidizing and nitrifying bacteria belonging to these genera in different hydrothermal environments of MG and RB vent fields [15, 29, 31, 32]. Also, the occurrence of endosymbiont-related sequences in the sediments under study (Table S1) is consistent with the presence of macrofaunal hosts in both MG and RB hydrothermal vent fields, typically colonized by B. azoricus mussels. B. azoricus endosymbionts are known to belong to the Gammaproteobacteria class and to use the CBB cycle for carbon fixation [44, 45]. Furthermore, a host-independent life-style for Bathymodiolus endosymbionts has already been reported [46,47,48,49].


a The key gene encoding proteins involved in energy-yielding processes and carbon fixation pathways suggested by metagenome-derived information. Each bubble represents an identified gene in the respective metagenomic dataset and its size the relative abundance in percentage of all genes detected in the sample. Gene encoding proteins were grouped based on their respective metabolic categories inferred from the KEGG Orthology (KO) database. b Assignment of metabolic roles to major taxonomic groups based on detection of key gene-encoding proteins in Menez Gwen (MG) and Rainbow (RB) metagenomes
A minimal number of both RuBisCO and prk gene sequences were also detected in RB metagenome associated to archaeal representatives (Table S1). Despite the CBB cycle has not been so far reported in the archaeal domain, a recently published study [50] suggests that the key enzymes of this cycle might participate in an alternative CO2 fixation pathway in Archaea—the reductive hexulose-phosphate (RHP) pathway. The archaeal RHP pathway only differs in a few steps from the CBB cycle and might be a primitive carbon metabolic pathway utilizing RuBisCO that later evolved to the photosynthetic Calvin–Benson cycle [50].
The presence of gene sequences coding for 2-oxoglutarate:ferredoxin oxidoreductase (kor EC:1.2.7.3), fumarate reductase (frd EC:1.3.5.4), and pyruvate:ferredoxin oxidoreductase (por EC:1.2.7.1) in our metagenomic datasets suggested that the microbial communities of both MG and RB sites might use rTCA cycle to fix CO2. However, the key enzyme of this cycle, ATP citrate lyase (acl EC:2.3.3.8), was remarkably overrepresented in the MG metagenome (Fig. 5). The prevalence of acl sequences in MG dataset might be the reflection of the dominant sulfur-oxidizing Epsilonproteobacteria members in the MG site (Fig. 5 and Table S1). In contrast, sequences found in RB metagenome affiliated most with Nitrospira and Methanosaeta genera (Table S1). These findings are consistent with previous studies that report hydrothermal Epsilonproteobacteria and nitrite-oxidizing Nitrospirae employing the rTCA cycle for CO2 fixation [51]. The rTCA cycle might be an advantage in an energy limiting environment since this pathway requires significantly less ATP and reducing equivalent compared with other cycles [40].
The reductive acetyl-CoA or Wood–Ljungdahl (WL) pathway is considered a significant mechanism of CO2 fixation under anaerobic and reducing conditions in both Bacteria and Archaea domains [40]. Markers of this pathway were detected in both sites: cdhA coding for carbon monoxide dehydrogenase (EC 1.2.7.4) and acsB coding for acetyl-CoA synthase (EC:2.3.1.169), apparently in higher proportions in the RB metagenome (Fig. 5). Carbon monoxide dehydrogenase is the key enzyme of the WL pathway, which may also couple CO oxidation to sulfate reduction, or reduce CO2 to either acetate (acetogenesis) or methane (methanogenesis) [52, 53]. In this study, markers of the WL pathway were mostly related to deltaproteobacterial sulfate reducers and archaeal methanogens (Table S1). The results are in line with previous studies that reported WL pathway in acetogenic members, in autotrophic sulfate-reducing bacteria and archaea as well as in methanogenic Archaea [54, 55].
Similarly, sequences encoding for propionyl-CoA synthetase (prpE EC:6.2.1.17) and malyl-CoA lyase (mcl EC:4.1.3.24), key enzymes in 3-hydroxypropionate (3-HP) bicycle [40], were found across RB metagenome. The marker genes were found to be mostly related to Gammaproteobacteria and Alphaproteobacteria members, which is in accordance with previous reports that identified single genes of this pathway in various strains of these classes [18, 56]. However, one of the key enzymes of the 3-HP pathway could not be identified in the metagenomic datasets, the malonyl-coA reductase (EC:1.2.1.75), which presumably indicates the absence of the 3-HP operating in the microbial communities of both MG and RB sites.
The 3-hydroxypropionate/4-hydroxybutyrate (3-HP/4-HB) cycle is mainly known to be conducted by autotrophic thermoacidophilic members of the Thaumarchaeota phylum [40]. The most significant enzyme of this cycle, 4-hydroxybutyryl-CoA dehydratase (abfD EC:4.2.1.120), was detected in RB metagenome specifically related to the genus Nitrosopumilus and Candidatus Nitrosotenuis (Fig. 5 and Table S1). These aerobic chemolithoautotrophs are known to be ammonia oxidizers, and the energy gained likely harnessed to fix carbon via the 3-HP/4-HB carbon fixation pathway [57]. The detection of the critical genes of this pathway in our metagenomic databases further confirms the presence of Nitrosopumilus representatives in RB hydrothermal sediments as previously suggested [32].
Sulfur Metabolism
Sulfur oxidation is reported as dominant energy-generating pathways for chemolithoautotrophic growth in hydrothermal vent environments, where hydrogen sulfide is presumably the main electron donor used by chemolithotrophic microbes [58, 59]. Queries to the metagenomic datasets identified genes encoding proteins from diverse pathways of sulfur energy metabolism, including the dissimilatory oxidation of sulfide to elemental sulfur, the sulfur oxidation Sox system responsible for thiosulfate oxidation, and the reverse dissimilatory sulfite reduction pathway for oxidation of sulfur to sulfate.
Evidence of periplasmic sulfide oxidation was detected in both MG and RB metagenomes suggested by the presence of genes coding for sulfide-oxidizing flavoproteins: the sulfide-quinone oxidoreductase (sqr EC:1.8.5.4) and, in a lesser extent, the flavocytochrome-C sulfide dehydrogenase (fccAB EC:1.8.2.3) (Fig. 5). These enzymes mediate the oxidation of sulfide to polysulfide and are responsible for starting the electron flow from sulfide to the transport chain. The taxonomic identities of the matched genes in the metagenomic datasets indicated relatedness to diverse genera of sulfur-oxidizing Epsilon- and Gammaproteobacteria (Table S2).
The full set of genes that constitute the thiosulfate oxidizing multienzyme system of the SOX pathway, including a core set (soxABXYZ) and supplementary genes (soxCD), capable of oxidizing various reduced sulfur compounds to sulfate (SO42−) [60], was overrepresented in the MG metagenome (Fig. 5). Genes encoding the Sox enzyme complex (soxABXYZ) for oxidation of thiosulfate (S2O32−) to elemental sulfur (S0) and the supplementary soxCD genes for the further oxidation of thiosulfate to sulfate (SO42−) were mainly found related to the epsilonproteobacterial genera Sulfurovum, Sulfurimonas, Arcobacter, and Nitratiruptor. The presence of this Sox multienzyme system in MG site is in accordance with the sulfur compound-oxidizing oxygen/nitrate-reducing pathway predicted for deep-sea chemoautotrophic Epsilonproteobacteria [61].
Additionally, evidence of polysulfide reduction to sulfide catalyzed by polysulfide reductase (psrA), an important process in deep-sea vent environments [62], was also detected in MG metagenomic dataset (Fig. 5) and mainly associated with representatives of the Campylobacterales order (Table S2). The detection of the Epsilonproteobacteria genera linked to both polysulfide reduction and Sox system pathways was previously identified in deep-sea hydrothermal isolates: Sulfurovum sp. [63], Nitratiruptor sp. [64], Sulfurimonas autotrophica [65], and Sulfurimonas denitrificans [43].
The set of genes encoding sulfur oxidation proteins detected in RB metagenome was mostly related to Chromatiales order and other unclassified Gammaproteobacteria (Table S2).
Indeed, the reduced sulfur compounds in the sediments of RB site might likely be oxidized to sulfate via the reverse sulfate reduction pathway, reported before in hydrothermal microbial communities [18, 19, 66]. The complete repertoire of genes involved in this pathway encode the enzymes thiosulfate sulfurtransferase (tst EC 2.8.1.1) that oxidize thiosulfate to sulfite (SO32−), reverse dissimilatory sulfite reductase complex (dsrAB EC:1.8.99.5 and supplementary shuttle molecules dsrEFH and dsrC) responsible for oxidation of elemental sulfur to sulfite, adenylylsulfate reductase (aprAB EC:1.8.99.2), and sulfate adenylyltransferase (sat EC:2.7.7.4) responsible for oxidation of sulfite to sulfate [67] (Fig. 5). The majority of the corresponding sequences in the metagenomic datasets were associated with sulfur-oxidizing Gammaproteobacteria (e.g., Thioalkalivibrio, Thiohalomonas, Beggiatoa, Sedimenticola, Thiothrix), Betaproteobacteria (e.g., Thiobacillus, Sulfuricella), as well as some Epsilonproteobacteria representatives (Table S2). The use of the reverse sulfate reduction pathway instead of the Sox pathway by hydrothermal Gammaproteobacteria has been previously reported [39, 61] and is potentially overrepresented in RB metagenome (Fig. 5).
The dissimilatory sulfate reduction pathway, occurring in sulfate-reducing prokaryotes, is carried out by the same reversible enzymes, sulfate adenylyltransferase (sat), adenylylsulfate reductase (apr), and dissimilatory sulfite reductase complex (dsr). In total, only about one-third of the sequences coding for these enzymes were affiliated to sulfate-reducing Deltaproteobacteria (e.g., Desulfobulbus, Desulfopila, Desulfovibrio), Nitrospirae (e.g., Thermodesulfovibrio), and Archaea (e.g., Archaeoglobus) representatives (Table S2). Together, these data suggest that the enzymes are most likely involved in the reverse process, the oxidation of sulfite to sulfate, in RB microbial communities.
Additionally, the co-occurrence of sulfur-oxidizing and sulfur-reducing activities could point to a reciprocal exchange of sulfur compounds and thereby an increase in the overall energy efficiency of the systems.
The metagenomic data suggest sulfur-oxidizing pathways as being the most representative energy sources supporting chemolithoautotrophy in both MG and RB sites, where the diverse populations of chemolithoautotrophs are potentially using different electron acceptors such as oxygen, nitrate, and nitrite. These results are in line with the reducing conditions and elevated levels of sulfide associated with both MG and RB hydrothermal fluids (Table 1).
Nitrogen Metabolism
Nitrogen cycle has been previously shown to play a crucial role in the establishment of the geochemical environment in hydrothermal vent fields. Dinitrogen (N2) is the most abundant form of nitrogen in seawater but only accessible to nitrogen-fixing microbes. Inorganic nitrogen compounds such as nitrite and nitrate are important terminal electron acceptors for chemolithoautotrophic growth in different hydrothermal vent environments [19, 38, 68].
Our results showed that microbial communities from both MG and RB sites possess a diversity of metabolic genes involved in nitrogen cycling (Fig. 5).
MG metagenome was found to be highly enriched with genes required for denitrification, suggesting that nitrate could be an important electron acceptor in addition to oxygen in MG hydrothermal sediments, possibly coupled to the oxidation of sulfur or organic matter. Denitrification involves the stepwise reduction of nitrate (NO3−) to nitrite (NO2−), nitric oxide (NO), nitrous oxide (N2O), and dinitrogen (N2) by the enzymes nitrate reductase (narGH or napA EC:1.7.99.4), nitrite reductase (nirK EC:1.7.2.1), nitric oxide reductase (norB EC:1.7.2.5), and nitrous oxide reductase (nosZ EC:1.7.2.4), respectively [69]. The marker genes of these enzymes were all identified in the metagenomic datasets being overrepresented in MG site (Fig. 5), and most of the hits related to members of Epsilonproteobacteria (e.g., Sulfurovum, Sulfurimonas, and Nitratifactor genera) (Table S3). This is consistent with the fact that the mesophilic Epsilonproteobacteria isolates collected from deep-sea hydrothermal vents, Sulfurovum lithotrophicum [70], Sulfurimonas paralvinellae [71], and Nitratifactor salsuginis [72] reduce nitrate to dinitrogen gas, coupling sulfur oxidation to denitrification pathway [73]. Furthermore, our results indicate that sulfur oxidation linked to denitrification is likely an important energy-generating pathway fueling the microbial community associated with MG site. In contrast, denitrification seems to be less usual across the microbial communities inhabiting the RB sediments since the genes encoding some enzymes of this pathway were scarcely found within RB metagenome (Fig. 5).
Despite the low number of ammonia monooxygenase (amoA EC:1.14.18.3) sequence markers observed in both datasets (Fig. 5), members of the genera Nitrosospira and Nitrosopumilus seemed to be the primary ammonia oxidizers at RB site (Table S3). Representatives of these taxa are known as ubiquitous members of the deep-sea, playing a pivotal role in the overall nitrogen cycle, and already found associated with Rainbow hydrothermal environments [11, 31, 32]. Besides ammonia oxidation, ammonia monooxygenase is also a key enzyme for nitrification, where it catalyzes the oxidation of ammonia to nitrite together with hydroxylamine dehydrogenase (hao EC:1.7.2.6). Here, the sequences related to hao were only observed in RB metagenome and related to Nitrosospira genus and novel Candidatus lineages of Planctomycetes phylum (Table S3). These novel Planctomycetes are likely anaerobic ammonia oxidizers [74], suggesting that hydroxylamine might be an important intermediate for RB-associated bacteria that use ammonia as an energy source.
Members of the bacterial phyla Nitrospirae and Nitrospinae, mainly associated with RB dataset, were potentially involved in nitrite oxidation, as suggested by the presence of genes encoding enzymes involved in the nitrification process, namely narG and narH (Table S3) [75, 76].
In turn, the amo sequence markers detected in the MG dataset were found related to the family Methylococcaceae (Table S3). In aerobic methane-oxidizing bacteria, the same gene encodes for the particulate methane monooxygenase (pMMO) responsible for oxidizing ethane to methanol (see "Methane metabolism" section).
Although in low number, other marker genes encoding enzymes involved in dissimilatory nitrate reduction to ammonium (DNRA) (nap EC:1.7.99.4, nirB EC:1.7.1.15, and nrfA EC:1.7.2.2) and in nitrogen fixation (nifD, nifK, and nifH EC:1.18.6.1) were identified in our data mostly related to RB metagenome (Fig. 5 and Table S3). The evidence of DNRA and nitrogen fixation pathways in hydrothermal vent environments has been previously reported [19, 77, 78].
Together, these results suggested that RB communities showed a broader diversity of metabolic genes involved in nitrogen metabolism, likely ammonia oxidation, denitrification, nitrification, nitrogen fixation, and dissimilatory nitrate reduction to ammonia, which contrasted with the MG communities where denitrification activity seems to dominate.
The TDN concentration previously measured in MG fluids (Table 1) gives evidence supporting the poor nitrogen supply for the microbial communities provided by MG fluids and might be reflected in a scarce nitrifying community at MG site.
Methane Metabolism
Despite the high concentrations of CH4, reported before in the hydrothermal vent fluids from both “Woody” vent in MG (1.4 mM) and “Regner” vent in RB (1.9 mM) (Table 1), in the present study, genes encoding enzymes involved in methane oxidation were scarcely found and being almost exclusive to MG dataset. Some evidence of methanotrophy (i.e., methane as sole carbon and energy source) was identified in MG metagenome and possibly restricted to gammaproteobacterial Methylococcaceae lineages. Specifically, the marker genes detected here, methanol dehydrogenase (mdh1 EC:1.1.2.7) and particulate methane monooxygenase (pmo EC:1.14.13.25), were related to Methylococcaceae members (Fig. 5 and Table S4). The presence of these methane oxidation proteins in the Menez Gwen hydrothermal sediments is consistent with the presence of both CH4 and O2 at this site and with a previous survey reporting methanotrophic members inhabiting these sediments [29].
Marker genes for methanogenesis (methyl-CoM reductase, mcr EC:2.8.4.1), involved in methane formation, were absent in both metagenomes as were other methane or hydrocarbon monooxygenase genes. Thus, the high levels of CH4 at these hydrothermal sites might have an abiogenic origin, as a consequence of the interaction between H2 and CO2 carbon dioxide dissolved in the ocean water. This has been reported particularly for Rainbow hydrothermal system, where conditions for abiotic synthesis of CH4 (and other hydrocarbons) were found to be promoted by the serpentinization reactions of the ultramafic rocks [23].
The metagenomic results indicate that other strategies different from methane-oxidizing pathways likely operate in the sediments associated with MG and RB deep-sea hydrothermal vents for energy generation.
Alternative Electron Donors
In addition to the oxidation of reduced sulfur compounds, nitrogen compounds, and methane, taxonomy annotation also indicates the presence of taxa involved in hydrogen oxidation (e.g., Aquificae, Hydrogenimonas) particularly in the MG site (Tables S1 and S2) and iron oxidation (e.g., Acidithiobacillus, Leptospirillum, Thiobacillus, Sideroxydans) mainly in the RB site (Tables S3 and S5). However, marker genes for Ni-Fe hydrogenases and for enzymes involved in iron oxidation were barely detected in both metagenomes (data not shown). Still, the acidic nature of RB hydrothermal fluids [24, 79], together with the elevated Fe content detected in RB sediments [32], provides the conditions for iron oxidizers as well as acidophilic lineages to inhabit the RB hydrothermal sediments.
Oxygen as Electron Acceptor
Vent microorganisms are known to thrive aerobically and anaerobically in the deep-sea hydrothermal environments in low-oxygen waters.
Evidence that oxygen is one important electron acceptor in energy metabolisms occurring within both sites was provided by the presence of the set of genes in the MG and RB metagenomes encoding for both enzyme complexes: cytochrome c oxidase (coxABC) and cbb3-type terminal cytochrome c oxidase (ccoNOP) (Fig. 5 and Table S5). Representatives of Beta-, Delta-, and Gammaproteobacteria, as well as Nitrospirae, Nitrospinae, Chloroflexi, Gemmatimonadetes, and Actinobacteria, are apparently going through aerobic respiration in RB site. On the other hand, Alpha-, Beta-, Gamma-, and Epsilonproteobacteria representatives are the main aerobic microorganisms in MG site (Table S5).
Conclusion
The microbial community structure of hydrothermal sediments from Menez Gwen and Rainbow vent fields was evaluated by a metagenomic sequencing approach. Community members’ phylogeny and function were inferred by protein-coding genes analysis. For the first time, key genes of sulfur, nitrogen, and methane energy-yielding processes were analyzed together with key genes of different carbon fixation pathways in order to characterize the genetic capabilities of the chemolithoautotrophic primary producers inhabiting Menez Gwen and Rainbow deep-sea hydrothermal sediments. The results evidenced versatile energy-yielding pathways for the microbial communities associated with the two geochemically contrasting hydrothermal environments. Some similarities with previously studied hydrothermal vent habitats have been observed, namely the high abundance of genes encoding for enzymes of reduced sulfur compounds oxidation and carbon fixation pathways.
Sulfur-oxidizing Epsilonproteobacteria were the predominant microorganisms in the Menez Gwen metagenome. These bacteria were mainly predicted to undergo sulfur oxidation coupled to denitrification to provide energy sources for carbon fixation, generally occurring via rTCA cycle. Apart from sulfur-oxidizing pathways, some genes potentially involved in methane and hydrogen oxidation were found in MG metagenome but apparently contributing less to chemolithoautotrophy.
From the Rainbow metagenomic dataset, we could outline a higher level of taxonomic diversity in the microbial communities. Thioalkalivibrio, Beggiatoa, Thiohalomonas, Sedimenticola, and other gammaproteobacterial genera were likely undergoing sulfur oxidation by the reverse sulfate reduction pathway. Moreover, oxidation of nitrogen compounds was favored in RB sediments where members of Nitrospira and Nitrospina were likely involved in nitrification and Nitrosospira and Nitrosopumilus genera in ammonia oxidation. Additionally, the metagenomic data indicated the potential for different carbon fixation pathways in RB site, where the CBB cycle was found to be the most significant one.
We anticipated that the high concentrations of dissolved hydrogen and carbon in hydrothermal fluids of peridotite-hosted Rainbow would be reflected in the RB community structure, likely carrying abundant methane- and hydrogen-metabolizing chemolithoautotrophs. However, in the metagenomes here analyzed, only a few hits related to key enzymes involved in methanotrophy were encountered. A possible interpretation is that an active mixing of Rainbow fluids with seawater might occur in the surface sediments, which would result in low H2 and CH4 available to support hydrogen and methane-oxidizing communities at RB site. On the other hand, the high concentration and availability of sulfur species in the circulating seawater within the sediments might favor the growth of distinct autotrophic microbes at RB site.
Overall, this study pointed to autotrophic carbon fixation being mainly driven by the rTCA and CBB cycles predictably supported by the oxidation of reduced sulfur compounds. Thioautotrophic microorganisms were seen to encode sulfur oxidation genes involved in different pathways, suggesting the utilization of multiple sulfur oxidation pathways according to substrate availability in the two geochemically distinct habitats. The presence of chemolithoautotrophs using different electron donors, such as hydrogen and methane in MG metagenome and nitrogen compounds in RB metagenome, points to a metabolic versatility in the microbial communities thriving in both sites.
Since the data presented here only reveals the genetic potential of the microbial communities in RB and MG sediments, we are unable to determine whether these metabolisms are actively being used by the microbes. This work provides a basis for future functional omics approaches, such as transcriptomics and proteomics studies that would further confirm and accurately quantify the functional capacity of microbial communities in these remote and understudied habitats. Nevertheless, the genomic data presented here highlights the microbial ecology of Menez Gwen and Rainbow hydrothermal sediments and the opportunity for future studies concerning non-conventional enzymes with potential biotechnological interest.
References
Jannasch HW, Mottl MJ (1985) Geomicrobiology of deep-sea hydrothermal vents. Science 229:717–725. https://doi.org/10.1126/science.229.4715.717
Orcutt BN, Sylvan JB, Knab NJ, Edwards KJ (2011) Microbial ecology of the dark ocean above, at, and below the seafloor. Microbiol Mol Biol Rev 75:361–422. https://doi.org/10.1128/MMBR.00039-10
Reysenbach AL, Shock E (2002) Merging genomes with geochemistry in hydrothermal ecosystems. Science 296:1077–1082. https://doi.org/10.1126/science.1072483
Teske A, Hinrichs KU, Edgcomb V, de Vera Gomez A, Kysela D, Sylva SP, Sogin ML, Jannasch HW (2002) Microbial diversity of hydrothermal sediments in the Guaymas Basin: evidence for anaerobic methanotrophic communities. Appl Environ Microb 68:1994–2007. https://doi.org/10.1128/AEM.68.4.1994-2007.2002
Nunoura T, Oida H, Nakaseama M, Kosaka A, Ohkubo SB, Kikuchi T, Kazama H, Hosoi-Tanabe S, Nakamura K, Kinoshita M, Hirayama H, Inagaki F, Tsunogai U, Ishibashi J, Takai K (2010) Archaeal diversity and distribution along thermal and geochemical gradients in hydrothermal sediments at the Yonaguni Knoll IV hydrothermal field in the Southern Okinawa trough. Appl Environ Microb 76:1198–1211. https://doi.org/10.1128/Aem.00924-09
Reveillaud J, Reddington E, McDermott J, Algar C, Meyer JL, Sylva S, Seewald J, German CR, Huber JA (2016) Subseafloor microbial communities in hydrogen-rich vent fluids from hydrothermal systems along the Mid-Cayman Rise. Environ. Microbiol 18:1970–1987. https://doi.org/10.1111/1462-2920.13173
Huber JA, Cantin HV, Huse SM, Welch DB, Sogin ML, Butterfield DA (2010) Isolated communities of Epsilonproteobacteria in hydrothermal vent fluids of the Mariana Arc seamounts. FEMS Microbiol. Ecol. 73:538–549. https://doi.org/10.1111/j.1574-6941.2010.00910.x
Huber JA, Welch DBM, Morrison HG, Huse SM, Neal PR, Butterfield DA, Sogin ML (2007) Microbial population structures in the deep marine biosphere. Science 318:97–100. https://doi.org/10.1126/science.1146689
Akerman NH, Butterfield DA, Huber JA (2013) Phylogenetic diversity and functional gene patterns of sulfur-oxidizing subseafloor Epsilonproteobacteria in diffuse hydrothermal vent fluids. Front Microbiol 4:185. https://doi.org/10.3389/Fmicb.2013.00185
Brazelton WJ, Ludwig KA, Sogin ML, Andreishcheva EN, Kelley DS, Shen C-C, Edwards RL, Baross JA (2010) Archaea and bacteria with surprising microdiversity show shifts in dominance over 1,000-year time scales in hydrothermal chimneys. PNAS 107:1612–1617. https://doi.org/10.1073/pnas.0905369107
Roussel EG, Konn C, Charlou JL, Donval JP, Fouquet Y, Querellou J, Prieur D, Bonavita MAC (2011) Comparison of microbial communities associated with three Atlantic ultramafic hydrothermal systems. FEMS Microbiol. Ecol. 77:647–665. https://doi.org/10.1111/j.1574-6941.2011.01161.x
Voordeckers JW, Do MH, Hugler M, Ko V, Sievert SM, Vetriani C (2008) Culture dependent and independent analyses of 16S rRNA and ATP citrate lyase genes: a comparison of microbial communities from different black smoker chimneys on the Mid-Atlantic Ridge. Extremophiles: life under extreme conditions 12:627–640. https://doi.org/10.1007/s00792-008-0167-5
Dick GJ, Anantharaman K, Baker BJ, Li M, Reed DC, Sheik CS (2013) The microbiology of deep-sea hydrothermal vent plumes: ecological and biogeographic linkages to seafloor and water column habitats. Front. Microbiol. 4:124. https://doi.org/10.3389/fmicb.2013.00124
Li M, Jain S, Dick GJ (2016) Genomic and transcriptomic resolution of organic matter utilization among deep-sea bacteria in Guaymas Basin hydrothermal plumes. Front Microbiol 7:1125. https://doi.org/10.3389/Fmicb.2016.01125
Flores GE, Campbell JH, Kirshtein JD, Meneghin J, Podar M, Steinberg JI, Seewald JS, Tivey MK, Voytek MA, Yang ZK, Reysenbach AL (2011) Microbial community structure of hydrothermal deposits from geochemically different vent fields along the Mid-Atlantic Ridge. Environ. Microbiol. 13:2158–2171. https://doi.org/10.1111/j.1462-2920.2011.02463.x
Flores GE, Shakya M, Meneghin J, Yang ZK, Seewald JS, Wheat CG, Podar M, Reysenbach AL (2012) Inter-field variability in the microbial communities of hydrothermal vent deposits from a back-arc basin. Geobiology 10:333–346. https://doi.org/10.1111/j.1472-4669.2012.00325.x
Anantharaman K, Breier JA, Dick GJ (2016) Metagenomic resolution of microbial functions in deep-sea hydrothermal plumes across the Eastern Lau Spreading Center. Isme J 10:225–239. https://doi.org/10.1038/ismej.2015.81
Wang H-l, Sun L (2016) Comparative metagenomic analysis of the microbial communities in the surroundings of Iheya north and Iheya ridge hydrothermal fields reveals insights into the survival strategy of microorganisms in deep-sea environments. J Mar Syst in press. doi:https://doi.org/10.1016/j.jmarsys.2016.10.006
Oulas A, Polymenakou PN, Seshadri R, Tripp HJ, Mandalakis M, Paez-Espino AD, Pati A, Chain P, Nomikou P, Carey S, Kilias S, Christakis C, Kotoulas G, Magoulas A, Ivanova NN, Kyrpides NC (2016) Metagenomic investigation of the geologically unique Hellenic Volcanic Arc reveals a distinctive ecosystem with unexpected physiology. Environ. Microbiol. 18:1122–1136. https://doi.org/10.1111/1462-2920.13095
Von Damm KL (1995) Controls on the chemistry and temporal variability of seafloor hydrothermal fluids. In: Seafloor Hydrothermal Systems: Physical, Chemical, Biological, and Geological Interactions (222–247). https://doi.org/10.1029/GM091
Charlou JL, Donval JP, Douville E, Jean-Baptiste P, Radford-Knoery J, Fouquet Y, Dapoigny A, Stievenard M (2000) Compared geochemical signatures and the evolution of Menez Gwen (37 degrees 50 ' N) and Lucky Strike (37 degrees 17 ' N) hydrothermal fluids, south of the Azores Triple Junction on the Mid-Atlantic Ridge. Chem. Geol. 171:49–75. https://doi.org/10.1016/S0009-2541(00)00244-8
Mügler C, Jean-Baptiste P, Perez F, Charlou J-L (2016) Modeling of hydrogen production by serpentinization in ultramafic-hosted hydrothermal systems: application to the Rainbow field. Geofluids 16:476–489. https://doi.org/10.1111/gfl.12169
Charlou JL, Donval JP, Konn C, Ondréas H, Fouquet Y, Jean-Baptiste P, Fourré E (2010) High production and fluxes of H2 and CH4 and evidence of abiotic hydrocarbon synthesis by serpentinization in ultramafic-hosted hydrothermal systems on the Mid-Atlantic Ridge. In: in Diversity of hydrothermal systems on slow spreading ocean ridges (eds Rona PA, Devey CW, Dyment J, and Murton BJ), American Geophysical Union, Washington, D. C.. https://doi.org/10.1029/2008GM000752
Charlou JL, Donval JP, Fouquet Y, Jean-Baptiste P, Holm N (2002) Geochemistry of high H2 and CH4 vent fluids issuing from ultramafic rocks at the Rainbow hydrothermal field (36°14′N, MAR). Chem. Geol. 191:345–359. https://doi.org/10.1016/S0009-2541(02)00134-1
Meier DV, Bach W, Girguis PR, Gruber-Vodicka HR, Reeves EP, Richter M, Vidoudez C, Amann R, Meyerdierks A (2016) Heterotrophic Proteobacteria in the vicinity of diffuse hydrothermal venting. Environ. Microbiol. 18:4348–4368. https://doi.org/10.1111/1462-2920.13304
Rossel PE, Stubbins A, Hach PF, Dittmar T (2015) Bioavailability and molecular composition of dissolved organic matter from a diffuse hydrothermal system. Mar. Chem. 177:257–266. https://doi.org/10.1016/j.marchem.2015.07.002
Mills R, Elderfield H, Thomson J (1993) A dual origin for the hydrothermal component in a metalliferous sediment core from the Mid-Atlantic Ridge. Journal of Geophysical Research: Solid Earth 98:9671–9681
Amend JP, McCollom TM, Hentscher M, Bach W (2011) Catabolic and anabolic energy for chemolithoautotrophs in deep-sea hydrothermal systems hosted in different rock types. Geochim Cosmochim Ac 75:5736–5748. https://doi.org/10.1016/j.gca.2011.07.041
Cerqueira T, Pinho D, Egas C, Froufe H, Altermark B, Candeias C, Santos RS, Bettencourt R (2015) Microbial diversity in deep-sea sediments from the Menez Gwen hydrothermal vent system of the Mid-Atlantic Ridge. Marine genomics 24, Part 3: 343–355. doi:https://doi.org/10.1016/j.margen.2015.09.001
Lopez-Garcia P, Duperron S, Philippot P, Foriel J, Susini J, Moreira D (2003) Bacterial diversity in hydrothermal sediment and epsilonproteobacterial dominance in experimental microcolonizers at the Mid-Atlantic Ridge. Environ. Microbiol. 5:961–976. https://doi.org/10.1046/j.1462-2920.2003.00495.x
Nercessian O, Fouquet Y, Pierre C, Prieur D, Jeanthon C (2005) Diversity of Bacteria and Archaea associated with a carbonate-rich metalliferous sediment sample from the rainbow vent field on the Mid-Atlantic Ridge. Environ. Microbiol. 7:698–714. https://doi.org/10.1111/j.1462-2920.2005.00744.x
Cerqueira T, Pinho D, Froufe H, Santos RS, Bettencourt R, Egas C (2017) Sediment microbial diversity of three deep-sea hydrothermal vents southwest of the Azores. Microb. Ecol. 74:332–349. https://doi.org/10.1007/s00248-017-0943-9
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19:455–477. https://doi.org/10.1089/cmb.2012.0021
Markowitz VM, Chen I-MA, Chu K, Szeto E, Palaniappan K, Pillay M, Ratner A, Huang J, Pagani I, Tringe S (2014) IMG/M 4 version of the integrated metagenome comparative analysis system. Nucleic Acids Res. 42:D568–D573. https://doi.org/10.1093/nar/gkt919
Meyer F, Paarmann D, D'Souza M, Olson R, Glass EM, Kubal M, Paczian T, Rodriguez A, Stevens R, Wilke A (2008) The metagenomics RAST server—a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform 9(1):386. https://doi.org/10.1186/1471-2105-9-386
Seyfried W, Pester NJ, Ding K, Rough M (2011) Vent fluid chemistry of the Rainbow hydrothermal system (36 N, MAR): phase equilibria and in situ pH controls on subseafloor alteration processes. Geochim Cosmochim Ac 75:1574–1593
McCollom TM, Seewald JS, German CR (2015) Investigation of extractable organic compounds in deep-sea hydrothermal vent fluids along the Mid-Atlantic Ridge. Geochim Cosmochim Ac 156:122–144. https://doi.org/10.1016/j.gca.2015.02.022
Nakagawa S, Takai K (2008) Deep-sea vent chemoautotrophs: diversity, biochemistry and ecological significance. FEMS Microbiol. Ecol. 65:1–14. https://doi.org/10.1111/j.1574-6941.2008.00502.x
Hügler M, Gärtner A, Imhoff JF (2010) Functional genes as markers for sulfur cycling and CO2 fixation in microbial communities of hydrothermal vents of the Logatchev field. FEMS Microbiol. Ecol. 73:526–537. https://doi.org/10.1111/j.1574-6941.2010.00919.x
Hugler M, Sievert SM (2011) Beyond the Calvin cycle: autotrophic carbon fixation in the ocean. Annu. Rev. Mar. Sci. 3:261–289. https://doi.org/10.1146/annurev-marine-120709-142712
Campbell BJ, Engel AS, Porter ML, Takai K (2006) The versatile epsilon-proteobacteria: key players in sulphidic habitats. Nat. Rev. Microbiol. 4:458–468. https://doi.org/10.1038/nrmicro1414
Lanzen A, Jorgensen SL, Bengtsson MM, Jonassen I, Ovreas L, Urich T (2011) Exploring the composition and diversity of microbial communities at the Jan Mayen hydrothermal vent field using RNA and DNA. Fems Microbiol Ecol 77:577–589. https://doi.org/10.1111/j.1574-6941.2011.01138.x
Sievert SM, Hugler M, Taylor CD, Wirsen CO (2008) Sulfur oxidation at deep-sea hydrothermal vents. In: Dahl C, Friedrich CG (eds). Microbial Sulfur Metabolism:238-258. https://doi.org/10.1007/978-3-540-72682-1_19
Petersen JM, Zielinski FU, Pape T, Seifert R, Moraru C, Amann R, Hourdez S, Girguis PR, Wankel SD, Barbe V, Pelletier E, Fink D, Borowski C, Bach W, Dubilier N (2011) Hydrogen is an energy source for hydrothermal vent symbioses. Nature 476:176–180. https://doi.org/10.1038/nature10325
Dubilier N, Bergin C, Lott C (2008) Symbiotic diversity in marine animals: the art of harnessing chemosynthesis. Nat. Rev. Microbiol. 6:725–740. https://doi.org/10.1038/nrmicro1992
Won YJ, Hallam SJ, O'Mullan GD, Pan IL, Buck KR, Vrijenhoek RC (2003) Environmental acquisition of thiotrophic endosymbionts by deep-sea mussels of the genus bathymodiolus. Appl Environ Microb 69:6785–6792. https://doi.org/10.1128/AEM.69.11.6785-6792.2003
Egas C, Pinheiro M, Gomes P, Barroso C, Bettencourt R (2012) The transcriptome of Bathymodiolus azoricus gill reveals expression of genes from endosymbionts and free-living deep-sea bacteria. Marine drugs 10:1765–1783. https://doi.org/10.3390/md10081765
Ponnudurai R, Kleiner M, Sayavedra L, Petersen JM, Moche M, Otto A, Becher D, Takeuchi T, Satoh N, Dubilier N, Schweder T, Markert S (2017) Metabolic and physiological interdependencies in the Bathymodiolus azoricus symbiosis. Isme J 11:463–477. https://doi.org/10.1038/ismej.2016.124
Wentrup C, Wendeberg A, Schimak M, Borowski C, Dubilier N (2014) Forever competent: deep-sea bivalves are colonized by their chemosynthetic symbionts throughout their lifetime. Environ. Microbiol. 16:3699–3713. https://doi.org/10.1111/1462-2920.12597
Kono T, Mehrotra S, Endo C, Kizu N, Matusda M, Kimura H, Mizohata E, Inoue T, Hasunuma T, Yokota A, Matsumura H, Ashida H (2017) A RuBisCO-mediated carbon metabolic pathway in methanogenic archaea 8:14007. https://doi.org/10.1038/ncomms14007
Hugler M, Wirsen CO, Fuchs G, Taylor CD, Sievert SM (2005) Evidence for autotrophic CO2 fixation via the reductive tricarboxylic acid cycle by members of the epsilon subdivision of proteobacteria. J. Bacteriol. 187:3020–3027. https://doi.org/10.1128/Jb.187.9.3020-3027.2005
King GM, Weber CF (2007) Distribution, diversity and ecology of aerobic CO-oxidizing bacteria. Nat. Rev. Microbiol. 5:107–118. https://doi.org/10.1038/nrmicro1595
Wu M, Ren QH, Durkin AS, Daugherty SC, Brinkac LM, Dodson RJ, Madupu R, Sullivan SA, Kolonay JF, Nelson WC, Tallon LJ, Jones KM, Ulrich LE, Gonzalez JM, Zhulin IB, Robb FT, Eisen JA (2005) Life in hot carbon monoxide: the complete genome sequence of Carboxydothermus hydrogenoformans Z-2901. PLoS Genet. 1:563–574. https://doi.org/10.1371/journal.pgen.0010065
Fuchs G (1994) Variations of the acetyl-CoA pathway in diversely related microorganisms that are not acetogens. In: Drake HL (ed) Acetogenesis. Springer US, Boston, MA, pp 507–520. https://doi.org/10.1007/978-1-4615-1777-1_19
Jansen K, Thauer RK, Widdel F, Fuchs G (1984) Carbon assimilation pathways in sulfate reducing bacteria. Formate, carbon dioxide, carbon monoxide, and acetate assimilation by Desulfovibrio baarsii. Arch. Microbiol. 138:257–262. https://doi.org/10.1007/bf00402132
Zarzycki J, Brecht V, Muller M, Fuchs G (2009) Identifying the missing steps of the autotrophic 3-hydroxypropionate CO2 fixation cycle in Chloroflexus aurantiacus. PNAS 106:21317–21322. https://doi.org/10.1073/pnas.0908356106
Herbold CW, Lebedeva E, Palatinszky M, Wagner M (2015) Candidatus Nitrosotenuis. Bergey's manual of systematics of archaea and bacteria. John Wiley & Sons, Ltd. https://doi.org/10.1002/9781118960608.gbm01291
Reinthaler T, van Aken HM, Herndl GJ (2010) Major contribution of autotrophy to microbial carbon cycling in the deep North Atlantic's interior. Deep-Sea Res Pt Ii 57:1572–1580. https://doi.org/10.1016/j.dsr2.2010.02.023
Anantharaman K, Breier JA, Sheik CS, Dick GJ (2013) Evidence for hydrogen oxidation and metabolic plasticity in widespread deep-sea sulfur-oxidizing bacteria. PNAS 110:330–335. https://doi.org/10.1073/pnas.1215340110
Friedrich CG, Bardischewsky F, Rother D, Quentmeier A, Fischer J (2005) Prokaryotic sulfur oxidation. Curr. Opin. Microbiol. 8:253–259. https://doi.org/10.1016/j.mib.2005.04.005
Yamamoto M, Takai K (2011) Sulfur metabolisms in epsilon- and gamma-Proteobacteria in deep-sea hydrothermal fields. Front Microbiol 2:192. https://doi.org/10.3389/Fmicb.2011.00192
Jormakka M, Yokoyama K, Yano T, Tamakoshi M, Akimoto S, Shimamura T, Curmi P, Iwata S (2008) Molecular mechanism of energy conservation in polysulfide respiration. Nat. Struct. Mol. Biol. 15:730–737. https://doi.org/10.1038/nsmb.1434
Nakagawa S, Takaki Y, Shimamura S, Reysenbach A-L, Takai K, Horikoshi K (2007) Deep-sea vent ε-proteobacterial genomes provide insights into emergence of pathogens. PNAS 104:12146–12150. https://doi.org/10.1073/pnas.0700687104
Nakagawa S, Takai K, Inagaki F, Hirayama H, Nunoura T, Horikoshi K, Sako Y (2005) Distribution, phylogenetic diversity and physiological characteristics of epsilon-Proteobacteria in a deep-sea hydrothermal field. Environ. Microbiol. 7:1619–1632. https://doi.org/10.1111/j.1462-2920.2005.00856.x
Inagaki F, Takai K, Kobayashi H, Nealson KH, Horikoshi K (2003) Sulfurimonas autotrophica gen. nov., sp. nov., a novel sulfur-oxidizing epsilon-proteobacterium isolated from hydrothermal sediments in the Mid-Okinawa Trough. Int. J. Syst. Evol. Microbiol. 53:1801–1805. https://doi.org/10.1099/ijs.0.02682-0
Xie W, Wang F, Guo L, Chen Z, Sievert SM, Meng J, Huang G, Li Y, Yan Q, Wu S, Wang X, Chen S, He G, Xiao X, Xu A (2011) Comparative metagenomics of microbial communities inhabiting deep-sea hydrothermal vent chimneys with contrasting chemistries. Isme J 5:414–426. https://doi.org/10.1038/ismej.2010.144
Meyer B, Kuever J (2007) Molecular analysis of the distribution and phylogeny of dissimilatory adenosine-5′-phosphosulfate reductase-encoding genes (aprBA) among sulfuroxidizing prokaryotes. Microbiology-Sgm 153:3478–3498. https://doi.org/10.1099/mic.0.2007/008250-0
Lam P, Cowen JP, Jones RD (2004) Autotrophic ammonia oxidation in a deep-sea hydrothermal plume. FEMS Microbiol. Ecol. 47:191–206. https://doi.org/10.1016/S0168-6496(03)00256-3
Stein LY (2011) Heterotrophic nitrification and nitrifier denitrification. Nitrification. American Society of Microbiology:95-114. https://doi.org/10.1128/9781555817145.ch5
Inagaki F, Takai K, Nealson KH, Horikoshi K (2004) Sulfurovum lithotrophicum gen. nov., sp. nov., a novel sulfur-oxidizing chemolithoautotroph within the ε-Proteobacteria isolated from Okinawa Trough hydrothermal sediments. Int. J. Syst. Evol. Microbiol. 54:1477–1482. https://doi.org/10.1099/ijs.0.03042-0
Takai K, Suzuki M, Nakagawa S, Miyazaki M, Suzuki Y, Inagaki F, Horikoshi K (2006) Sulfurimonas paralvinellae sp. nov., a novel mesophilic, hydrogen-and sulfur-oxidizing chemolithoautotroph within the Epsilonproteobacteria isolated from a deep-sea hydrothermal vent polychaete nest, reclassification of Thiomicrospira denitrificans as Sulfurimonas denitrificans comb. nov. and emended description of the genus Sulfurimonas. Int. J. Syst. Evol. Microbiol. 56:1725–1733. https://doi.org/10.1099/ijs.0.64255-0
Nakagawa S, Takai K, Inagaki F, Horikoshi K, Sako Y (2005) Nitratiruptor tergarcus gen. nov., sp. nov. and nitratifractor salsuginis gen. nov., sp. nov., nitrate-reducing chemolithoautotrophs of the epsilon-Proteobacteria isolated from a deep-sea hydrothermal system in the Mid-Okinawa Trough. Int. J. Syst. Evol. Microbiol. 55:925–933. https://doi.org/10.1099/ijs.0.63480-0
Vetriani C, Voordeckers JW, Crespo-Medina M, O'Brien CE, Giovannelli D, Lutz RA (2014) Deep-sea hydrothermal vent Epsilonproteobacteria encode a conserved and widespread nitrate reduction pathway (Nap). Isme J 8:1510–1521. https://doi.org/10.1038/ismej.2013.246
Byrne N, Strous M, Crepeau V, Kartal B, Birrien JL, Schmid M, Lesongeur F, Schouten S, Jaeschke A, Jetten M, Prieur D, Godfroy A (2009) Presence and activity of anaerobic ammonium-oxidizing bacteria at deep-sea hydrothermal vents. Isme J 3:117–123. https://doi.org/10.1038/ismej.2008.72
Lücker S, Nowka B, Rattei T, Spieck E, Daims H (2013) The genome of Nitrospina gracilis illuminates the metabolism and evolution of the major marine nitrite oxidizer. Front Microbiol 4:27. https://doi.org/10.3389/fmicb.2013.00027
Baker BJ, Sheik CS, Taylor CA, Jain S, Bhasi A, Cavalcoli JD, Dick GJ (2013) Community transcriptomic assembly reveals microbes that contribute to deep-sea carbon and nitrogen cycling. Isme J 7:1962. https://doi.org/10.1038/ismej.2013.85
Mehta MP, Butterfield DA, Baross JA (2003) Phylogenetic diversity of nitrogenase (nifH) genes in deep-sea and hydrothermal vent environments of the Juan de Fuca Ridge. Appl Environ Microb 69:960–970. https://doi.org/10.1128/AEM.69.2.960-970.2003
Voss M, Bange HW, Dippner JW, Middelburg JJ, Montoya JP, Ward B (2013) The marine nitrogen cycle: recent discoveries, uncertainties and the potential relevance of climate change. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 368:20130121. https://doi.org/10.1098/rstb.2013.0121
Douville E, Charlou J, Oelkers E, Bienvenu P, Colon CJ, Donval J, Fouquet Y, Prieur D, Appriou P (2002) The rainbow vent fluids (36 14′ N, MAR): the influence of ultramafic rocks and phase separation on trace metal content in Mid-Atlantic Ridge hydrothermal fluids. Chem. Geol. 184:37–48
Marcon Y, Sahling H, Borowski C, Ferreira CD, Thal J, Bohrmann G (2013) Megafaunal distribution and assessment of total methane and sulfide consumption by mussel beds at Menez Gwen hydrothermal vent, based on geo-referenced photomosaics. Deep-Sea Res Pt I 75:93–109. https://doi.org/10.1016/j.dsr.2013.01.008
Acknowledgements
We are thankful to the scientific parties of the BioBaz 2013 cruise, in particular to the crew members of the RV “Pourquoi Pas?,” the ROV “Victor6000” team (Ifremer, France) for their assistance in obtaining the sediment samples, Eva Martins and Cátia Cardoso for sample handling on board, Tomás Melo for map and figure design, and Valentina Costa and Diogo Pinho for all technical assistance in the laboratory and with data sequencing. We also acknowledge IMAR-Centre management unit of the University of the Azores. This study was supported by the “Direção Regional da Ciência e Tecnologia” (DRCT) (TC doctoral grant—M3.1.2/F/052/2011) and “Fundação para a Ciência e Tecnologia” (FCT), through the strategic project UID/MAR/04292/2013 granted to MARE.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
ESM 1
(DOCX 28 kb)
Rights and permissions
About this article

Cite this article
Cerqueira, T., Barroso, C., Froufe, H. et al. Metagenomic Signatures of Microbial Communities in Deep-Sea Hydrothermal Sediments of Azores Vent Fields. Microb Ecol 76, 387–403 (2018). https://doi.org/10.1007/s00248-018-1144-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-018-1144-x